

Abstract
Treatment of intrauterine infection is likely key to preventing a significant proportion of preterm deliveries before 32 weeks of gestation. Azithromycin (AZ) may be an effective antimicrobial in pregnancy; however, few gestation age-approriate data are available to inform the design of AZ-based treatment regimens in early pregnancy. We aimed to determine whether a single intra-amniotic AZ dose or repeated maternal intravenous (i.v.) AZ doses would safely yield therapeutic levels of AZ in an 80-day-gestation (term is 150 days) ovine fetus. Fifty sheep carrying single pregnancies at 80 days gestation were randomized to receive either: (i) a single intra-amniotic AZ administration or (ii) maternal intravenous AZ administration every 12 h. Amniotic fluid, maternal plasma, and fetal AZ concentrations were determined over a 5-day treatment regimen. Markers of liver injury and amniotic fluid inflammation were measured to assess fetal injury in response to drug exposure. A single intra-amniotic administration yielded significant AZ accumulation in the amniotic fluid and fetal lung. In contrast, repeated maternal intravenous administrations achieved high levels of AZ accumulation in the fetal lung and liver and a statistically significant increase in the fetal plasma drug concentration at 120 h. There was no evidence of fetal injury in response to drug exposure. These data suggest that (i) repeated maternal i.v. AZ dosing yields substantial fetal tissue uptake, although fetal plasma drug levels remain low; (ii) transfer of AZ from the amniotic fluid is less than transplacental transfer; and (iii) exposure to high concentrations of AZ did not elicit overt changes in fetal white blood cell counts, amniotic fluid monocyte chemoattractant protein 1 concentrations, or hepatotoxicity, all consistent with an absence of fetal injury.
INTRODUCTION
Preterm birth (PTB) (delivery before 37 weeks of completed gestation) remains a leading cause of neonatal death and disease (1, 2). Intrauterine infection is a primary cause of high-risk deliveries prior to 32 weeks of gestation (3), and a substantial body of clinical data demonstrate an inverse relationship between the incidence of intrauterine infection (2, 4, 5) and histologic chorioamnionitis (6, 7) and the gestational age at delivery. PTB is estimated to account for about 10% of total child health care expenditure in the United States (8, 9). Accordingly, developing effective antimicrobial treatments for intrauterine infection is likely a key step in preventing a significant proportion of PTBs.
Antibiotics are commonly used in pregnancy; however, their application is not without controversy. There is a wealth of conflicting data regarding the efficacy of antibiotics in treating bacterial vaginosis in an attempt to prevent PTB (for a review, see reference 10). A recent Cochrane review of 21 trials (7,847 women) assessing the efficacy of such treatments concluded that although antibiotic therapy was effective in resolving bacterial vaginosis and reduced the risk of late miscarriage, it did not reduce the risk of PTB or preterm prelabor rupture of membranes (PPROM) (11). Data from the ORACLE I trial demonstrated that, relative to placebo, erythromycin treatment of women with PPROM resulted in an improvement in composite neonatal outcomes and prolonged pregnancy but no overall reduction in PTB (12). In contrast, the authors of the ORACLE II trial of erythromycin treatment for women with spontaneous preterm labor concluded that antibiotics should not be routinely prescribed without evidence of clinical infection (13). These data suggest that the use of antibiotics in pregnancy should take into account the clinical presentation, the target organisms, and the physiological changes that result from pregnancy, as well as a host of factors, including ethnicity, body weight, gestational age, and drug coadministration (14).
Azithromycin (AZ) is a dibasic azalide class macrolide antibiotic commonly prescribed in pregnancy for treatment of chlamydia (15), scrub typhus (16), Q fever (17), and malaria (18) (in combination with chloroquinone). AZ is also active against the Ureaplasma spp. (19), which are frequently identified in cases of chorioamnionitis (20). AZ has a number of pharmacokinetic properties that make it well suited to use in pregnancy infections (21); it has an extended tissue half-life (∼68 h) and has been shown to accumulate in leukocytes, potentially amplifying its efficacy in treating infection (21). AZ has an excellent safety profile; reports of hepatotoxicity following AZ administration (22) are uncommon, its use is not associated with fetal abnormalities or malformations (23, 24), and it is well tolerated in patients with compromised renal function (21). A recent population level study in Denmark concluded that the small absolute risk of tachyarrhythmia derived from potential Q-T period elongation in high-risk individuals (25) is not significant in young and middle-aged adults (i.e., the normal pregnant population) (26).
Estimates suggest that 2% of U.S. women are prescribed AZ in the first trimester of pregnancy (27); in one cohort of U.S. women with a diagnosis associated with PTB, 46.1% were prescribed antimicrobial agents (28). However, there is a lack of data describing the maternal-fetal drug transfer and the safety of AZ in early pregnancy. Previous data from pregnant sheep in late pregnancy suggest low rates of maternal-fetal and amniotic fluid (AF)-fetal AZ transfer (29). In early to midpregnancy, the fetal skin is structurally immature (30), and as such, may pose less of a barrier to AZ uptake from the AF. We hypothesized that the structurally immature second trimester fetal skin would allow greater fetal AZ uptake from the amniotic fluid than is observed later in pregnancy.
In the present study, we used a sheep model of second trimester pregnancy to determine maternal plasma (MP), fetal plasma (from cord arterial blood) (FP), AF, cerebrospinal fluid (CSF), fetal liver, and fetal lung azithromycin concentrations resulting from single intra-amniotic (IA) or repeated maternal i.v. AZ administrations. We assessed the fetal uptake achieved by the two treatment regimens against MIC data for the treatment of Ureaplasma spp., which are commonly isolated from the AF and chorioamnion in cases of PPROM and PTB (31) and are associated with a host of neonatal morbidities, including bronchopulmonary dysplasia (32). Serum markers of liver injury, white blood cell counts and AF monocyte chemoattractant protein 1 (MCP-1) concentrations, were also measured over the 5-day regimen to assess potential fetal injury in response to drug exposure.
MATERIALS AND METHODS
Animals.
All animal work was reviewed and approved by the University of Western Australia's Animal Ethics Committee. Fifty sheep, each carrying a single fetus at 80 days gestational age (GA), were randomized to receive either (i) a single IA injection of 6.4 mg AZ (Zithromax i.v.; Pfizer, New York, NY) under ultrasound guidance using a Philips CX-50 ultrasound system (Philips Healthcare, Andover, MA) (n = 25) or (ii) repeated maternal intravenous (i.v.) AZ (Pfizer) doses, 10 mg/kg maternal body weight every 12 h as a bolus over 5 min (n = 25). The bolus AZ concentration was 13.5 mg/ml in a mean volume of 43.1 ± 3.7 ml. Sterile saline was used as a vehicle in all animals. All the animals underwent amniocentesis only twice, in order to minimize the confounding risk from infection and/or fetal loss. As the early-GA fetuses in this study were not able to be chronically catheterized, the animals in each treatment group were euthanized with an intravenous bolus of pentobarbital after either (i) 4 h (with AF and MP only collected at 2 h), (ii) 12 h (with AF and MP only collected at 6 h), (iii) 36 h (with AF and MP only collected at 24 h), (iv) 72 h (with AF and MP only collected at 48 h), or (v) 120 h (with AF and MP only collected at 96 h) (n = 5/time point for each group). Arterial cord blood, FP, CSF, AF, MP, fetal lung, and fetal liver tissues were collected at autopsy. Arterial cord blood pH, partial O2 (pO2), and pCO2 values were determined using a Siemens Rapidlab 1265 blood gas analyzer (Siemens, Victoria, Australia). The peak maternal-fetal AZ transfer time after repeated dosing was unknown at the time of the study. As such, sampling after 12 h of treatment in the repeated maternal intravenous administration group was performed immediately before the next scheduled AZ dose in order to yield stable minimum (trough) AZ concentrations. Fetuses were weighed, scored for the presence of meconium, and sexed at autopsy.
Hepatotoxicity analysis.
Fetal arterial cord blood for liver function tests (aspartate aminotransferase [AST], gamma glutamyl transpeptidase [GGT], glutamate dehydrogenase [GLDH], albumin [ALB], and total bilirubin [TB]) was collected in a 10-ml SST clot-activating Vacutainer (BD, Franklin Lakes, NJ). Analyses were performed by Vetpath (Perth, Western Australia [WA], Australia).
Inflammatory analysis.
Fetal arterial cord blood for leukocyte analysis (complete and differential counts) was collected in a 10-ml Vacutainer (BD) containing EDTA. Analyses were performed by Vetpath (Perth, WA, Australia). AF MCP-1 concentrations, a marker of intrauterine inflammation in sheep, were measured using an MCP-1 ELISA VetSet kit (VS0083B-002; Kingfisher Biotech, Inc., Saint Paul, MN), with washing performed on a Biosan plate washer (Intelliwasher 3D-IW8; Biosan, Riga, Latvia). Standards (calibration curve, R2 > 0.98) were assayed in triplicate (average coefficient of variation [CV], 7%), and unknown samples were assayed in duplicate. The assay limit of detection was <4 pg/ml. One hundred microliters of each standard or sample was incubated overnight (16 h) at 4°C before the assay was completed following the manufacturer's instructions, with absorbance at 450 nm read on an Anthos 2010 microplate reader (Biochrom Ltd., Cambridge, United Kingdom).
Azithromycin quantification.
Fluid samples (MP, AF, and CSF) were processed for analysis as described previously (29). Briefly, AZ standards were constructed in saline as a series of seven 10-fold dilutions, from 20,000 ng/ml to 0.2 ng/ml. One hundred microliters of thoroughly mixed unknown samples or standard was added to 320 μl of liquid chromatography-mass spectrometry (LC-MS) grade methanol containing 50 ng roxithromycin (R4393; Sigma-Aldrich, St. Louis, MO) as an internal standard. Samples were vortexed for 20 s and centrifuged at 1,700 × g for 20 min at 24°C in an Eppendorf 5810R benchtop centrifuge (Eppendorf, Hamburg, Germany). The supernatant was transferred to a glass tube and dried for 3 h under nitrogen at room temperature. The precipitate was resuspended by vortexing for 1 min in 100 μl of LC-MS grade methanol before being cleared in a 0.22-μm spin-X cellulose-acetate centrifuge filter tube (CLS-8160; Sigma-Aldrich) in accordance with the manufacturer's instructions. Assay controls were constructed by spiking control (no AZ exposure) AF and MP samples with 50 ng roxithromycin and AZ to give a final AZ concentration of either 5,000 ng/ml or 50 ng/ml. Assay controls were processed at the beginning and end of each sample preparation run. AZ solution (0.5 μl; 20 μg/ml) and roxithromycin solution (0.5 μl; 2.5 μg/ml) were injected at the beginning of each LC-MS run and used to confirm peak transitions. The interassay CV for pooled 5,000-ng/ml assay control samples was determined to be 9.2% (n = 6). The CV for pooled 50-ng/ml assay control samples was determined to be 16.4% (n = 7). The extraction efficiencies for pooled 5,000- and 50-ng/ml assay control samples were 88.8% (n = 6) and 74.6% (n = 7), respectively. The limit of detection for fluid samples was determined to be <2 ng/ml by constructing a one-sided 95% confidence interval from the mean value of blank values based on the method described by Burd (33). Samples measured at less than the limit of AZ detection were assigned an arbitrary concentration value of 0 ng/ml.
Tissue samples were processed for analysis based on the method described by Hunter and colleagues (34). The tissue samples were defrosted on wet ice, and 50 ng roxithromycin and 100 mg of tissue were added to a Precellys MK28-R lysing tube (Bertin Corp., Rockville, MD) containing LC-MS grade acetonitrile (Honeywell-Burdick and Jackson, Morristown, NJ) to a final volume of 1,120 μl. The tissue was homogenized using two cycles of 6,500 rpm for 30 s. Samples were incubated at 50°C for 1 h before being centrifuged at 1,700 × g in an Eppendorf 5810R benchtop centrifuge for 20 min at 24°C. Supernatant (700 μl) was transferred into a glass tube and dried under nitrogen for 3 h. The precipitate was resuspended in 100 μl of acetonitrile at 50°C, vortexed for 1 min, and cleared in a 0.22-μm spin-X cellulose-acetate centrifuge filter tube (Sigma-Aldrich) in accordance with the manufacturer's instructions. AZ standards were constructed in saline as a series of seven 10-fold dilutions from 20,000 ng/ml to 0.2 ng/ml. One hundred microliters of thoroughly mixed standard was added to 1,020 μl LC-MS grade acetonitrile containing 50 ng roxithromycin and 100 mg control (no AZ exposure) tissue and processed as described above. AZ standards were run in triplicate for fetal lung and fetal liver analyses. AZ solution (0.5 μl; 20,000 ng/ml) and roxithromycin solution (0.5 μl; 2.5 μg/ml) were injected at the beginning of each LC-MS run and used to confirm peak transitions. The extraction efficiencies for the 20,000-ng/ml and 2,000-ng/ml lung AZ standards were 99% (n = 6) and 96% (n = 6), respectively. The extraction efficiencies for the 20,000-ng/ml and 2,000-ng/ml liver AZ standards were 99% (n = 5) and 99% (n = 3), respectively. The CVs for the 20,000-ng/ml and 2,000-ng/ml lung AZ standards were 90% (n = 6) and 84% (n = 6), respectively. The CVs for the 20,000-ng/ml and 2,000-ng/ml liver AZ standards were 89% (n = 5) and 80% (n = 3), respectively. The limit of detection for both liver and lung samples was determined to be <200 ng/mg by constructing a one-sided 95% confidence interval from the mean blank values based on the method described by Burd (33). Samples measured at less than the limit of AZ detection were assigned an arbitrary concentration value of 0 ng/ml. The concentrations of the extracts were back-calculated to derive levels in tissues as nanograms per gram tissue.
Separation was performed on a Kinetex (Phenomenex, Torrance, CA) 2.6-μm, 150- by 3.0-mm C18 column. For the solvents, “A” was water (Optima; ThermoFisher Scientific, Waltham, MA) plus 0.1% formic acid and “B” was acetonitrile plus 0.1% formic acid (Honeywell-Burdick and Jackson). Both solvents were LC-MS grade. The flow rate was 0.3 ml/min with the following gradient: 0 min at 0% B, 7 min at 75% B, 8 min at 100% B, 9 min at 100% B, 9.3 min at 0% B, and 12 min at 0% B. The total run time was 12 min. The LC-MS system was an Agilent 1290 UPLC coupled to an Agilent 6460 triple-quadrupole mass spectrometer (Agilent Technologies Inc., Santa Clara, CA). The mass spectrometer was operated under the following conditions: gas temperature, 250°C; gas flow, 8 ml/min; nebulizer, 35 lb/in2; sheath gas temperature, 300°C; sheath gas flow, 10 liters/min; capillary, 4,000 V, The instrument was operated in positive mode. The transitions monitored were 749.5 to 591.4 for AZ and 837.5 to 679.0 for roxithromycin, and in both cases, the collision energy was 15. Extracted sample (0.1 μl) was injected onto the LC-MS. The data for unknown, standard, and assay control samples (both AZ and roxithromycin peak area values) were blank corrected. The AZ peak value was then divided by the roxithromycin internal-standard peak value for that sample. Standard sample ratio values were fitted to calibration curves (R2 > 0.98), which were used to determine the concentrations of unknown samples.
Pharmacokinetic analysis of amniotic fluid AZ data.
Means and standard deviations (SD) of AZ concentrations from each time point were calculated. AF AZ values from the single IA AZ administration group were subjected to pharmacokinetic analysis using PKSolver (35). Model selection (noncompartmental analysis or one- or two-compartment analysis) was based on the calculation of the delta second-order Akaike's information criterion (ΔAIC) (36). A two-compartment analysis was found to best describe the peak AF data (0 to 120 h) from the single IA AZ administration group. Fetal lung and liver (ng/g)/plasma (ng/ml) AZ concentration ratios were calculated for FP, MP, and AF values.
Statistics.
All values represent means and SD. Analyses were performed using IBM SPSS Statistics for Windows, version 20.0 (IBM Corporation, Armonk, NY). Data were assessed for normality with Shapiro-Wilk tests. Putative outliers were assessed with Dixon's Q test. Nonparametric data were subjected to log10 transformation before distributions were reanalyzed. Mean differences between groups were tested for significance with univariate analysis of variance (ANOVA). A P value of ≤0.05 was accepted as significant. Homogeneity of variances was assessed using Levene's test and by plotting standardized residuals against a normal distribution curve. Interactions and main effects were assessed for treatment (single IA versus repeated maternal i.v. AZ), time (post-first dose), and treatment time.
RESULTS
Delivery data.
Forty-nine of 50 fetuses survived their protocols. One fetal death occurred after 60 h of repeated maternal i.v. AZ treatment. No abnormalities were identified in any of the other 9 animals, which received 60 h or more of repeated maternal i.v. AZ treatment. There was little difference in delivery weights across treatment regimens or between treatment groups (Table 1). Small but statistically significant differences in arterial cord blood pH, pO2, and pCO2 were observed in the IA AZ group. Only one animal had light meconium staining, possibly suggesting fetal distress.
TABLE 1.
Fetal delivery data
| AZ route | Autopsy time (h) group | % Male fetuses | Delivery wt (kg) | Arterial cord blood |
No. with meconium/total | ||
|---|---|---|---|---|---|---|---|
| pH | pO2 | pCO2 | |||||
| Single IA | 4 (n = 5) | 60 | 0.33 ± 0.05 | 7.3 ± 0.05 | 6.2 ± 0.7 | 54.6 ± 3.7 | 0/5 |
| 12 (n = 5) | 0 | 0.36 ± 0.10 | 7.4 ± 0.04a | 7.4 ± 1.3a | 52.2 ± 3.8a | 0/5 | |
| 36 (n = 5) | 40 | 0.32 ± 0.04 | 7.3 ± 0.06 | 8.9 ± 1.5a | 58.0 ± 7.7 | 0/5 | |
| 72 (n = 5) | 40 | 0.34 ± 0.03 | 7.3 ± 0.04 | 6.8 ± 2.3 | 56.2 ± 4.2 | 0/5 | |
| 120 (n = 5) | 80 | 0.43 ± 0.06 | 7.3 ± 0.04 | 7.5 ± 1.0a | 61.0 ± 3.3 | 0/5 | |
| 12-h maternal i.v. | 4 (n = 5) | 40 | 0.33 ± 0.03 | 7.3 ± 0.05 | 6.8 ± 0.8 | 58.0 ± 6.4 | 0/5 |
| 12 (n = 5) | 40 | 0.31 ± 0.01 | 7.3 ± 0.08 | 4.6 ± 0.2 | 63.0 ± 11.0 | 1/5 | |
| 36 (n = 5) | 20 | 0.35 ± 0.02 | 7.3 ± 0.06 | 5.4 ± 0.4 | 59.4 ± 6.6 | 0/5 | |
| 72 (n = 5) | 60 | 0.34 ± 0.05 | 7.3 ± 0.04 | 6.9 ± 1.2 | 62.3 ± 6.6 | 0/5 | |
| 120 (n = 4) | 66b | 0.42 ± 0.04 | 7.3 ± 0.05 | 5.1 ± 2.2 | 64.0 ± 7.9 | 0/4 | |
Significant difference (P < 0.05) between time groups.
Only 3 of the 4 fetuses in this group were sexed at autopsy.
Inflammation data.
Complete and differential blood counts remained broadly constant across the duration of individual treatments and between the two treatment groups. Analysis of fetal arterial cord blood neutrophil concentrations demonstrated significant differences between treatment times (P = 0.029) and treatment groups (P = 0.027) but no interaction between the two variables (P = 0.351). Pairwise comparisons demonstrated a significant difference (5.3 × 107 ± 4 × 107 versus 14.4 × 107 ± 10 × 107; P = 0.029) in the neutrophil concentrations of fetal arterial cord blood at 120 h after treatment initiation between the single IA AZ and repeated maternal i.v. AZ groups, respectively. The fetal arterial cord blood neutrophil concentration was also significantly increased (14.4 × 107 ± 10 × 107 versus 5.4 × 107 ± 3.8 × 107; P = 0.031) at 120 h relative to the concentration at 36 h in the repeated maternal i.v. AZ group. No other significant differences were identified between fetal arterial cord blood lymphocyte, monocyte, or eosinophil concentrations at any time point in either treatment group (Fig. 1). No MCP-1 protein was detected in the AF of animals in either the single IA or repeated maternal i.v. AZ group.
FIG 1.
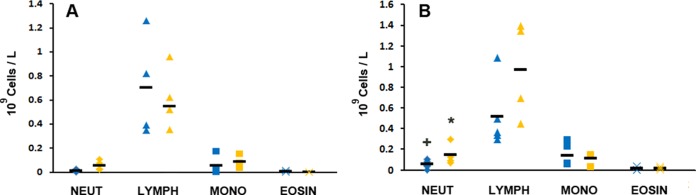
Concentrations of leukocytes in fetal arterial cord blood at 36 h (A) and 120 h (B). Blue symbols, single IA AZ administration; orange symbols, repeated maternal i.v. AZ administrations. The horizontal bars represent group means. *, significant difference (P < 0.05) in neutrophil concentrations in fetal arterial cord blood at 120 h after treatment initiation between the single IA AZ and repeated maternal i.v. AZ groups; +, significant difference (P < 0.05) in the neutrophil concentration at 120 h relative to the concentration at 36 h in the repeated maternal i.v. AZ group. NEUT, neutrophils; LYMPH, lymphocytes; MONO, monocytes; EOSIN, eosinophils.
Hepatotoxicity data.
Little difference was observed in the concentrations of selected liver injury markers across the duration of treatments and between the two treatment groups. Analysis of markers of hepatotoxicity (Fig. 2) in fetal arterial cord blood demonstrated a statistically significant difference between treatment times for AST (P = 0.002) and a significant interaction between treatment group and treatment time for AST (P = 0.013), GGT (P = 0.012), and TB (P = 0.016). Pairwise comparisons demonstrated small but significant increases in AST at 12 h (30.8 ± 5.1 versus 17.2 ± 2.3 U/liter; P = 0.002), GGT at 4 h (7.4 ± 1.5 versus 3.4 ± 0.6 U/liter; P = 0.004), and TB at 4 h (12.0 ± 3.0 versus 7.0 ± 3.1 μmol/liter; P = 0.013) and 36 h (7.0 ± 4.0 versus 11.2 ± 4.2 μmol/liter; P = 0.015) between single IA AZ and repeated maternal i.v. treatment groups, respectively. There was no overt liver pathology identified at autopsy, suggesting that the small differences identified in serum injury markers are unlikely to be of biological relevance.
FIG 2.

Concentrations of markers of liver injury in fetal arterial cord blood following single IA AZ (blue) and repeated maternal i.v. AZ (orange) administrations. *, significant differences (P < 0.05) were identified in AST at 12 h, GGT at 4 h, and TB at 4 h and 36 h between single IA AZ and repeated maternal i.v. treatment groups, respectively. The error bars indicate 1 SD.
Amniotic fluid azithromycin concentration.
The pharmacokinetic parameters for peak AF AZ concentrations (0 to 120 h) in the single IA AZ group are given in Table 2. Single IA AZ administration achieved high AF concentrations of AZ, with a Cmax (maximum concentration of drug in serum) of 25.6 μg/ml and a Tmax (time to maximum concentration of drug in serum) of 0.16 h (Fig. 3A). AF AZ concentrations remained above the AZ MIC for Ureaplasma spp. for at least 72 h. In contrast, repeated maternal i.v. AZ administration resulted in little initial AF AZ accumulation. After 24 h of treatment, an apparent accumulation of AZ in the AF was observed over the next 4 days of treatment to yield a trough value of 111.0 ± 13.1 ng/ml AZ at 120 h (Fig. 3A). Analysis of AZ concentrations demonstrated significant differences between treatment groups (P = 0.000) and treatment times (P = 0.000) and significant interaction (P = 0.000) between the two variables. Pairwise analysis demonstrated a statistically significant (P < 0.015) difference in AF AZ concentrations between treatment groups from 2 h to 24 h but no significant difference in AF AZ concentrations at later time points.
TABLE 2.
Amniotic fluid pharmacokinetic data following administration of a single intra-amniotic bolusa
| Parameterb | Unit | Value |
|---|---|---|
| Dose | mg | 6.40 |
| A | ng/ml | 24,611.34 |
| α | 1iter/h | 0.90 |
| B | ng/ml | 4,934.30 |
| β | 1iter/h | 0.03 |
| ka | 1iters/h | 21.30 |
| k10 | 1iter/h | 0.14 |
| k12 | 1iter/h | 0.61 |
| k21 | 1iter/h | 0.18 |
| t1/2α | h | 0.77 |
| t1/2β | h | 25.02 |
| t1/2ka | h | 0.032 |
| V/F | (mg)/(ng/ml) | 0.00,022 |
| CL/F | (mg)/(ng/ml)/h | 3.14E-05 |
| V2/F | (mg)/(ng/ml) | 0.00,077 |
| CL2/F | (mg)/(ng/ml)/h | 0.00,014 |
| Tmax | h | 0.16 |
| Cmax | μg/m · liter | 25.62 |
| AUC0–t | μg/ml · h | 198.0 |
| AUC0–∞ | μg/ml · h | 204.0 |
| AUMC | μg/ml · h2 | 6,460.50 |
| MRT | h | 31.70 |
Calculated from a two-compartment analysis using peak value data from 10 time points (2 to 120 h).
A, zero time intercept for α phase; α, distribution phase; B, zero time intercept for β phase; β, elimination phase; kα, first-order absorption rate constant; k10, first-order elimination rate constant; k12, first-order distribution rate constant; t1/2α, half-life at α phase; t1/2β, half-life at β phase; V/F, apparent volume of distribution; CL/F, apparent clearance; CL2/F, intercompartmental clearance; AUC0–t, area under the concentration-time curve from 0 h to t; AUC0–∞, area under the concentration-time curve from 0 h to infinity; AUMC, area under the first moment of the concentration-time curve; MRT, mean residence time.
FIG 3.

Concentrations of AZ in AF (A), MP (B), and FP (C) following single IA AZ (blue) and repeated maternal i.v. AZ (orange) administrations. *, significant differences (P < 0.05) in AZ concentrations between single IA AZ and repeated maternal i.v. AZ groups; +, significant differences (P < 0.05) between FP AZ concentrations in the repeated maternal i.v. AZ group at 120 h and the repeated maternal i.v. AZ group at 4 to 72 h. The error bars indicate SD.
Maternal plasma azithromycin concentration.
Assuming a MIC of 0.5 μg/ml against Ureaplasma spp. (19), single IA AZ administration did not achieve AZ concentrations of greater than or equal to the MIC in the MP at any time point (Fig. 3B). The highest MP AZ concentration achieved was 10.0 ± 11.4 ng/ml at 6 h (0.23% of a comparable AF AZ concentration), suggesting that the rate of transfer between the AF and MP was insufficient to meaningfully increase MP AZ levels at the time points analyzed. Repeated maternal i.v. AZ administrations resulted in transient therapeutic concentrations in the MP. All MP AZ concentrations after 12 h represent trough concentrations (12 h after the last i.v. dose, taken immediately before the next i.v. AZ administration) (Fig. 3B). Maternal i.v. AZ dosing every 12 h achieved AZ concentrations greater than or equal to the MIC in the MP at 2 h, 6 h, and 24 h and between 96 and 120 h of treatment. Trough concentrations of 558.9 ± 637.2 ng/ml at 96 h and 581.6 ± 389.7 ng/ml at 120 h (compared with a peak Cmax of 739.2 ± 683.6 ng/ml at 2 h) are suggestive of AZ accumulation over the treatment duration. Analysis of AZ concentrations demonstrated significant differences for treatment time (P = 0.013) and treatment group (P = 0.000) and an interaction (P = 0.013) between the two variables. Pairwise comparisons demonstrated a significant difference in MP AZ concentrations at 2 h to 120 h (P < 0.05) between single IA AZ and repeated maternal i.v. AZ administrations.
Fetal plasma azithromycin concentration.
Despite achieving high peak values in targeted compartments (AF/MP), FP AZ concentrations remained low in both treatment groups (Fig. 3C). The highest measured FP concentration in the single IA AZ administration group (36 h; 10.9 ± 11.5 ng/ml) was just 0.73% of the comparative AF AZ concentration. The highest measured FP concentration in the repeated maternal i.v. AZ administration group (120 h; 74.2 ± 26.8 ng/ml) was 13% of the comparative MP AZ concentration. No observed FP AZ concentration from either treatment group approached the MIC for Ureaplasma spp. Univariate analysis of AZ concentrations demonstrated statistically significant differences between treatment groups (P = 0.000) and treatment times (P = 0.000) and a significant interaction between the two variables (P = 0.000). Pairwise analysis demonstrated statistically significant differences in FP AZ concentrations at 72 h (7.3 ± 8.5 versus 46.2 ± 25.5 ng/ml; P = 0.000) and 120 h (2.0 ± 3.0 versus 74.2 ± 26.8 ng/ml; P = 0.000) between single IA AZ and repeated maternal i.v. AZ treatments, respectively. The FP AZ concentration at 120 h in the repeated maternal i.v. AZ group was significantly different from earlier FP AZ concentrations achieved at 4 h, 12 h, 36 h, and 72 h (P = 0.000).
Fetal cerebrospinal fluid azithromycin concentration.
Concentrations of AZ in the fetal CSF did not approach MIC levels at any observed time point for either treatment group. Data from 12 h and 72 h were not available for both groups and were excluded from the analysis. No AZ was detected in the fetal CSF concentration in the single IA AZ group. The highest observed value in the repeated maternal i.v. AZ group was 11.4 ± 9.4 ng/ml at 120 h.
Fetal lung azithromycin concentration.
Significant AZ accumulation was observed in fetal lung tissue from both treatment groups (Fig. 4A); the highest observed value in the single IA AZ treatment group was 1,086.0 ± 582.8 ng/g at 36 h, and the highest observed value in the repeated maternal i.v. AZ treatment group was 3,088.3 ± 556.1 ng/g at 72 h. Analysis of AZ concentrations demonstrated statistically significant differences between treatment groups (P = 0.000) and treatment times (P = 0.000) and a significant interaction between the two variables. Pairwise analysis demonstrated statistically significant differences in fetal lung AZ concentrations at 12 h (1,063.0 ± 252.2 ng/g versus 108.0 ± 125.3 ng/g; P = 0.000), 72 h (654.0 ± 259.5 ng/g versus 3,088.3 ± 556.1 ng/g; P = 0.000), and 120 h (390.4 ± 68.1 ng/g versus,2883.4 ± 793.8 ng/g; P = 0.000) between single IA AZ and repeated maternal i.v. AZ treatments, respectively. The ratios of the fetal lung AZ concentration to AF and FP (single IA AZ treatment animals) and AF, FP, and MP (repeated maternal i.v. AZ animals) concentrations are given in Table 3.
FIG 4.
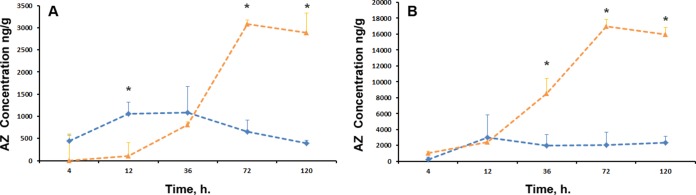
Concentrations of AZ in fetal lung (A) and liver (B) following single IA AZ (blue) and repeated maternal i.v. AZ (orange) administrations. *, significant difference (P < 0.05) in AZ concentrations between the single IA AZ and repeated maternal i.v. AZ groups. The error bars indicate SD.
TABLE 3.
Ratios of fetal lung AZ concentrations to AF, FP, and MP AZ concentrations
| Time (h) | Ratioa |
||||
|---|---|---|---|---|---|
| Single IA AZ |
Repeated maternal i.v. AZ |
||||
| AF | FP | AF | FP | MP | |
| 4 | 0.1 | 96.1 | 1.1 | ||
| 12 | 0.3 | 130.3 | 1.1 | ||
| 36 | 0.7 | 99.7 | 57.2 | 85.9 | 1.9 |
| 72 | 0.7 | 89.8 | 30.6 | 66.8 | 13.2 |
| 120 | 2.0 | 199.6 | 26.0 | 38.9 | 5.0 |
Fetal lung AZ concentrations (ng/g) and AF, FP, and MP AZ concentrations (ng/ml). The 36-, 72-, and 120-h values are minimum AZ concentrations for repeated maternal i.v. AZ animals.
Fetal liver azithromycin concentration.
As identified in the fetal lung, significant AZ accumulation was observed in fetal liver tissue from both treatment groups (Fig. 4B); the highest observed value in the IA AZ treatment group was 3,030.0 ± 2,813.0 ng/g at 12 h, and the highest observed value in the repeated maternal i.v. AZ treatment group was 16,969.0 ± 904.4 ng/g at 72 h. Analysis of AZ concentrations demonstrated statistically significant differences between treatment groups (P = 0.000) and treatment times (P = 0.000) and a significant interaction between the two variables. Pairwise analysis demonstrated statistically significant differences in fetal liver AZ concentrations at 36 h (2,004.0 ± 1,345.7 versus 8,554.0 ± 1,907.7 ng/g; P = 0.000), 72 h (2,067 ± 1,628.3 versus 16,969.0 ± 904.4 ng/g; P = 0.000), and 120 h (2,318.0 ± 814.0 versus 15,958.0 ± 848.6 ng/g; P = 0.000) between single IA AZ and repeated maternal i.v. AZ treatments, respectively. The ratios of the fetal liver AZ concentration to AF and FP concentrations are given in Table 4.
TABLE 4.
Ratios of fetal liver AZ concentrations to AF, FP, and MP AZ concentrations
| Time (h) | Ratioa |
||||
|---|---|---|---|---|---|
| Single IA AZ |
Repeated maternal i.v. AZ |
||||
| AF | FP | AF | FP | MP | |
| 4 | 0.1 | 52.3 | 96.3 | 5.5 | |
| 12 | 0.8 | 371.3 | 14.4 | ||
| 36 | 0.7 | 184.1 | 598.3 | 897.8 | 20.3 |
| 72 | 2.2 | 283.9 | 168.3 | 366.9 | 72.3 |
| 120 | 11.7 | 1,185.1 | 143.8 | 215.2 | 27.4 |
Fetal liver AZ concentrations (ng/g) and AF, FP, and MP AZ concentrations (ng/ml). The 36-, 72-, and 120-h values are minimum AZ concentrations for repeated maternal i.v. AZ animals.
DISCUSSION
Based on our data from 5 days of single IA AZ or repeated maternal i.v. AZ treatment, the primary findings of this study are (i) that the rate of AZ transfer from the AF to the fetus following a single IA AZ dose of 6.4 mg is insufficient to yield FP AZ levels greater than or equal to the MIC for Ureaplasma spp.; (ii) although evidence of FP accumulation emerged after 120 h of treatment, the rate of AZ transfer from the maternal circulation to the fetus is insufficient to yield fetal plasma AZ levels greater than or equal to the MIC for Ureaplasma spp.; (iii) significant and sustained accumulation of AZ was observed in the fetal lung and liver in the repeated maternal i.v. AZ treatment group and, to a lesser extent, in the single IA AZ treatment group; and (iv) the substantial accumulation of AZ in the fetal liver and lung in repeated maternal i.v. AZ animals suggests that AZ transfer across the immature second trimester fetal skin is limited relative to transplacental AZ transfer.
Intrauterine infection is most commonly identified in cases of PPROM and PTB prior to 32 weeks gestation (2, 6). Work in nonhuman primates by Novy et al. and Grigsby et al. have underscored the importance of Ureaplasma spp. in preterm birth (37) and demonstrated the potential utility of AZ for treatment of intrauterine infection in pregnancy (38). Current evidence suggests that polymicrobial (39) infections are common and that the Ureaplasma spp. are among the microorganisms most frequently identified in the AF (40). Given its ready availability, broad spectrum of activity, low cost, and limited adverse side effect profile, AZ is thus an attractive candidate antibiotic for use in early pregnancy. AZ's status as a generic drug also makes it an attractive option for developing and low-income countries, the nations that report some of the highest rates of preterm birth (1).
As such, a number of investigators have assessed the pharmacokinetics of AZ in gravid (29, 41) and nongravid (34, 42–44) animal models, as well as in humans (14, 45, 46). Although the MP pharmacokinetics of AZ in humans at term (45) and in the late second trimester (46) are well described, there remains a lack of data describing the fetal uptake of AZ in the second trimester of pregnancy. This lack of data, combined with debate as to whether AZ can cross the placenta in sufficient quantities to treat intra-amniotic infection (41), has hindered the development of optimal treatment regimens for common pregnancy pathogens, including the Ureaplasma spp. In the present study, we aimed to determine whether a single IA AZ dose or repeated maternal i.v. AZ doses would yield therapeutic levels for the Ureaplasma spp. in an 80-day GA ovine fetus.
Earlier in vitro studies using a noncirculating placental-perfusion model to simulate chronic oral drug treatment suggested a steady-state transplacental transfer rate of 2.6% (47). Noting that the Tmax for a single 0.5-g oral dose of AZ is 2.5 h, analysis of placental tissues and sera from 20 women with term pregnancies given 1 g oral AZ demonstrated AF/maternal-serum and fetal-serum/maternal-serum ratios of 49% and 6%, respectively, at 6 h (45). The AF AZ pharmacokinetic data from the single IA AZ treatment group in the present study were broadly in agreement with our earlier data from third trimester sheep pregnancies, albeit with a longer half-life (t1/2β) of 25.2 h and a mean residence time of 31.7 h (29). With the unsurprising exception of AF in the single IA AZ treatment group, AF and FP AZ levels remained low and well below calculated MIC breakpoints for the Ureaplasma spp. at all time points. However, there are strong data to suggest that both tissue and fluid AZ concentrations should, where possible, be taken into account when estimating maternal-fetal transfer and that MIC values determined in vitro may not accurately estimate drug efficacy in vivo.
AZ pharmacokinetics are characterized by rapid and extensive tissue accumulation in conjunction with low plasma concentrations (44, 48). Ramsey and colleagues have suggested the importance of determining fetal tissue AZ concentrations to assist in determining whether AZ should be recommended for use in treating perinatal infections (45). Davila and colleagues reported that tissue (including lung and liver) AZ concentrations were higher than serum concentrations in both rabbits and rats following oral dosing (49). Work by Girard et al. suggests that AZ is effective against localized infections by Gram-positive and Gram-negative bacteria in rodent models even when serum AZ concentrations are below the MIC (50). In light of this, Escudero and colleagues have noted that relying on serum AZ concentrations markedly underestimates therapeutic concentrations at the site of infection (48).
Recent work by Acosta et al. suggests that a similar pattern may hold true for AF AZ concentrations. Using a late-pregnancy macaque model, they used pharmacokinetic and pharmacodynamic data to determine that an AF AZ concentration of 39 ng/ml (well below the MIC of 0.5 μg/ml) was sufficient to eradicate 95% of the 107 CFU of the Ureaplasma parvum serovar 1 isolate used to establish intrauterine infection (41). Although the authors speculated that differences between the AF pH and protein binding may explain this discrepancy with in vitro broth microdilution MIC assays, it is equally possible that significant AZ tissue accumulation (potentially assisted by AZ's ability to concentrate in invading immunocytes [44]) also contributes to effective treatment at sub-MIC AF concentrations. Extrapolating from these data, therapeutic AZ concentrations for Acosta and colleagues' serovar 1 isolate of U. parvum would have been achieved in the AF and FP in repeated maternal i.v. AZ treatment animals in the present study by 48 h and 72 h, respectively. For the single IA AZ treatment group, predicted therapeutic concentrations of AZ in the AF would have been maintained across the 120-h study, although FP AZ concentrations would have failed to reach the estimated 39 ng/ml necessary to eradicate 95% of the inoculum.
The fetal liver and lung data in the present study suggest that significant accumulation in fetal tissue rapidly occurs in the second trimester in ovine pregnancy following maternal i.v. and, to a lesser extent, IA AZ administration. In keeping with previous studies (29, 45), accumulation in tissue was observed in tandem with low apparent AZ levels in the fetal plasma. In the present study, the concentrations of AZ in the fetal liver were between 52.3 and 1,185.1 times higher than fetal plasma AZ concentrations in single IA AZ treatment animals and between 215.2 and 897.8 times higher than fetal plasma concentrations in repeated maternal i.v. AZ treatment animals (Table 3). A similar pattern was identified in the fetal lung; concentrations of AZ were between 96.1 and 199.6 times higher than plasma AZ concentrations in single IA AZ treatment animals and between 38.9 and 85.9 times higher than plasma AZ concentrations in repeated maternal i.v. AZ treatment animals (Table 4). Similar values have been reported by other investigators. For example, Escudero and colleagues (48) cited work by Davila et al. (49), noting that “tissue/serum ratios in liver, kidney, lung and ileum of 1381, 1171, 1261 and 1682, respectively, have been reported in rats after a single (200 mg/kg) oral administration of azithromycin.” Similarly, Foulds and colleagues have reported that AZ concentrations in tissues commonly range from 10 to 100 times the serum AZ concentrations (51).
Whether the significant accumulation of AZ in tissue identified in the present study also occurs at later gestation ages is a key question. AZ clearance is known to be mediated, at least in part, by two drug efflux transporters, P-glycoprotein (P-gp) (ABCB1/MDR1) and multidrug resistance protein 2 (MRP2) (ABCC2) (14, 52). P-gp is an ATP-dependent efflux pump that has been detected in both the rodent and the human placenta. MRP2 is a transmembrane ATP-binding cassette transport protein that is found on the apical membrane of syncytiotrophoblasts and the endothelium of fetal blood vessels (52). It is hypothesized to act as an important protective mechanism in the human placenta, limiting fetal exposure to xenobiotics. The regulation of placental drug transport pumps in pregnancy is both complex and likely dependent on a host of variables, including tissue distribution, gestation, species, hormonal levels, and the presence of infection (52). Work by Novotna and colleagues (53) has demonstrated that, in the rat placenta, P-gp is first detectable at 13 days GA, with significantly higher expression peaking (in terms of both intensity and distribution) close to term at 22 days GA. Interestingly, these data are in contrast to findings from studies in human placental tissue (54, 55). A recent study demonstrated that term (>37 weeks GA) human placenta expressed approximately 4.4 times more MRP2 mRNA than early (<32 weeks GA) placenta (56). It is tempting to speculate that changes in the expression of placental drug efflux pumps may contribute to different rates of maternal-fetal AZ transfer at different gestational ages. In any case, these data add further weight to the argument for employing gestation age-approriate animal models when assessing drug therapies in pregnancy.
Despite achieving elevated concentrations in fetal tissue, neither AZ treatment group was associated with marked differences in serum markers of liver injury, increases in AF MCP-1 protein concentrations, or overt changes in circulating immunocyte populations. These data suggest that there were no overt toxic responses following exposure of the fetus to sustained and elevated concentrations of AZ early in pregnancy. The death of one fetus (out of a study population of 50 animals) is well within the normal, expected pregnancy loss rate for our sheep model.
In this study, we used a sheep model of early pregnancy to investigate the maternal-fetal transfer of AZ in early pregnancy (80 days GA). The data suggest (i) that FP AZ concentrations remain low following both single IA and repeated maternal i.v. administrations; (ii) that AZ can cross the AF-exposed second trimester epithelium (fetal skin, lung, and gut) to a limited extent, accumulating in both the fetal lung and liver; (iii) that AZ accumulates at high levels in the fetal lung and liver following maternal i.v. administration, suggesting that maternal-fetal AZ transfer is more effective than AF-fetal AZ transfer; and (iv) that when assessing maternal-fetal drug transfer in pregnancy, fetal tissue accumulation, as well as fluid (FP, AF, or CSF) levels, should be studied, especially for agents such as AZ with a known ability to accumulate in tissues at high levels.
In extrapolating our findings, it is important to take into account the limitations of this study. First, ovine placentation is rather different from that of humans (57). Second, the early GA of the fetuses under investigation and a desire to assess tissue AZ concentrations prevented us from utilizing chronically catheterized preparations, necessitating a cross-sectional approach. Third, comparing fluid and tissue concentrations with MIC values to determine therapeutic efficacy must be done with some caution due to differences in drug availability (58).
With these limitations in mind, we conclude that of the two AZ delivery regimens assessed, repeated maternal i.v. AZ administration is likely the most useful and effective means of delivering AZ to the fetus in the second trimester of pregnancy. IA AZ administration did result in sustained AZ levels greater than or equal to the MIC in the AF and fetal lung and liver uptake; however, repeated maternal i.v. AZ dosing gave equivalent or higher AZ levels in the fetal lung, fetal liver, FP, and AF after 36 h of treatment, in addition to AZ concentrations greater than or equal to the MIC in the MP without the need for amniocentesis. Given reports of AZ accumulation in leukocytes, it would be of interest to determine the impact of active uterine infection on maternal-fetal AZ transfer. It would also be important to determine if the 48 to 72 h required for maternal i.v. AZ administration to achieve therapeutic concentrations in the AF and FP is fast enough to effectively treat intrauterine infection and prevent fetal injury.
In conclusion, we suggest that additional studies are required to better characterize in vivo AZ treatment efficacy, especially against Ureaplasma sp. isolates that show AZ resistance in vitro, and also in cases of polymicrobial infections of the amniotic environment. Furthermore, studies investigating the optimal maternal dosing required to safely minimize the time necessary to achieve therapeutic fetal AZ concentrations would also be of benefit.
ACKNOWLEDGMENTS
This work was supported by National Health and Medical Research Council project grant APP1049148 (M.W.K.). Bilateral travel between Australian and United Kingdom laboratories was funded by an international exchange Royal Society Grant (IE130066 to O.B.S. and M.W.K.). O.B.S. is supported by the Microbiology and Infection Translational Research Group (MITReG) and the Children and Young People's Research Network (CYPRN) as part of the Welsh Government initiative to support research. Y.M., J.A.K., and M.W.K. are supported by the Women and Infants Research Foundation (Perth, Western Australia). The UWA Centre for Metabolomics is funded by the National Collaborative Research Infrastructure Strategy (NCRIS) and the WA State Government ICP funding program.
We gratefully acknowledge technical assistance (laboratory assistance, animal handling, and husbandry) provided by E. Woodward, A. Armitage, M. Davies, A. Wilson, K. Muthasm, and P. Cowl (all University of Western Australia, Perth, Western Australia, Australia). We thank Sara and Andrew Ritchie (Icon Agriculture, Darkan, Western Australia) for their expertise in providing date-mated sheep, Liz Nathan (Women and Infants Research Foundation, Perth, Western Australia) for assistance with statistical analyses, and Siemens Australia for generously providing the Rapidlab 1200 reagents and consumables used in this study.
We report no conflict of interest.
Footnotes
Published ahead of print 25 August 2014
REFERENCES
- 1.Althabe F, Howson CP, Kinney M, Lawn J, WHO 2012. Born too soon: the gobal action report on preterm birth. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Goldenberg RL, Culhane JF, Iams JD, Romero R. 2008. Epidemiology and causes of preterm birth. Lancet 371:75–84. 10.1016/S0140-6736(08)60074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Romero R, Espinoza J, Chaiworapongsa T, Kalache K. 2002. Infection and prematurity and the role of preventive strategies. Semin. Neonatol. 7:259–274. 10.1053/siny.2002.0121. [DOI] [PubMed] [Google Scholar]
- 4.Goldenberg RL, Andrews WW, Faye-Petersen OM, Goepfert AR, Cliver SP, Hauth JC. 2006. The Alabama Preterm Birth Study: intrauterine infection and placental histologic findings in preterm births of males and females less than 32 weeks. Am. J. Obstet. Gynecol. 195:1533–1537. 10.1016/j.ajog.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 5.Romero R, Espinoza J, Goncalves LF, Kusanovic JP, Friel L, Hassan S. 2007. The role of inflammation and infection in preterm birth. Semin. Reprod. Med. 25:21–39. 10.1055/s-2006-956773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lahra MM, Jeffery HE. 2004. A fetal response to chorioamnionitis is associated with early survival after preterm birth. Am. J. Obstet. Gynecol. 190:147–151. 10.1016/j.ajog.2003.07.012. [DOI] [PubMed] [Google Scholar]
- 7.Hillier SL, Witkin SS, Krohn MA, Kiviat NB, Eschenbach DA. 1993. The relationship of amniotic fluid cytokines and preterm delivery, amniotic fluid infection, histologic chorioamnionitis, and chorioamnion infection. Obstet. Gynecol. 81:941–948. [PubMed] [Google Scholar]
- 8.Lewit EM, Baker LS, Corman H, Shiono PH. 1995. The direct cost of low birth weight. Future Child. 5:35–56. 10.2307/1602506. [DOI] [PubMed] [Google Scholar]
- 9.NIH. 2007. Preterm birth: causes, consequences, and prevention. NIH, Bethesda, MD. [PubMed] [Google Scholar]
- 10.Iams JD, Romero R, Culhane JF, Goldenberg RL. 2008. Primary, secondary, and tertiary interventions to reduce the morbidity and mortality of preterm birth. Lancet 371:164–175. 10.1016/S0140-6736(08)60108-7. [DOI] [PubMed] [Google Scholar]
- 11.Brocklehurst P, Gordon A, Heatley E, Milan SJ. 2013. Antibiotics for treating bacterial vaginosis in pregnancy. Cochrane Database Syst. Rev. 1:CD000262. 10.1002/14651858.CD000262.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kenyon SL, Taylor DJ, Tarnow-Mordi W. 2001. Broad-spectrum antibiotics for preterm, prelabour rupture of fetal membranes: the ORACLE I randomised trial. Lancet 357:979–988. 10.1016/S0140-6736(00)04233-1. [DOI] [PubMed] [Google Scholar]
- 13.Kenyon SL, Taylor DJ, Tarnow-Mordi W. 2001. Broad-spectrum antibiotics for spontaneous preterm labour: The ORACLE II randomised trial. Lancet 357:989–994. 10.1016/S0140-6736(00)04234-3. [DOI] [PubMed] [Google Scholar]
- 14.Fischer JH, Sarto GE, Habibi M, Kilpatrick SJ, Tuomala RE, Shier JM, Wollett L, Fischer PA, Khorana KS, Rodvold KA. 2012. Influence of body weight, ethnicity, oral contraceptives, and pregnancy on the pharmacokinetics of azithromycin in women of childbearing age. Antimicrob. Agents Chemother. 56:715–724. 10.1128/AAC.00717-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pitsouni E, Iavazzo C, Athanasiou S, Falagas ME. 2007. Single-dose azithromycin versus erythromycin or amoxicillin for Chlamydia trachomatis infection during pregnancy: a meta-analysis of randomised controlled trials. Int. J. Antimicrob. Agents 30:213–221. 10.1016/j.ijantimicag.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 16.Kim YS, Hyo JL, Chang M, Sung KS, Yun ER, Soo KS. 2006. Scrub typhus during pregnancy and its treatment: a case series and review of the literature. Am. J. Trop. Med. Hyg. 75:955–959. [PubMed] [Google Scholar]
- 17.Cerar D, Karner P, Avšič-Županc T, Strle F. 2009. Azithromycin for acute Q. fever in pregnancy. Wien. Klin. Wochenschr. 121:469–472. 10.1007/s00508-009-1180-0. [DOI] [PubMed] [Google Scholar]
- 18.Chandra RS, Orazem J, Ubben D, Duparc S, Robbins J, Vandenbroucke P. 2013. Creative solutions to extraordinary challenges in clinical trials: Methodology of a phase III trial of azithromycin and chloroquine fixed-dose combination in pregnant women in Africa. Malar. J. 12:122. 10.1186/1475-2875-12-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waites KB, Katz B, Schelonka RL. 2005. Mycoplasmas and ureaplasmas as neonatal pathogens. Clin. Microbiol. Rev. 18:757–789. 10.1128/CMR.18.4.757-789.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tita ATN, Andrews WW. 2010. Diagnosis and management of clinical chorioamnionitis. Clin. Perinatol. 37:339–354. 10.1016/j.clp.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amsden GW. 1996. Erythromycin, clarithromycin, and azithromycin: are the differences real? Clin. Ther. 18:56–72. [DOI] [PubMed] [Google Scholar]
- 22.Kim M, Hynicka LM. 2012. Azithromycin-induced hepatotoxicity. Hosp. Pharm. 47:946–949. 10.1310/hpj4712-946. [DOI] [Google Scholar]
- 23.Lin KJ, Mitchell AA, Yau WP, Louik C, Hernández-Díaz S. 2013. Safety of macrolides during pregnancy. Am. J. Obstet. Gynecol. 208: 221.e1–8. 10.1016/j.ajog.2012.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romøren M, Lindbæk M, Nordeng H. 2012. Pregnancy outcome after gestational exposure to erythromycin: a population-based register study from Norway. Br. J. Clin. Pharmacol. 74:1053–1062. 10.1111/j.1365-2125.2012.04286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ray WA, Murray KT, Hall K, Arbogast PG, Stein CM. 2012. Azithromycin and the risk of cardiovascular death. N. Engl. J. Med. 366:1881–1890. 10.1056/NEJMoa1003833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Svanström H, Pasternak B, Hviid A. 2013. Use of azithromycin and death from cardiovascular causes. N. Engl. J. Med. 368:1704–1712. 10.1056/NEJMoa1300799. [DOI] [PubMed] [Google Scholar]
- 27.Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, Hernández-Díaz S. 2011. Medication use during pregnancy, with particular focus on prescription drugs: 1976–2008. Am. J. Obstet. Gynecol. 205:51.e1–8. 10.1016/j.ajog.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andrade SE, Gurwitz JH, Davis RL, Chan KA, Finkelstein JA, Fortman K, McPhillips H, Raebel MA, Roblin D, Smith DH, Yood MU, Morse AN, Platt R. 2004. Prescription drug use in pregnancy. Am. J. Obstet. Gynecol. 191:398–407. 10.1016/j.ajog.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 29.Keelan JA, Nitsos I, Saito M, Musk GC, Kemp MW, Timmins M, Li S, Yaegashi N, Newnham JP. 2011. Maternal-amniotic-fetal distribution of macrolide antibiotics following intravenous, intramuscular, and intra-amniotic administration in late pregnant sheep. Am. J. Obstet. Gynecol. 204:546.e10–7. 10.1016/j.ajog.2011.02.035. [DOI] [PubMed] [Google Scholar]
- 30.Holbrook KA, Odland GF. 1975. The fine structure of developing human epidermis: light, scanning, and transmission electron microscopy of the periderm. J. Invest. Dermatol. 65:16–38. 10.1111/1523-1747.ep12598029. [DOI] [PubMed] [Google Scholar]
- 31.Viscardi RM. 2010. Ureaplasma species: role in diseases of prematurity. Clin. Perinatol. 37:393–409. 10.1016/j.clp.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viscardi RM. 2014. Ureaplasma species: role in neonatal morbidities and outcomes. Arch. Dis. Child. Fetal Neonatal Ed. 99:F87–F92. 10.1136/archdischild-2012-303351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burd EM. 2010. Validation of laboratory-developed molecular assays for infectious diseases. Clin. Microbiol. Rev. 23:550–576. 10.1128/CMR.00074-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunter RP, Koch DE, Coke RL, Goatley MA, Isaza R. 2003. Azithromycin metabolite identification in plasma, bile, and tissues of the ball python (Python regius). J. Vet. Pharmacol. Ther. 26:117–121. 10.1046/j.1365-2885.2003.00464.x. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, Huo M, Zhou J, Xie S. 2010. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 99:306–314. 10.1016/j.cmpb.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 36.Ludden TM, Beal SL, Sheiner LB. 1994. Comparison of the Akaike Information Criterion, the Schwarz criterion and the F test as guides to model selection. J. Pharmacokinet. Biopharm. 22:431–445. 10.1007/BF02353864. [DOI] [PubMed] [Google Scholar]
- 37.Novy MJ, Duffy L, Axthelm MK, Sadowsky DW, Witkin SS, Gravett MG, Cassell GH, Waites KB. 2009. Ureaplasma parvum or Mycoplasma hominis as sole pathogens cause chorioamnionitis, preterm delivery, and fetal pneumonia in rhesus macaques. Reprod. Sci. 16:56–70. 10.1177/1933719108325508. [DOI] [PubMed] [Google Scholar]
- 38.Grigsby PL, Novy MJ, Sadowsky DW, Morgan TK, Long M, Acosta E, Duffy LB, Waites KB. 2012. Maternal azithromycin therapy for Ureaplasma intra-amniotic infection delays preterm delivery and reduces fetal lung injury in a primate model. Am. J. Obstet. Gynecol. 207:475.e1–475.e14. 10.1016/j.ajog.2012.10.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones HE, Harris KA, Azizia M, Bank L, Carpenter B, Hartley JC, Klein N, Peebles D. 2009. Differing prevalence and diversity of bacterial species in fetal membranes from very preterm and term labor. PLoS One 4:e8205. 10.1371/journal.pone.0008205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DiGiulio DB, Romero R, Kusanovic JP, Gómez R, Kim CJ, Seok KS, Gotsch F, Mazaki-Tovi S, Vaisbuch E, Sanders K, Bik EM, Chaiworapongsa T, Oyarzún E, Relman DA. 2010. Prevalence and diversity of microbes in the amniotic fluid, the fetal inflammatory response, and pregnancy outcome in women with preterm pre-labor rupture of membranes. Am. J. Reprod. Immunol. 64:38–57. 10.1111/j.1600-0897.2010.00830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Acosta EP, Grigsby PL, Larson KB, James AM, Long MC, Duffy LB, Waites KB, Novy MJ. 2014. Transplacental transfer of azithromycin and its use for eradicating intra-amniotic ureaplasma infection in a primate model. J. Infect. Dis. 209:898–904. 10.1093/infdis/jit578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cárceles CM, Fernández-Varón E, Marín P, Escudero E. 2007. Tissue disposition of azithromycin after intravenous and intramuscular administration to rabbits. Vet. J. 174:154–159. 10.1016/j.tvjl.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 43.Cárceles CM, Font A, Escudero E, Espuny A, Marín P, Fernández-Varón E. 2005. Pharmacokinetics of azithromycin after i.v. and i.m. administration to sheep. J. Vet. Pharmacol. Ther. 28:475–479. 10.1111/j.1365-2885.2005.00680.x. [DOI] [PubMed] [Google Scholar]
- 44.Stamler DA, Edelstein MAC, Edelstein PH. 1994. Azithromycin pharmacokinetics and intracellular concentrations in Legionella pneumophila-infected and uninfected guinea pigs and their alveolar macrophages. Antimicrob. Agents Chemother. 38:217–222. 10.1128/AAC.38.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramsey PS, Vaules MB, Vasdev GM, Andrews WW, Ramin KD. 2003. Maternal and transplacental pharmacokinetics of azithromycin. Am. J. Obstet. Gynecol. 188:714–718. 10.1067/mob.2003.141. [DOI] [PubMed] [Google Scholar]
- 46.Salman S, Rogerson SJ, Kose K, Griffin S, Gomorai S, Baiwog F, Winmai J, Kandai J, Karunajeewa HA, O'Halloran SJ, Siba P, Ilett KF, Mueller I, Davis TME. 2010. Pharmacokinetic properties of azithromycin in pregnancy. Antimicrob. Agents Chemother. 54:360–366. 10.1128/AAC.00771-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heikkinen T, Laine K, Neuvonen PJ, Ekblad U. 2000. The transplacental transfer of the macrolide antibiotics erythromycin, roxithromycin and azithromycin. Br. J. Obstet. Gynaecol. 107:770–775. 10.1111/j.1471-0528.2000.tb13339.x. [DOI] [PubMed] [Google Scholar]
- 48.Escudero E, Fernández-Varón E, Marín P, Espuny A, Nájera MD, Cárceles CM. 2006. Pharmacokinetics and tissue tolerance of azithromycin after intramuscular administration to rabbits. Res. Vet. Sci. 81:366–372. 10.1016/j.rvsc.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 49.Davila D, Kolacny-Babic L, Plavsic F. 1991. Pharmacokinetics of azithromycin after single oral dosing of experimental animals. Biopharm. Drug Dispos. 12:505–514. 10.1002/bdd.2510120704. [DOI] [PubMed] [Google Scholar]
- 50.Girard AE, Girard D, Retsema JA. 1990. Correlation of the extravascular pharmacokinetics of azithromycin with in-vivo efficacy in models of localized infection. J. Antimicrob. Chemother. 25:61–71. 10.1093/jac/25.suppl_A.61. [DOI] [PubMed] [Google Scholar]
- 51.Foulds G, Shepard RM, Johnson RB. 1990. The pharmacokinetics of azithromycin in human serum and tissues. J. Antimicrob. Chemother. 25:73–82. [DOI] [PubMed] [Google Scholar]
- 52.Vähäkangas K, Myllynen P. 2009. Drug transporters in the human blood-placental barrier. Br. J. Pharmacol. 158:665–678. 10.1111/j.1476-5381.2009.00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Novotna M, Libra A, Kopecky M, Pavek P, Fendrich Z, Semecky V, Staud F. 2004. P-glycoprotein expression and distribution in the rat placenta during pregnancy. Reprod. Toxicol. 18:785–792. 10.1016/j.reprotox.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 54.Mathias AA, Hitti J, Unadkat JD. 2005. P-glycoprotein and breast cancer resistance protein expression in human placentae of various gestational ages. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289:R963–R969. 10.1152/ajpregu.00173.2005. [DOI] [PubMed] [Google Scholar]
- 55.Sun M, Kingdom J, Baczyk D, Lye SJ, Matthews SG, Gibb W. 2006. Expression of the multidrug resistance P-glycoprotein (ABCB1 glycoprotein) in the human placenta decreases with advancing gestation. Placenta 27:602–609. 10.1016/j.placenta.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 56.Meyer Zu Schwabediss HE, Jedlitschky G, Gratz M, Haenisch S, Linnemann K, Fusch C, Cascorbi I, Kroemer HK. 2005. Variable expression of MRP2 (ABCC2) in human placenta: influence of gestational age and cellular differentiation. Drug Metab. Dispos. 33:896–904. 10.1124/dmd.104.003335. [DOI] [PubMed] [Google Scholar]
- 57.Carter AM. 2007. Animal models of human placentation: a review. Placenta 28:S41–S47. 10.1016/j.placenta.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 58.Mouton JW, Theuretzbacher U, Craig WA, Tulkens PM, Derendorf H, Cars O. 2008. Tissue concentrations: do we ever learn? J. Antimicrob. Chemother. 61:235–237. 10.1093/jac/dkm476. [DOI] [PubMed] [Google Scholar]