

Abstract
HIV-1 transcription is activated by the Tat protein, which recruits CDK9/cyclin T1 to the HIV-1 promoter. CDK9 is phosphorylated by CDK2, which facilitates formation of the high-molecular-weight positive transcription elongation factor b (P-TEFb) complex. We previously showed that chelation of intracellular iron inhibits CDK2 and CDK9 activities and suppresses HIV-1 transcription, but the mechanism of the inhibition was not understood. In the present study, we tested a set of novel iron chelators for the ability to inhibit HIV-1 transcription and elucidated their mechanism of action. Novel phenyl-1-pyridin-2yl-ethanone (PPY)-based iron chelators were synthesized and examined for their effects on cellular iron, HIV-1 inhibition, and cytotoxicity. Activities of CDK2 and CDK9, expression of CDK9-dependent and CDK2-inhibitory mRNAs, NF-κB expression, and HIV-1- and NF-κB-dependent transcription were determined. PPY-based iron chelators significantly inhibited HIV-1, with minimal cytotoxicity, in cultured and primary cells chronically or acutely infected with HIV-1 subtype B, but they had less of an effect on HIV-1 subtype C. Iron chelators upregulated the expression of IκB-α, with increased accumulation of cytoplasmic NF-κB. The iron chelators inhibited CDK2 activity and reduced the amount of CDK9/cyclin T1 in the large P-TEFb complex. Iron chelators reduced HIV-1 Gag and Env mRNA synthesis but had no effect on HIV-1 reverse transcription. In addition, iron chelators moderately inhibited basal HIV-1 transcription, equally affecting HIV-1 and Sp1- or NF-κB-driven transcription. By virtue of their involvement in targeting several key steps in HIV-1 transcription, these novel iron chelators have the potential for the development of new therapeutics for the treatment of HIV-1 infection.
INTRODUCTION
HIV-1 transcription is induced by the HIV-1 Tat protein, which recruits CDK9/cyclin T1, the kinase of positive transcription elongation factor b (P-TEFb), to TAR RNA, promoting processive elongation of HIV-1 transcription (reviewed in reference 1). Basal HIV-1 transcription is activated primarily by host cell Sp1 and NF-κB transcription factors, which bind to the HIV-1 long terminal repeat (LTR) and may also recruit CDK9/cyclin T1 independently of Tat (2). P-TEFb forms a high-molecular-weight complex (large P-TEFb complex) in which CDK9/cyclin T1 is associated with 7SK RNA and several additional proteins, including a hexamethylene bis-acetamide-inducible protein 1 (HEXIM1) dimer, La-related protein 7 (LARP7) (3–5), and the methylphosphatase capping enzyme (MePCE) (6, 7). In addition, Tat facilitates the formation of the superelongation complex (SEC), containing active P-TEFb and additional elongation factors and coactivators (8, 9). While the kinase activity of CDK9 in the large P-TEFb complex is suppressed (10, 11), this complex serves as the source of CDK9/cyclin T1 for recruitment by HIV-1 Tat (12). In a recent study, we demonstrated that HIV-1 transcription is regulated by CDK2, which phosphorylates the Ser90 amino acid residue of CDK9 (13). Dephosphorylation of this residue reduces the large P-TEFb complex and decreases HIV-1 transcription (13). Macrophages differentiated from induced pluripotent stem cells with stable CDK2 knockdown also exhibited the reduced susceptibility of these cells to HIV-1 infection (14), confirming our previous observation of CDK2 as a key regulator of HIV-1 transcription.
We previously described a role of iron chelators in the inhibition of HIV-1 transcription and replication, likely by reducing the activities of CDK2 and CDK9 (15, 16); however, the exact mechanism of action has remained unclear. Induction of p21 (CIP1/WAF1) expression by iron chelators was recently shown to inhibit CDK2 activity in 293T cells (17–19). Moreover, blocking of p21-mediated CDK9 and viral reverse transcriptase activities provides a potential protection barrier against HIV-1 infection (17). Since CDK2 phosphorylates the HIV Tat protein and also the host protein CDK9 (18), it may be possible that the induction of p21 by iron chelators inhibits CDK2 activity, leading to the suppression of CDK9-dependent HIV-1 transcription (19). HIV-1 Tat also recruits NF-κB along with CDK9/cyclin T1 (2), and this recruitment occurs in a cooperative manner (20, 21), as Tat interacts with the p65 subunit of NF-κB through NFBP (22). HIV-1 basal transcription is largely regulated by the Sp1 transcription factor (23), which recruits CDK9/cyclin T1 to the LTR in the absence of Tat (24). Tat also stimulates Sp1 phosphorylation by DNA-PK, which also contributes to the induction of HIV-1 transcription (25).
In the present study, we further analyzed the mechanism of HIV-1 inhibition by iron chelators by using several novel iron chelators which have a flexible scaffold compared to that of previously reported di-2-pyridylketone thiosemicarbazone (DpT)- and 2-benzoylpyridine thiosemicarbazone (BpT)-based chelators (15). We created novel phenyl-1-pyridin-2yl-ethanone (PPY)-based iron chelators and analyzed them for the ability to inhibit HIV-1. The iron chelators efficiently reduced cellular iron and also hampered cell cycle progression of the treated cells. The chelators inhibited HIV-1 subtype B infection in cultured and primary cells, and also in chronically infected T cells, at low or subnanomolar concentrations, without being cytotoxic. The chelators efficiently reduced HIV-1 mRNA expression but had no effect on reverse transcription. We observed increased expression of p21, cyclin A, and cyclin E and increased G1 cell cycle accumulation of the T cells treated with iron chelators. Iron chelators inhibited CDK2 activity and also reduced the amount of CDK9/cyclin T1 in the large P-TEFb complex. Analysis of CDK9-dependent genes showed increased expression of the NF-κB inhibitor IκB-α. In cells treated with iron chelators, NF-κB was found to accumulate in the cytoplasm, and the overall expression level of NF-κB was decreased. Iron chelators had a more profound effect on Tat-activated HIV-1 transcription than on basal HIV-1 or Sp1- or NF-κB-driven transcription. Our results indicate that PPY-based iron chelators markedly inhibit HIV-1 transcription by inhibiting CDK2 and CDK9 activities that are directly related to the upregulation of p21 expression and downregulation of NF-κB expression via the IκB-α induction mechanism.
MATERIALS AND METHODS
Cells and media.
293T, THP-1, and CD4+ T cells (CEM) were purchased from the American Type Culture Collection (Manassas, VA). The J1.1 and A3R5.7 cell lines were obtained from the NIH AIDS Research and Reference Reagent Program. Peripheral blood mononuclear cells (PBMCs) were purchased from Astra Biologics. All cells were cultured at 37°C in a 5% CO2 atmosphere. CEM and chronically HIV-1-infected J1.1 T cells, as well as the promonocytic THP-1 cell line, were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum and 1% antibiotic solution (penicillin and streptomycin; Invitrogen). The human T lymphoblastoid cell line A3R5.7, expressing the CD4, CXCR4, and CCR5 receptors, was cultured in complete RPMI 1640 medium supplemented with Geneticin (G418; 1 mg/ml). 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) containing 10% fetal bovine serum and 1% antibiotic solution (penicillin and streptomycin).
Plasmids.
The HIV-1 proviral clone NL4-3 (26) was kindly provided by Lee Ratner. The HIV-1 subtype C proviral clone HIV1084i (27) was a kind gift of Charles Wood. The HIV-1 proviral vector pNL4-3.Luc.R−E− (courtesy of Nathaniel Landau, NYU School of Medicine, New York, NY) was obtained from the NIH AIDS Research and Reference Reagent Program. HIV-1 LTR–luciferase expression vectors were kindly provided by Manuel López-Cabrera (Unidad de Biología Molecular, Madrid, Spain) (28) and carried the following sequences: the HIV-1 LTR (positions −105 to +77) followed by the luciferase reporter gene (HIV LTR 2×NFκB 3×SP1); the HIV-1 LTR (positions −105 to +77) with Sp1-inactivated sites, followed by the luciferase reporter gene (HIV LTR 2×NFκB ΔSP1); and HIV-1 (positions −81 to +77) with NF-κB-deleted sites, followed by the luciferase reporter gene (HIV LTR ΔNFκB 3×SP1).
Antibodies.
Antibodies for IκB-α, IκB-α phosphorylated on Ser32 (IκB-α-P), the NF-κB p65 subunit, and p21 were purchased from Cell Signaling Technology (Danvers, MA). Antibodies for α-tubulin were from Sigma. Antibodies for p21 were from BD Biosciences.
Synthesis of PPY.
Previously described Bp4eT and Bp4aT tridentate iron chelators (Fig. 1A) were modified by replacing the phenyl group with a benzyl moiety. The general steps for the synthesis of the PPY compound are shown in Fig. 1B. A solution of phenylacetonitrile (23.4 g; 0.2 mol) and methyl picolinate (30 g; 0.22 mol) in 100 ml of tetrahydrofuran (THF) was added dropwise to a suspension of NaH (16.8 g; 0.42 mol; 60% [wt/wt] in paraffin) in 300 ml of THF while stirring and cooling in an ice-water bath. The reaction mixture was stirred and cooled with a water bath for an additional 2 h. The water bath was then removed, and the reaction mixture was stirred overnight at room temperature. The next day, the dark solution was evaporated under reduced pressure and the solid residue was dissolved in a minimal amount of water and extracted twice with 200 ml chloroform. The water layer was acidified with concentrated hydrochloric acid (300 ml), refluxed for 1 day, cooled down to room temperature, and left at room temperature overnight. The hydrochloride precipitate was filtered, washed with acetone, and air dried. The hydrochloride salt was dissolved in 100 ml methanol. Sodium hydrocarbonate was added in portions, with stirring, until the production of gas stopped, and the mixture was further stirred for another 30 min. The solution was then filtered and evaporated. The obtained solid residue was treated with 50 ml water and 100 ml chloroform. The organic layer was removed, and the water layer was extracted with 100 ml chloroform. The combined chloroform solution was dried over anhydrous sodium sulfate and evaporated. After complete crystallization, the product was washed with a small amount of cold hexane and air dried. PPY was isolated in 9.8 g.
FIG 1.

Effects of iron chelators on cellular iron and cell cycle progression. (A) Chemical structures of Dp44mT, Bp4eT, and Bp4aT. (B to D) General steps for synthesis of PPY (control) and the PPYeT and PPYaT iron chelators. (E to G). Nano-LC-FTMS analyses of PPY, PPYeT, and PPYaT. TIC, total ion current; EIC, extract ion chromatogram; PA, peak area. As shown in the EICs, PPY could also be detected in the samples of PPYeT and PPYaT. The purities of PPYeT and PPYaT were 85% and 90%, respectively.
Synthesis of PPY analogs PPYeT and PPYaT.
An equimolar mixture of PPY (2.5 mmol) and the appropriate thiosemicarbazide was refluxed in 20 ml of a 1:1 water-ethanol mixture for 24 h. After cooling, a precipitated product was filtered off and dried under vacuum at 60°C. PPYeT was isolated in 0.42 g (56% yield), and PPYaT was isolated in 0.56 g (72% yield).
LC-MS analysis of PPY-based compounds.
PPY, PPYeT, and PPYaT were dissolved in dimethyl sulfoxide (DMSO) for preparation of stock solutions (10 mM), which were further diluted to 100 μM with a 0.1% formic acid aqueous solution for nano-liquid chromatography–Fourier transform mass spectrometry (LC-FTMS) analysis. The samples were loaded onto a nano-C18 column attached to a Shimadzu nano-LC coupled in-line to an LTQ Orbitrap XL tandem mass spectrometer (Thermo Fischer Scientific, GA). The injection volume was 0.2 μl. The mobile phase consisted of a 0.1% formic acid aqueous solution (A) and a 0.1% formic acid acetonitrile solution (B). The gradient elution program was as follows: 0 to 6.02 min, 1% B; 6.02 to 6.11 min, 1 to 2% B; 6.11 to 10 min, 2 to 98% B; and 10 to 30 min, 98% B (vol/vol). The flow rate was set at 600 nl/min. The compounds were ionized by electrospray ionization and detected by the Orbitrap instrument at a mass resolution of 30,000 (full scan, m/z 180 to 2,000). The spray voltage, capillary temperature, and capillary voltage were set to 2.0 kV, 200°C, and 39.5 V, respectively.
Determination of effects of iron chelators on LIP.
The effects of PPY-based iron chelators on the labile iron pool (LIP) in human acute monocytic leukemia (THP-1) cells was examined as described previously (29). Cells were loaded with calcein-AM, a nonfluorescent, hydrophobic compound that easily permeates intact, viable cells. Treatment of the cells with iron chelators removes the calcein-bound iron and produces calcein, a hydrophilic, strongly fluorescent compound that is well retained in the cell cytoplasm. The fluorescence intensity is related to the amount of cellular iron effectively removed by the chelators (29). The chelating efficiency was quantified using the following formula: efficiency = (F − F0)/F0, where F0 is the fluorescence intensity in the presence of chelator at time zero and F is the fluorescence intensity at a given time after the addition of the chelator. The values derived from the (F − F0)/F0 term are proportional to the concentrations of chelated iron present when reaction equilibrium is reached and reflect the chelating efficiencies of the iron chelators.
Cell viability assays.
CEM T cells were cultured in 96-well plates at 37°C and incubated with iron chelators for the indicated periods. Cell viability was determined using calcein-AM and a trypan blue-based assay. To assess cytotoxicity with calcein-AM, medium was removed and the cells were washed with Dulbecco's phosphate-buffered saline (PBS) to remove serum esterase activity, which may cause an increase in fluorescence through the hydrolysis of calcein-AM. Cells were then supplemented with 0.2 μM calcein-AM (Invitrogen) for 10 min at 37°C. A positive-control sample containing 100% dead cells was prepared by treating cells with Triton X-100 (1% [vol/vol]) and then incubating them with 0.2 μM calcein-AM. Fluorescence was measured using the luminescence spectrometer described above, implementing an excitation wavelength of 495 nm and emission filters at 515 nm. To measure cellular viability with trypan blue, the cells were supplemented with 0.2% trypan blue, transferred to a plastic disposable counting chamber, and counted on a TC10 automatic cell counter (Bio-Rad).
Luciferase assays.
CEM T cells were infected with vesicular stomatitis virus glycoprotein (VSV-G)-pseudotyped pNL4-3.Luc.R−E− virus, prepared as previously described (15), and then cultured at 0.5 × 106 cells/ml in 6-well plates at 37°C and 5% CO2. The cells were collected, washed with PBS, and resuspended in 100 μl of PBS. Next, 100 μl of reconstituted luciferase buffer (Luclite kit; PerkinElmer) was added to each well, and after 10 min of incubation, the lysates were transferred into white plates (PerkinElmer) and luminescence was measured using Labsystems Luminoscan RT equipment (PerkinElmer). PBMCs were purchased from Astra Biologics (Redmond, WA). Donors were negative for HIV-1 and -2, hepatitis B virus, hepatitis C virus, and human T-cell leukemia virus (HTLV-1). PBMCs were isolated from peripheral blood by apheresis, with additional purification by density gradient centrifugation, and were cryopreserved until used. PBMCs were activated with phytohemagglutinin (PHA) (0.5 mg/ml) and interleukin-2 (IL-2) (10 U/ml) for 24 h prior to infection with VSV-G-pseudotyped pNL4-3.Luc.R−E− (HIV-1 Luc) virus at approximately 1 ng of p24 per 5 × 106 cells. After 48 h, the cells were seeded in 96-well white plates and incubated with the iron chelators for 24 h at 37°C. Luciferase buffer was then added to each well, and luminescence was measured using a Labsystems Luminoscan RT instrument as described above.
To measure luciferase activity in 293T cells, the cells were cultured in 96-well plates, transfected with HIV-1 LTR–luciferase vectors, and cotransfected with cytomegalovirus enhanced green fluorescent protein (CMV EGFP) to control for the efficiency of transfection. At 48 h posttransfection, cells were washed three times with PBS and then resuspended in 100 μl of PBS. One hundred microliters of reconstituted Luclite buffer (Luclite kit; PerkinElmer) was added to each well. After 10 min of incubation, the cell lysates were transferred to white plates (PerkinElmer), and luminescence was measured on a Labsystems Luminoscan RT instrument (PerkinElmer).
Cell cycle analysis of 293T cells treated with iron chelators.
Approximately 1 million cells were fixed in 70% ethanol at −20°C for 2 h and stained with propidium iodide (PI) (10 mg/ml) containing RNase A (1 mg/ml) for 30 min. The data were acquired on a BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and analyzed using FlowJo software. The unpaired t test was used to determine statistical significance.
CDK2 and CDK9 phosphorylation assays.
293T cells were treated with iron chelators (1 μM) for 48 h. Cells were lysed in whole-cell lysis buffer (50 mM Tris-HCl, pH 7.5, 0.3 M NaCl, 1% NP-40, 0.1% SDS) supplemented with a protease cocktail (Sigma). CDK2 was immunoprecipitated using anti-CDK2 antibodies. Kinase assay was performed at 30°C for 20 min in kinase assay buffer containing 2 μg histone H1 as the substrate, 200 μM ATP, and 5 μCi of [γ-32P]ATP. At the end of the incubation, SDS-containing electrophoresis sample buffer was added to stop the reaction, and protein bands were resolved by 10% SDS-PAGE. Gels were dried, and protein bands were visualized by exposure to a PhosphorImager screen.
Analysis of IκB-α, cyclin A, cyclin E, and p21 mRNA expression.
293T cells were treated with iron chelators (1 μM) for 48 h. Total RNA was extracted from cultured 293T cells by use of TRIzol reagent according to the manufacturer's protocol (Invitrogen). Total RNA (100 ng) was reverse transcribed to cDNA by use of a Superscript reverse transcription-PCR (RT-PCR) kit (Invitrogen, Carlsbad, CA); hexamers and oligo(dT) were used as primers. For real-time PCR analysis, cDNA was amplified using a Roche LightCycler 480 machine (Roche Diagnostics) and SYBR Green1 master mix (Roche Diagnostics). PCR was carried out with denaturation at 95°C for 10 s, annealing at 60°C for 10 s, and extension at 72°C for 10 s, for 45 cycles. For quantification of mRNA levels for p21, CDK2, cyclin A, cyclin E, and IκB-α, β-actin was used as a housekeeping normalization standard. Primer sequences were as follows: for p21, GCCTTGCAGGAAACTGACTC (forward) and CTTGGCAGCAACTGGATTTT (reverse), for an amplicon size of 183 bp; for CDK2, TTTGCTGAGATGGTGACTCG (forward) and CTTCATCCAGGGGAGGTACA (reverse), for an amplicon size of 196 bp; for cyclin A, GAAACTGCAGCTCGTAGGAA (forward) and ACTTTCAGAAGCAAGTGTTCCA (reverse), for an amplicon size of 150 bp; for cyclin E, AGCACTTTCTTGAGCAACACC (forward) and CGCCATATACCGGTCAAAGA (reverse), for an amplicon size of 161 bp; for IκB-α, GCCTGGACTCCATGAAAGAC (forward) and GTCTGCTGCAGGTTGTTCTG (reverse), for an amplicon size of 179 bp; and for β-actin, AGGCTCAGAGCAAGAGAG (forward) and TACATGGCTGGTGTGTTGA (reverse), for an amplicon size of 229 bp. Mean crossing point (Cp) PCR reaction values for p21, CDK2, cyclin A, cyclin E, IκB, and β-actin were determined, and the ΔΔCT method was used to calculate relative expression levels. The unpaired t test was used to determine statistical significance.
Separation of large and small P-TEFb complexes by differential salt extraction.
Separation of large and small P-TEFb complexes was performed using the methodology described in our recent publication (13), with slight modifications. Briefly, 293T cells in DMEM containing 10% fetal bovine serum were cultured for 48 h in the absence or presence of 1 μM iron chelators. Cells were washed and resuspended in 500 μl/107 cells of buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 10 mM MgCl2, 1 mM EDTA, 250 μM sucrose, 1 mM dithiothreitol [DTT], 0.5% NP-40) supplemented with protease inhibitors. The mixture was incubated on ice for 10 min and centrifuged at 1,000 × g for 5 min. The supernatant, containing the large complex extract (LC), was collected and stored at −80°C until further analysis. The pellet was resuspended in 500 μl/107 cells of buffer B (20 mM HEPES-KOH [pH 7.9], 450 mM NaCl, 1.5 mM MgCl2, 0.5 mM EDTA, 1 mM DTT, and protease inhibitors), incubated on ice for 10 min, and then centrifuged at 10,000 × g for 1 h. The supernatant, containing the small complex extract (SC), was collected and stored at −80°C until further analysis (11). The LC and SC were resolved by SDS-PAGE, transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Allen, TX), and probed with anti-CDK9 and anti-cyclin T1 antibodies.
CDK9 kinase assay.
Kinase assay was performed at 30°C for 30 min in a kinase assay buffer (50 mM HEPES-KOH, pH 7.9, 10 mM MgCl2, 6 mM EGTA, 2.5 mM DTT) containing 100 ng of the glutathione S-transferase C-terminal domain (GST-CTD) as the substrate, 200 μM cold ATP, and 5 μCi of [γ-32P]ATP. The kinase reaction was terminated by addition of SDS-PAGE buffer, followed by electrophoresis on a 10% SDS-polyacrylamide gel. The gels were dried and exposed to a PhosphorImager screen to visualize the protein bands.
Immunoprecipitation.
For preparation of whole-cell lysates, 293T cells were harvested in whole-cell lysis buffer (50 mM Tris-HCl buffer, pH 7.5, containing 0.5 M NaCl, 1% NP-40, 0.1% SDS, and a cocktail of protease inhibitors) as described above. Immunoprecipitation of CDK9 by use of an anti-CDK9 polyclonal antibody (Santa Cruz) was performed as previously described (13). Briefly, 400 μg of lysate and 800 ng of antibody were incubated for 2 h at 4°C with 50 μl of 50% protein A/G agarose suspended in 50 mM Tris-HCl, pH 7.5, containing 150 mM NaCl and 1% NP-40 (TNN buffer). The agarose beads were recovered by centrifugation, washed with TNN buffer, resolved in a 10% Tris–glycine–0.1% SDS gel, transferred to PVDF membranes, and immunoblotted with antibodies against CDK9 or cyclin T1 (Santa Cruz).
Detection and quantification of IκB-α, phosphorylated IκB-α, and the NF-κB p65 subunit.
293T cells were cultured at 37°C in the presence or absence of 10 μM iron chelators for 48 h. For Western blot analysis, the cells were lysed in SDS-PAGE loading buffer, and proteins were resolved by 10% SDS-PAGE and immunoblotted using antibodies against IκB-α, IκB-α phosphorylated on Ser32 (IκB-α-P), or tubulin, as a loading control. To analyze NF-κB p65 subunit distribution between the nucleus and the cytoplasm, the iron chelator-treated cells were lysed in cytoplasm buffer (50 mM Tris-HCl buffer, pH 7.5, containing 0.1% NP-40 and 250 mM sucrose). The cytoplasm was separate from the nuclear material by spinning at 1,000 × g for 15 min. The cytoplasm and the precipitated nuclear material were separated, lysed in SDS-PAGE loading buffer, resolved by 10% SDS-PAGE, and immunoblotted with antibodies against the NF-κB p65 subunit or tubulin, as a loading control. For immunofluorescence analysis, 293T cells were cultured at 37°C on plastic slides (Nunc, Rochester, NY) in the presence or absence of 10 μM iron chelators for 48 h. Cells were fixed with 4% formaldehyde and incubated for 1 h with primary anti-p65 antibody (1:50; Santa Cruz). Slides were washed with PBS and then incubated for 1 h with secondary anti-rabbit antibodies conjugated to Alexa Fluor 488 (Invitrogen) at a dilution of 1:50. The NF-κB-positive cells were visualized by use of a fluorescence microscope (Olympus IX 510) at a magnification of ×20.
RT and quantitative PCR.
For quantitative analysis of HIV-1 RNA, total RNAs were isolated from various samples, including PBMC lysates and lysates of acutely HIV-1-infected (A3R5.7) and chronically HIV-1-infected (J1.1) cell lines. RNAs were purified using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. A total of 0.5 μg of RNA from the RNA fraction was treated with 0.25 mg/ml RNase-free DNase I (Roche, Mannheim, Germany) for 60 min in the presence of 5 mM MgCl2, followed by heat inactivation at 65°C for 15 min. A 200- to 250-ng aliquot of total RNA was used to generate cDNA with the GoScript reverse transcription system (Promega, Madison, WI), using an oligo(dT) reverse primer. Subsequent quantitative real-time PCR analysis was performed with 2 μl of undiluted and 10−1- and 10−2-diluted aliquots of RT reaction mixes. iQ SYBR green supermix (Bio-Rad, Hercules, CA) was used with the following primers, specific for the HIV-1 gag gene: Gag1483-F, 5′-AAGGGGAAGTGACATAGCAG-3′; and Gag1625-R, 5′-GCTGGTAGGGCTATACATTCTTAC-3′ (amplifying a 143-nucleotide [nt] fragment of the HIV-1 gag gene). Serial dilutions of DNA from 8E5 cells (a CEM cell line containing a single copy of HIV-1 LAV provirus per cell) were used as the quantitative standards. To normalize HIV-1 RNA quantifications in the human cells to the target cell DNA, the β-globin gene was quantified by real-time PCR, using the following set of β-globin-specific primers and probe: BGF1, 5′-CAACCTCAAACAGACACCATGG-3′; BGR1, 5′-TCCACGTTCACCTTGCCC-3′; and BGX1 probe, 5′-6-carboxyfluorescein (FAM)-CTCCTGAGGAGAAGTCTGCCGTTACTGCC-6-carboxytetramethylrhodamine (TAMRA)-3′. Real-time PCRs were carried out at least in triplicate, using a Peltier PTC-200 thermal cycler with a Chromo4 continuous-fluorescence detector (both from MJ Research) and Opticon Monitor 2.03 software.
For quantification of HIV-1 DNA, THP-1 cells were infected with HIV-1 Luc, treated with 1 μM iron chelators or 1 μM zidovudine (AZT), and further cultured for another 48 h. Total DNA was extracted from 4 × 106 cells by use of lysis buffer (10 mM Tris-HCl, pH 8, 10 mM EDTA, 5 mM NaCl, 200 μg/ml proteinase K). The cells were lysed for 20 to 30 min at room temperature, and proteinase K was inactivated by heating to 95°C for 5 min. For real-time PCR analysis, 100 ng DNA was amplified using a Roche Light Cycler 480 machine (Roche Diagnostics) and SYBR Green1 master mix (Roche Diagnostics). PCR was carried out with initial preincubation for 5 min at 45°C and then 3 min at 95°C, followed by 45 cycles of denaturation at 95°C for 15 s and annealing and extension at 60°C for 45 s, with a final extension at 72°C for 10 s. Quantification of early-LTR and late-LTR expression was carried out using β-globin DNA as a normalization standard. Primer sequences used were as follows: for early LTR, GGCTAACTAGGGAACCCACTG (forward) and CTGCTAGAGATTTTCCACACTGAC (reverse); for late LTR, TGTGTGCCCGTCTGTTGTGT (forward) and GAGTCCTGCGTCGAGAGATC (reverse); and for β-globin, CAACCTCAAACAGACACCATGG (forward) and TCCACGTTCACCTTGCCC (reverse) (see reference 30 for primer information). Mean Cp values for early LTR, late LTR, and β-globin were determined, and the ΔΔCT method was used to calculate relative expression levels. The unpaired t test was used to test statistical significance.
RESULTS
Chemical structures of PPY-based iron chelators.
In a previous study, we screened a number of di-2-pyridylketone thiosemicarbazone (DpT)- and 2-benzoylpyridine thiosemicarbazone (BpT)-based tridentate iron chelators for HIV-1 inhibition and identified the Dp44eT, Bp4aT, and Bp4eT chelators as the most efficient and suitable for further modifications (15) (Fig. 1A). All of these chelators were shown to be toxic in vivo (31). Since the diarylketone and thiosemicarbozone moieties are likely to adopt a planar conformation that could potentially increase their ability to intercalate DNA, we substituted a benzyl group for the phenyl group. The benzyl analogs PPYaT and PPYeT were synthesized (see Materials and Methods), along with the nonchelating PPY compound, which served as a treatment negative control. The chemical structures of PPY-based iron chelators are shown in Fig. 1B to D. The PPY-based iron chelators were analyzed for purity by high-resolution mass spectrometry (Fig. 1E to G) and also by nuclear magnetic resonance (NMR) analysis (not shown). Both PPYeT and PPYaT were found to contain PPY, and their purities were 85% and 90%, respectively (Fig. 1F and G).
PPYaT and PPYeT efficiently chelate cellular iron.
The ability of PPY-based compounds to chelate iron was tested in promonocytic THP-1 cells as previously described (29) and because of these cells' ability to handle iron. THP-1 cells were treated with nonfluorescent cell-permeative calcein-AM, which, after being transported into cells, is converted to calcein, a weakly iron-binding fluorescent compound whose fluorescence is quenched upon binding to iron. THP-1 cells were pretreated with FeS and then loaded with calcein-AM. After washing off the excess calcein-AM, the cells were treated with the nonchelating PPY compound, the iron chelators (PPYeT and PPYaT), and, as a positive control, salicylaldehyde isonicotinyl hydrazone (SIH) (32). As shown in Fig. 2A, the gains of fractional fluorescence [(F − F0)/F0], which are directly proportional to the amounts of chelatable iron, were similar for PPYeT and SIH but slightly smaller for PPYaT. The nonchelating PPY compound had no effect on the gain of fractional fluorescence. These results indicated that both PPYeT and PPYaT were able to chelate iron in cultured cells, to the same extent as that observed with SIH. Next, we analyzed the effects of PPY-based iron chelators on the levels of transferrin receptor (TFR) mRNA, which contains iron-responsive elements (IREs) in the 3′-untranslated region (3′-UTR) that, when bound to iron-responsive protein (IRP), stabilize RNA (33). Thus, the level of mRNA reflects the amount of cellular iron that assembles into the iron-sulfur cluster of IRP, prevents binding to IREs, and destabilizes TFR mRNA (33). Real-time PCR analysis revealed a significant increase in TFR mRNA levels in cells treated with PPYaT and PPYeT but not PPY or DMSO vehicle control (Fig. 2B), further indicating that PPY-based iron chelators reduce the intracellular iron content.
FIG 2.

Effects of iron chelators on cellular iron and cell cycle progression. (A) Labile iron pool depletion. THP-1 cells were treated with 25 μM ferric sulfate for 1 h at 37°C and then loaded with 0.1 μM calcein-AM for 10 min at 37°C. After washing with PBS, cells were treated with 30 μM SIH, 3 μM PPY, 3 μM PPYeT, or 3 μM PPYaT. Calcein fluorescence was measured in a Roche 4800 real-time PCR machine, as a function of time. Fractional fluorescence [(F − F0)/F0] proportional to the amount of chelatable iron was plotted on the y axis. (B) Expression of transferrin receptor (TFR) was measured in THP-1 cells treated with PPY-based iron chelators, the nonchelating PPY compound, and DMSO, as a control. RNAs were extracted, reverse transcribed, and analyzed by real-time PCR, using 18S RNA as the normalization control. (C to E) Effects of PPY-based iron chelators on cell cycle progression. THP-1 cells, CEM T cells, or 293T cells were treated with 1 μM iron chelators for 24 h and then fixed with 70% ethanol, stained with PI, and analyzed by fluorescence-activated cell sorting (FACS). Data were analyzed using BD FACSCalibur software. All results are shown as means ± standard deviations (SD) of three independent measurements. *, P ≤ 0.01.
Treatment with PPYaT or PPYeT leads to arrest of cells in G1/S phase.
Iron chelators are known to arrest the cell cycle (34, 35). Therefore, we examined the effects of PPY-based iron chelators on the cell cycle progression of THP-1, CEM, and 293T cells (Fig. 2C to E). Cells treated with DMSO or the control PPY compound showed about 50 to 55% of the cells in G1 phase of the cell cycle (Fig. 2C to E). THP-1 and CEM cells showed about 35% in G2/M phase and 10% in S phase, whereas 293T cells showed about 20% in G2/M phase and 20% in S phase (Fig. 2C to E). The different effects on different cell types are likely to reflect different growth properties of these cells. Treatment with the PPYeT or PPYaT iron chelator resulted in a sharp decrease of the cell number in G2/M phase and increased G1-phase accumulation (Fig. 2C to E).
PPYaT and PPYeT inhibit one round of HIV-1 infection in cultured T cells and PBMCs.
The effects of PPY-based iron chelators on one round of HIV-1 infection were analyzed in CEM T cells infected with VSV-G-pseudotyped HIV-1 pNL4-3 virus expressing luciferase in place of nef (HIV-1 Luc) (36). Luciferase activity was measured as an indicator of HIV-1 gene expression. HIV-1 gene expression was inhibited by the PPYaT or PPYeT chelator but not by the control compound PPY or DMSO, which was used as a normalization point, at nanomolar concentrations (50% effective concentration [EC50]= 14 nM) (Fig. 2A). The PPY-based iron chelators showed no toxicity below a concentration of 10 μM in CEM T cells incubated with the chelators for 24 h at 37°C (Fig. 2B). We also analyzed the effects of the PPYaT and PPYeT iron chelators on one round of HIV-1 infection in PBMCs. The cells were activated with PHA and IL-2 (described in Materials and Methods) and then infected with HIV-1 Luc. After 48 h, the cells were treated for 24 h with PPY-based iron chelators, and with PPY as a control, and then analyzed for luciferase activity. As shown in Fig. 3C, PPYaT and PPYeT, but not PPY, markedly inhibited HIV-1 replication (EC50, ∼1 μM). Cytotoxic effects of iron chelators in PBMCs, determined by trypan blue exclusion using an automatic cell counter, showed no significant cell death at concentrations up to 100 μM for PPYaT- or control PPY-treated cells (Fig. 3D). In contrast, PPYeT showed about a 50% reduction of viability at 96 μM. Thus, taken together, these results indicate that PPY-based iron chelators markedly inhibit HIV-1 infection of T cells and PBMCs. PPYaT showed less toxicity than PPYeT.
FIG 3.

Inhibition of one round of HIV-1 replication and toxicity in CEM T cells and PBMCs. (A and C) CEM T cells (A) or PBMCs activated with PHA and IL-2 (C) were infected with VSV-G-pseudotyped pNL4-3.Luc.R−E− (HIV-1 Luc) virus for 18 h at 37°C and then treated for 24 h at 37°C with the indicated concentrations of iron chelators. The cells were then lysed, and luciferase activity was measured. EC50s were determined with GraphPad Prism 6 software. (B) CEM T cells were treated with the indicated concentrations of iron chelators for 24 h at 37°C. CEM T cells were treated with 0.4 μM calcein-AM for 30 min, and calcein fluorescence was measured at a 485-nm excitation wavelength and 515-nm emission wavelength on a luminescence spectrometer equipped with a robotic arm (PerkinElmer LS 50B). EC50s were determined with GraphPad Prism 6 software. (D) Activated PBMCs were treated with the indicated concentrations of iron chelators for 24 h at 37°C, and the viability of cells was measured by the trypan blue exclusion method. Fifty percent cytotoxicity concentrations (CC50s) were determined with GraphPad Prism 6 software.
Effects of PPY-based iron chelators on HIV-1 transcription in acutely and chronically infected T cells and PBMCs.
We analyzed the effect of the least toxic iron chelator (PPYaT) on HIV-1 in A2R5.7 T cells acutely infected with HIV-1 subtype B or subtype C (Fig. 4A and B). Analysis of unspliced viral RNA showed an about 3-fold decrease of HIV-1 expression, even at the lowest concentration (0.3 μM) (Fig. 4A). In contrast, acute infection with HIV-1 subtype C showed no effect of PPYaT at 0.3 μM and about a 2-fold inhibition at 3 μM (Fig. 4B). We next investigated the effects of iron chelators on HIV-1 expression in chronically infected J1.1 T cells (Fig. 4C). The cells were pretreated with a combination antiretroviral therapy (cART) cocktail for 7 days to avoid HIV-1 replication and reinfection. Treatment with iron chelators reduced HIV-1 RNA production up to 3-fold (for PPYeT at 10 μM) (Fig. 4C). Finally, we tested the effects of iron chelators on HIV-1 transcription in acutely infected PBMCs. PBMCs obtained from three different donors were infected with dual-tropic HIV-1 strain 89.6 and then treated with iron chelators for 48 h. Analysis of HIV-1 unspliced RNA showed significant, 2- to 3-fold reductions of HIV-1 expression (Fig. 4D). Taken together, these results indicate that iron chelators strongly inhibit HIV-1 transcription of HIV-1 subtype B in chronically and acutely infected primary and cultured T cells. The effect of iron chelators on subtype C was less pronounced, suggesting that iron chelators may not be suitable for inhibiting all HIV-1 subtypes and indicating a subtype specificity of iron responsiveness.
FIG 4.

Effects of PPY-based iron chelators on HIV-1 transcription in acutely and chronically infected T cells and PBMCs. (A) Treatment of T cells acutely infected with a subtype B isolate of HIV-1 reduces viral transcription. A2R5.7 cells were incubated with the NL4-3 strain of HIV-1 (100 ng of p24 per ml) for 6 h at 37°C. The cells were washed twice with RPMI 1640 medium, incubated with fresh culture medium containing the indicated doses of PPY or PPYaT for 48 h, and harvested for RNA extraction and subsequent quantitative real-time RT-PCR with gag-specific primers. Results shown are means ± SD of three independent measurements. **, P ≤ 0.01. (B) T cells acutely infected with a subtype C isolate of HIV-1 display dose-dependent effects on viral transcription. A2R5.7 cells were inoculated with HIV1084i as described for panel A. The total RNA isolated from cells after 48 h of incubation with the indicated iron chelators was reverse transcribed and analyzed by real-time PCR with gag-specific primers. Results shown are means ± SD of three independent measurements. *, P ≤ 0.05. (C) Iron chelators inhibit viral transcription in chronically HIV-1-infected T cells, in a dose-dependent manner. J1.1 T cells (chronically HIV-1-infected cells derived from the Jurkat cell line) were preincubated with a cART cocktail (10 μM lamivudine, 10 μM emtricitabine, 10 μM tenofovir, and 10 μM indinavir) for 7 days to avoid HIV-1 replication and reinfection. After a double washing, the cells were incubated in fresh culture medium without drugs for 24 h and then for 48 h with the indicated doses of iron chelators. Subsequent isolation and quantitative analysis of HIV-1 RNA were performed as described for panel A. Results shown are means ± SD of three independent measurements. *, P ≤ 0.05; **, P ≤ 0.01. (D) Treatment with iron chelators inhibits HIV-1 transcription in acutely infected PBMCs. PBMCs from three different donors were inoculated with the dual-tropic HIV-1 strain 89.6 for 6 h and then, after a double washing, incubated in IL-2-supplemented RPMI medium for 72 h. The cells were washed again and then incubated with IL-2-containing medium supplemented with the indicated doses of iron chelators for 48 h. RNAs were isolated from cell lysates and analyzed as described for panel A. Results shown are means of values for triplicate samples. *, P ≤ 0.05; **, P ≤ 0.01.
Treatment with PPYaT or PPYeT leads to induction of IκB-α expression and redistribution of the NF-κB p65 subunit.
Because iron chelators were shown to result in accumulation of cells at G1 phase of the cell cycle, we examined the effects of PPY-based iron chelators on the expression levels of CDK2-associated cyclins and the CDK9-dependent genes for HLA and IκB-α. We used 18S RNA as a normalization housekeeping control, as it was shown to be the most reliable housekeeping control among several tested, including ACTB and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (37). Treatment with PPYaT or PPYeT induced expression of cyclin A and cyclin E (Fig. 5A). The slight increase in CDK2 expression was not statistically significant. There was no effect on HLA expression, while IκB-α expression showed a significant, >10-fold increase of the IκB-α protein level (Fig. 5B). No significant increase in IκB-α phosphorylation on the Ser32 residue was detected (Fig. 5B), suggesting that IκB-α was not undergoing degradation and that NF-κB levels might be modulated. We observed increased expression of p21 but no increase in its protein levels (data not shown), as previously reported by Le and Richardson (38). To determine whether the iron chelators affected the expression and cytoplasmic localization of NK-κB, we analyzed the level of NF-κB expression and its distribution between the nucleus and the cytoplasm. We found significantly higher levels of NF-κB localized in the cytoplasm in PPYeT- or PPYaT-treated cells compared to the PPY-treated controls (Fig. 5C). Analysis of NF-κB p65 expression in the cytoplasm and nucleus by immunoblotting showed localization of p65 in the cytoplasm in the chelator-treated cells, in contrast to the case for cells treated with the PPY control (Fig. 5D). Taken together, these results indicate that iron chelators have strong effects on NF-κB.
FIG 5.

PPY-based iron chelators induce expression of IκB-α and affect NF-κB cellular distribution. (A) 293T cells were treated with 10 μM PPY, PPYaT, or PPYeT. DMSO was used as a vehicle control. After 24 h of treatment, RNAs were extracted, reverse transcribed, and analyzed by real-time PCR for CDK2, cyclin A, cyclin E, HLA, and IκB-α, using 18S RNA as a housekeeping control gene. (B) 293T cells were treated as described for panel A and then lysed in SDS-PAGE loading buffer, resolved by 10% SDS-PAGE, and probed with antibodies against IκB-α, phosphorylated IκB-α, and tubulin, as a loading control. Results were quantified using ImageQuant software. Data are representative of two independent experiments. In the bottom panel, averages of results from two independent experiments are shown. (C) 293T cells were treated as described for panel A, fixed, and stained with primary antibodies against the NF-κB p65 subunit and with fluorescein isothiocyanate (FITC)-linked secondary antibodies. Photographs were taken on an Olympus IX 51 microscope at a magnification of ×200, and the pictures were scored for the distribution of NF-κB localized only in the cytoplasm or both in the nucleus and in the cytoplasm. Average data obtained from 6 separate fields are shown. (D) NF-κB p65 expression was analyzed by resolving cytoplasmic and nuclear extracts by 10% SDS-PAGE and immunoblotting with antibodies against the NF-κB p65 subunit and tubulin, as a loading control.
Iron chelators inhibit CDK2 activity and reduce the formation of large P-TEFb complex.
While we could not detect changes in the p21 expression levels, our previous studies suggested that CDK2 activity is reduced by chelator-treated cells (15, 16). To determine whether the PPY-based iron chelators inhibit CDK2 activity, 293T cells were cultured for 48 h at 37°C in the presence or absence of the iron chelators, using the PPY compound as a control. CDK2 was immunoprecipitated from cell lysates by use of anti-CDK2 antibodies and assayed using histone H1 as the substrate (see Materials and Methods for details). Treatment of cells with iron chelators reduced the CDK2 activity about 3-fold (Fig. 6A). To further examine the effects of PPY-based iron chelators on CDK9 activity, Flag-tagged CDK9 and cyclin T1 were expressed in 293T cells and treated with the iron chelators, CDK9 was immunoprecipitated from cell lysates by use of anti-Flag antibodies, and its activity was assayed with Rb-CTF peptide as the substrate. The CDK9 activity was increased in PPYeT-treated cells, with a slight decrease in PPYaT-treated cells (Fig. 6B). While an increase in CDK9 activity was unexpected, it reflects the increase of low-molecular-weight CDK9/cyclin T1, disfavoring HIV-1 transcription, which uses high-molecular-weight P-TEFb as a source of CDK9/cyclin T1. Thus, we investigated the effects of iron chelators on large and small P-TEFb complexes.
FIG 6.

Iron chelators inhibit CDK2 activity but increase CDK9 activity. (A) 293T cells were seeded into a 6-well plate and treated with 10 μM PPY or PPY-based iron chelators for 48 h. Cells were lysed, and CDK2 was immunoprecipitated using anti-CDK2 antibodies. Kinase assay was performed using histone H1 as the substrate. The bottom panel shows quantification results from two independent experiments. (B) 293T cells were treated with iron chelators as described for panel A. CDK9 was precipitated from cell lysates by use of anti-CDK9 antibodies and incubated with recombinant Rb-CTF in the presence of [32P]ATP. The reaction mixtures were resolved in a 10% SDS-Tris-glycine gel and analyzed by immunoblotting (WB) or on a PhosphorImager device (top panel). Quantification from the PhosphorImager is shown in the bottom panel, which shows average results from two separate experiments. *, P ≤ 0.01.
To determine the effects of iron chelators on CDK9/cyclin T1, we used a salt extraction procedure that we previously utilized to determine the effect of CDK2 knockdown on P-TEFb (13). CDK9 and cyclin T1 were expressed in 293T cells, and then the large and small P-TEFb complexes were extracted using low- and high-salt lysis buffers. As shown in Fig. 7A to C, the large P-TEFb complex isolated from the iron chelator-treated cells contained substantially smaller amounts of both CDK9 and cyclin T1. In contrast, the level of cyclin T1 was significantly increased in the small complex (Fig. 7A and C). These results suggest that CDK9/cyclin T1 associates less efficiently with the large P-TEFb complex in iron chelator-treated cells. This result is consistent with our previous finding that CDK2 knockdown decreases the amount of the large P-TEFb complex (13).
FIG 7.
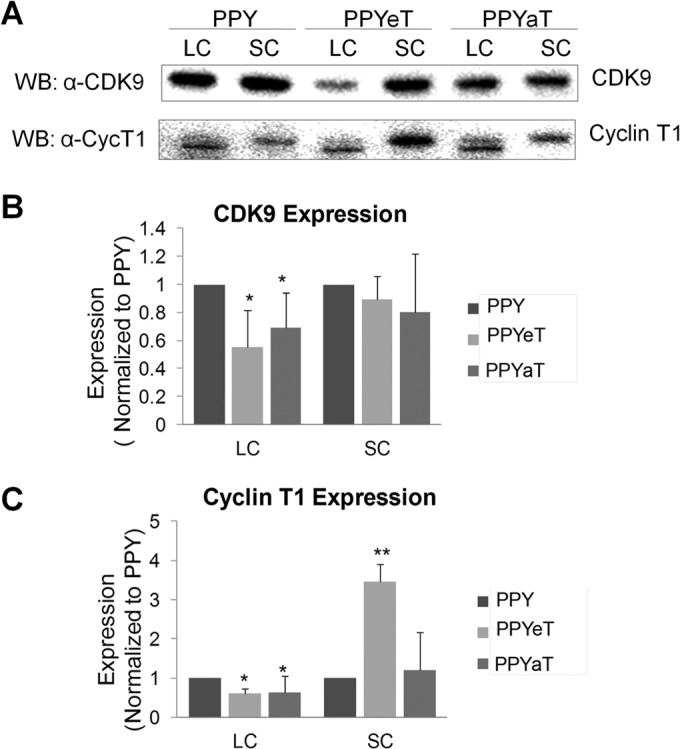
Treatment with iron chelators reduces the large P-TEFb complex. 293T cells were treated with 10 μM PPY or PPY-based iron chelators and then extracted sequentially to obtain low-salt and high-salt extracts, as described in Materials and Methods. Low-salt extracts contained the large P-TEFb complex (LC), and high-salt extracts contained the small P-TEFb complex (SC). The amount of material was normalized to the total protein amounts in the extracts, resolved by 10% SDS-PAGE, and analyzed by immunoblotting for CDK9 and cyclin T1. (A) Representative immunoblotting results. (B and C) CDK9 (B) and cyclin T1 (C) expression, shown as averages of results from two separate experiments. *, P ≤ 0.05; **, P ≤ 0.01.
Iron chelators reduce HIV-1 mRNA expression but do not inhibit HIV-1 reverse transcription.
To further elucidate the mechanism of HIV-1 inhibition by PPY-based iron chelators, we analyzed HIV-1 mRNA expression in CEM T cells infected with HIV-1 Luc virus. Expression of Gag- and Env-encoding mRNAs was significantly reduced by treatment with PPYeT or PPYaT in comparison to the case for DMSO- or PPY-treated controls (Fig. 8A). We noticed that DMSO treatment induced Gag and Env expression (Fig. 8A), in accord with previous observations (39). We also analyzed whether iron chelators have an effect on HIV-1 reverse transcription. We analyzed early RT by quantifying HIV-1 DNA for the early LTR (30). The established HIV-1 inhibitor AZT showed a statistically significant effect on HIV-1 LTR expression, whereas iron chelators showed no effect (Fig. 8B). Thus, iron chelators had no inhibitory effect on HIV-1 RT while reducing HIV-1 transcription, as evidenced by the reduction of HIV-1 Gag and Env gene expression.
FIG 8.

Effects of PPY-based iron chelators on HIV-1 mRNA expression, HIV-1 reverse transcription, and basal HIV-1 transcription. (A and B) THP-1 cells were left uninfected or infected with HIV-1 Luc and then left untreated or treated with DMSO, AZT, 1 μM PPY (control compound), 1 μM PPYeT, or 1 μM PPYaT, as indicated, for 48 h (A) or 6 h (B). RNA (A) or DNA (B) was extracted. RNA was reverse transcribed and analyzed with primers for the HIV-1 gag and env genes by real-time PCR on a Roche 4800 machine, using 18S RNA as a reference. DNA was analyzed by real-time PCR on a Roche 4800 machine, using primers for early and late LTRs and with the β-globin gene as a reference. (C and D) Effects of PPY-based iron chelators on basal HIV-1 transcription. 293T cells were transiently transfected with vectors expressing the HIV LTR followed by the luciferase reporter gene (WT HIV LTR 2×NFκB 3×SP1 [C], HIV LTR ΔNFκB 3×SP1 [D], or HIV LTR 2×NFκB ΔSP1 [E]; see Materials and Methods for details on the vectors). For normalization, the cells were also cotransfected with a GFP-expressing vector. At 24 h posttransfection, the cells were treated with 10 μM PPY-based iron chelators or the PPY control for 24 h. The cells were then lysed, and luciferase activity was measured. GFP fluorescence was measured in parallel and used for normalization. *, P ≤ 0.01.
Inhibition of basal HIV-1 transcription by PPY-based iron chelators.
To further investigate the effects of iron chelators on HIV-1 transcription, we examined their effects in 293T cells transfected with vectors expressing luciferase under the control of various HIV-1 LTR mutants (see Materials and Methods for details). Treatment with the PPYaT or PPYeT iron chelator had a moderate (<2-fold) but statistically significant inhibitory effect on wild-type (WT) HIV-1 LTR-driven transcription (Fig. 8C). We also observed similar effects on the HIV-1 LTR with deletions of NF-κB or Sp1 sites (Fig. 8D and E). HIV-1 basal transcription is regulated by Sp1 and NF-κB (23, 25), in conjunction with NF-κB that cooperates with Tat in Tat-activated HIV-1 transcription (2). Thus, the relatively moderate effect of iron chelators on basal HIV-1 transcription could be amplified because of the inability of HIV-1 Tat to engage Sp1 or NF-κB for the induction of HIV-1 transcription.
DISCUSSION
Our results presented here demonstrate that synthetically modified novel PPY-based iron chelators with a benzyl group substituted for the phenyl group are effective at chelating intracellular iron and inhibiting HIV-1 transcription. The PPY-based compounds chelated cellular labile iron with an efficiency similar to that of SIH. They also promoted synthesis of transferrin receptor mRNA, suggesting that cellular iron was substantially reduced. The PPY-based iron chelators are similar to previously studied HIV-1-inhibitory benzoylpyridine thiosemicarbazone compounds that also chelate cellular labile iron similarly to SIH (40). Only one aromatic ring was shown to be required to coordinate the binding of iron (31). Therefore, the benzyl moiety of PPY-based compounds can be utilized further for optimization of other properties, such as adjustment of the ADME-Tox (absorption, distribution, metabolism, excretion, and toxicity) profile. We observed a good therapeutic window for the PPYaT chelator in CEM T cells and PBMCs over the concentration range tested (Fig. 3). PPYeT, on the other hand, showed some toxicity in both CEM T cells and PBMCs and had a less favorable therapeutic window, consistent with its higher potency for chelating iron.
Previously, administration of Dp44mT in mice showed little alteration in hematological and biochemical indices (0.4 to 0.75 mg/kg of body weight/day) but induced antitumor activity (5). The BpT-based iron chelators showed more antineoplastic activity than their DpT homologs in vitro (6). Recently, Dp44mT showed a significant amount of methemoglobin (metHb) formation in intact red blood cells and in mice (31). A modified analog, di-2-pyridylketone-4-cyclohexyl-4-methyl-3-thiosemicarbazone, did not generate production of metHb and thus was proposed to be suitable for further optimization (31). Also, BpT was shown to induce metHb, whereas its analog, the lipophilic t-BuBpT chelator, was a less potent inducer of metHb (41). Thus, PPY-based iron chelators may also potentially induce MetHb formation, and further optimization may be needed to alleviate this effect. The benzyl moiety of PPY-based compounds can be used for further optimization, as mentioned above.
In early studies, iron chelators were shown to inhibit cell cycle progression, which coincided with the inhibition of CDK2 enzymatic activity (34, 35). Iron chelators were shown to increase p21 mRNA and protein expression (42). The expression of p21 was also shown to be increased by HIF-1α, which displaced c-myc on the p21 promoter (43). We previously demonstrated that the iron chelators 311, ICL670, Bp4eT, and Dp4eT inhibit the activity of CDK2 (15, 16). Here we showed that PPY-based iron chelators inhibited cell cycle progression of promonocytic THP-1 cells, CEM T cells, and epithelial 293T cells. We were not able to detect increased p21 protein expression. However, reduced p21 expression can also be inhibitory for CDK2/cyclin E, which requires low levels of p21 to be used as a scaffold for the assembly of CDK2/cyclin E complexes (44). In agreement with our previous studies, CDK2 activity was significantly reduced. Whether the decreased CDK2 activity might be mediated by a deregulation of p21 remains to be determined. The decrease in the cell cycle progression coincided with the increased expression of cyclin A and cyclin E, whereas the expression of CDK2 was unchanged. CDK9 activity was increased in the PPYeT-treated cells, likely due to the shift of CDK9/cyclin T1 from the large to the small P-TEFb complex, which was more prominent in the PPYeT-treated cells. We observed a similar effect in CDK2 knockdown cells (not shown). We recently showed that CDK2 phosphorylates CDK9 Ser90 and that a lack of this phosphorylation disrupts formation of the large P-TEFb complex (13). Thus, the reduction of the large P-TEFb complex was likely due to the reduction of CDK2 activity and CDK9 phosphorylation.
We observed a strong inhibitory effect of PPYaT on cultured T cells infected with HIV-1 subtype B, but less so with HIV subtype C. Previously, subtype C was shown to exhibit slower replication due to a different enzymatic activity of its reverse transcriptase (45). Thus, iron chelators may not be efficient against all HIV subtypes, and whether they have an effect on other HIV subtypes remains to be determined. We also observed an inhibition of HIV-1 gene expression in latently infected T cells and also an inhibition of acute HIV-1 infection of primary PBMCs by HIV-1 subtype B. These results indicate that PPY-based iron chelators are likely to inhibit HIV-1 transcription. The EC50s varied depending on the cell types used. The lowest EC50s were observed in CEM T cells, in accord with our previous study of HIV-1 inhibition by BpT and DpT iron chelators, which showed low nanomolar EC50s in CEM T cells (15). These low EC50s likely reflect an unusual sensitivity of this leukemia cell line to iron chelators, which is reflected in the relatively high toxicity of the chelators in this cell line. In PBMCs, the chelators showed much higher EC50s for HIV-1 inhibition but also had much less toxicity. Ultimately, in vivo testing of the chelators in an animal model, such as HIV-1-infected humanized mice, will determine their in vivo toxicity and efficacy for HIV-1 inhibition.
CDK9/cyclin T1 induces expression of IL-8 and Gro-β (46) and represses expression of major histocompatibility complex (MHC) class II genes, such as HLA-DRA (47). We have previously shown that hypoxia decreases the expression of IκB-α but not that of Gro-β or HLA-DRA (48). Data presented in this report suggest that inhibition of CDK9-depedent genes by PPY-based iron chelators could lead to the overexpression of IκB-α but have no effect on HLA expression. To our knowledge, this is the first demonstration of IκB-α expression being linked to the reduction of cellular iron. HIV-1 basal transcription is largely regulated by the Sp1 transcription factor (23), whereas in Tat-activated transcription, NF-κB plays an important regulatory role by acting in concert with Tat and CDK9/cyclin T1 (2). NF-κB might recruit CDK9/cyclin T1 to the HIV-1 LTR in a cooperative manner (20, 21), in part because of the interaction of Tat with the p65 subunit of NF-κB through NFBP (22). In the absence of TAR RNA, cyclin T1 can be recruited to the LTR by Sp1 (24). NF-κB regulates the expression of a large number of genes that are critical for induction of apoptosis, viral replication, tumorigenesis, inflammation, and various autoimmune diseases (49). NF-κB is composed of homo- and heterodimers of five members of the Rel family, including NF-κB 1 (p50), NF-κB 2 (p52), RelA, RelB, and c-Rel (Rel) (49). Inactive NF-κB is sequestered in the cytoplasm, bound by members of the IκB family of inhibitory proteins, which include IκB-α, IκB-β, IκB-γ, and IκB-δ (49). IκB phosphorylation by IκB kinases (IKKs) leads to their ubiquitination and subsequent degradation, which expose the nuclear localization signal of NF-κB and help to translocate it to the nucleus (50). Our analysis showed that increased expression of IκB-α coincides with the accumulation of NF-κB in the cytoplasm and a reduction of NF-κB in the nucleus. Thus, in the PPY-based iron chelator-treated cells, NF-κB is unavailable for HIV-1 transcription activation, contributing to the inhibition of HIV-1. Similarly, IκB-α expression was increased in stable CDK2 knockdown cell lines (data not shown), suggesting that CDK2 negatively regulates its expression.
In addition to the inhibition of HIV-1 transcription, iron depletion may affect other steps in HIV-1 replication (reviewed in reference 51). During viral entry, HIV-1 replication is dependent on the activity of a host cell ribonucleotide reductase (RNR) that contains nonheme iron, which is important for its enzymatic activity (52). Recently, expression of p21 was shown to downregulate the expression of the RNR2 subunit, which decreased the intracellular deoxynucleoside triphosphate (dNTP) pool and impaired HIV-1 and simian immunodeficiency virus (SIV) reverse transcription (53). Expression of p21 inhibited RNR2 transcription by repressing the E2F1 transcription factor which activates RNR2 transcription (53). While we have not analyzed the dNTP pools, we have not seen a reduction in HIV-1 reverse transcription, so PPY-based iron chelators are not likely to affect RNR2. Export of unspliced HIV-1 mRNA requires the HIV-1 Rev protein and host elongation factor 5A (eIF5α), which contains N-epsilon-4-amino-2-hydroxybutyl-lysine (hypusine), produced by deoxyhypusine hydroxylase (DOHH), an iron-containing enzyme (54). The topical fungicide ciclopirox and the iron chelator deferiprone were shown to inhibit HIV-1 gene expression by interfering with the hydroxylation step in the hypusine modification of eIF5α (55). More recently, ciclopirox and deferiprone were shown to induce apoptosis though mitochondrial membrane depolarization in HIV-infected T cells, thus promoting selective elimination of HIV-1-infected cells in long-term culture (56). Whether PPY-based iron chelators can specifically eliminate HIV-1-infected cells remains to be determined.
Finally, it must be noted that the very different mechanism of action of these chelators from those of other established antiviral therapies may present an advantage for the future use of iron chelators. They may be potentially useful for treatment of resistant viral infections, which are a major problem for the existing antiretroviral drugs. Many common chemotherapeutic drugs currently in clinical use, such as doxorubicin, display marked cytotoxicity profiles in vitro (57) but are well tolerated at appropriate doses in vivo and have led to vast improvements in cancer treatment. Thus, as discussed above, PPY-based iron chelators can be modified further to reduce their potential pharmacological toxicity in vivo and to improve their potential future use as anti-HIV-1 therapeutic agents.
Taken together, the low EC50s of PPY-based iron chelators and their good therapeutic window indicate their application in future potentially useful antiretroviral therapeutics. Hence, these compounds may pave the way for treatment of subjects infected with HIV-1 with a high iron load, a condition shown to be related to the progression of HIV-1 infection (58).
ACKNOWLEDGMENTS
This work was supported by NIH research grants (1SC1GM082325, 1P50HL118006-01, 1R01HL125005-01, U19AI109664-01, R01AI043894, R21AI114490, R21AI113140, and 5G12MD007597) and a District of Columbia Developmental Center for AIDS Research grant (P30AI087714).
We thank the NIH AIDS Research and Reference Reagent Program for the pHEF-VSVG expression vector (courtesy of Lung-Ji Chang) and pNL4-3.Luc.R−E− (courtesy of Nathaniel Landau).
No financial competing interests are declared.
N.K. conducted the research, analyzed results, and wrote the manuscript. S.I. conducted experiments with chronically and acutely infected cultured and primary cells and participated in manuscript preparation. D.B., X.N., and M.X. conducted the research and analyzed results. D.K. and K.G. designed and synthesized iron chelators and contributed to the preparation of the manuscript. X.L. analyzed the purity of iron chelators by using LC-MS. S.D. and F.K. participated in the study design and writing of the manuscript. S.N. takes primary responsibility for the paper. S.N. conducted the design of and performed some experiments, analyzed results, and drafted the manuscript.
The findings and conclusions in this paper have not been disseminated formally by the Food and Drug Administration and should not be construed to represent any Agency determination or policy.
Footnotes
Published ahead of print 25 August 2014
REFERENCES
- 1.Lu H, Li Z, Xue Y, Zhou Q. 2013. Viral-host interactions that control HIV-1 transcriptional elongation. Chem. Rev. 113:8567–8582. 10.1021/cr400120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.West MJ, Lowe AD, Karn J. 2001. Activation of human immunodeficiency virus transcription in T cells revisited: NF-kappaB p65 stimulates transcriptional elongation. J. Virol. 75:8524–8537. 10.1128/JVI.75.18.8524-8537.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He N, Jahchan NS, Hong E, Li Q, Bayfield MA, Maraia RJ, Luo K, Zhou Q. 2008. A La-related protein modulates 7SK snRNP integrity to suppress P-TEFb-dependent transcriptional elongation and tumorigenesis. Mol. Cell 29:588–599. 10.1016/j.molcel.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krueger BJ, Jeronimo C, Roy BB, Bouchard A, Barrandon C, Byers SA, Searcey CE, Cooper JJ, Bensaude O, Cohen EA, Coulombe B, Price DH. 2008. LARP7 is a stable component of the 7SK snRNP while P-TEFb, HEXIM1 and hnRNP A1 are reversibly associated. Nucleic Acids Res. 36:2219–2229. 10.1093/nar/gkn061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Markert A, Grimm M, Martinez J, Wiesner J, Meyerhans A, Meyuhas O, Sickmann A, Fischer U. 2008. The La-related protein LARP7 is a component of the 7SK ribonucleoprotein and affects transcription of cellular and viral polymerase II genes. EMBO Rep. 9:569–575. 10.1038/embor.2008.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barboric M, Lenasi T, Chen H, Johansen EB, Guo S, Peterlin BM. 2009. 7SK snRNP/P-TEFb couples transcription elongation with alternative splicing and is essential for vertebrate development. Proc. Natl. Acad. Sci. U. S. A. 106:7798–7803. 10.1073/pnas.0903188106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeronimo C, Forget D, Bouchard A, Li Q, Chua G, Poitras C, Therien C, Bergeron D, Bourassa S, Greenblatt J, Chabot B, Poirier GG, Hughes TR, Blanchette M, Price DH, Coulombe B. 2007. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol. Cell 27:262–274. 10.1016/j.molcel.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M. 2010. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 38:439–451. 10.1016/j.molcel.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, Zhou Q. 2010. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 38:428–438. 10.1016/j.molcel.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Z, Zhu Q, Luo K, Zhou Q. 2001. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 414:317–322. 10.1038/35104575. [DOI] [PubMed] [Google Scholar]
- 11.Nguyen VT, Kiss T, Michels AA, Bensaude O. 2001. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 414:322–325. 10.1038/35104581. [DOI] [PubMed] [Google Scholar]
- 12.Sedore SC, Byers SA, Biglione S, Price JP, Maury WJ, Price DH. 2007. Manipulation of P-TEFb control machinery by HIV: recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 35:4347–4358. 10.1093/nar/gkm443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Breuer D, Kotelkin A, Ammosova T, Kumari N, Ivanov A, Ilatovskiy AV, Beullens M, Roane PR, Bollen M, Petukhov MG, Kashanchi F, Nekhai S. 2012. CDK2 regulates HIV-1 transcription by phosphorylation of CDK9 on serine 90. Retrovirology 9:94. 10.1186/1742-4690-9-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jerebtsova M, Kumari N, Xu M, de Melo GB, Niu X, Jeang KT, Nekhai S. 2012. HIV-1 resistant CDK2-knockdown macrophage-like cells generated from 293T cell-derived human induced pluripotent stem cells. Biology (Basel) 1:175–195. 10.3390/biology1020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Debebe Z, Ammosova T, Breuer D, Lovejoy DB, Kalinowski DS, Kumar K, Jerebtsova M, Ray P, Kashanchi F, Gordeuk VR, Richardson DR, Nekhai S. 2011. Iron chelators of the di-2-pyridylketone thiosemicarbazone and 2-benzoylpyridine thiosemicarbazone series inhibit HIV-1 transcription: identification of novel cellular targets—iron, cyclin-dependent kinase (CDK) 2, and CDK9. Mol. Pharmacol. 79:185–196. 10.1124/mol.110.069062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Debebe Z, Ammosova T, Jerebtsova M, Kurantsin-Mills J, Niu X, Charles S, Richardson DR, Ray PE, Gordeuk VR, Nekhai S. 2007. Iron chelators ICL670 and 311 inhibit HIV-1 transcription. Virology 367:324–333. 10.1016/j.virol.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H, Li C, Huang J, Cung T, Seiss K, Beamon J, Carrington MF, Porter LC, Burke PS, Yang Y, Ryan BJ, Liu R, Weiss RH, Pereyra F, Cress WD, Brass AL, Rosenberg ES, Walker BD, Yu XG, Lichterfeld M. 2011. CD4+ T cells from elite controllers resist HIV-1 infection by selective upregulation of p21. J. Clin. Invest. 121:1549–1560. 10.1172/JCI44539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ammosova T, Berro R, Jerebtsova M, Jackson A, Charles S, Klase Z, Southerland W, Gordeuk VR, Kashanchi F, Nekhai S. 2006. Phosphorylation of HIV-1 Tat by CDK2 in HIV-1 transcription. Retrovirology 3:78. 10.1186/1742-4690-3-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adesina SK, Holly A, Kramer-Marek G, Capala J, Akala EO. 2014. Polylactide-based paclitaxel-loaded nanoparticles fabricated by dispersion polymerization: characterization, evaluation in cancer cell lines, and preliminary biodistribution studies. J. Pharm. Sci. 103:2546–2555. 10.1002/jps.24061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM. 2001. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol. Cell 8:327–337. 10.1016/S1097-2765(01)00314-8. [DOI] [PubMed] [Google Scholar]
- 21.Deng L, de la Fuente C, Fu P, Wang L, Donnelly R, Wade JD, Lambert P, Li H, Lee CG, Kashanchi F. 2000. Acetylation of HIV-1 Tat by CBP/P300 increases transcription of integrated HIV-1 genome and enhances binding to core histones. Virology 277:278–295. 10.1006/viro.2000.0593. [DOI] [PubMed] [Google Scholar]
- 22.Sweet T, Sawaya BE, Khalili K, Amini S. 2005. Interplay between NFBP and NF-kappaB modulates tat activation of the LTR. J. Cell. Physiol. 204:375–380. 10.1002/jcp.20419. [DOI] [PubMed] [Google Scholar]
- 23.Jochmann R, Thurau M, Jung S, Hofmann C, Naschberger E, Kremmer E, Harrer T, Miller M, Schaft N, Sturzl M. 2009. O-linked N-acetylglucosaminylation of Sp1 inhibits the human immunodeficiency virus type 1 promoter. J. Virol. 83:3704–3718. 10.1128/JVI.01384-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yedavalli VS, Benkirane M, Jeang KT. 2003. Tat and trans-activation-responsive (TAR) RNA-independent induction of HIV-1 long terminal repeat by human and murine cyclin T1 requires Sp1. J. Biol. Chem. 278:6404–6410. 10.1074/jbc.M209162200. [DOI] [PubMed] [Google Scholar]
- 25.Chun RF, Semmes OJ, Neuveut C, Jeang KT. 1998. Modulation of Sp1 phosphorylation by human immunodeficiency virus type 1 Tat. J. Virol. 72:2615–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westervelt P, Trowbridge DB, Epstein LG, Blumberg BM, Li Y, Hahn BH, Shaw GM, Price RW, Ratner L. 1992. Macrophage tropism determinants of human immunodeficiency virus type 1 in vivo. J. Virol. 66:2577–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grisson RD, Chenine AL, Yeh LY, He J, Wood C, Bhat GJ, Xu W, Kankasa C, Ruprecht RM. 2004. Infectious molecular clone of a recently transmitted pediatric human immunodeficiency virus clade C isolate from Africa: evidence of intraclade recombination. J. Virol. 78:14066–14069. 10.1128/JVI.78.24.14066-14069.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gomez-Gonzalo M, Carretero M, Rullas J, Lara-Pezzi E, Aramburu J, Berkhout B, Alcami J, Lopez-Cabrera M. 2001. The hepatitis B virus X protein induces HIV-1 replication and transcription in synergy with T-cell activation signals: functional roles of NF-kappaB/NF-AT and SP1-binding sites in the HIV-1 long terminal repeat promoter. J. Biol. Chem. 276:35435–35443. 10.1074/jbc.M103020200. [DOI] [PubMed] [Google Scholar]
- 29.Epsztejn S, Kakhlon O, Glickstein H, Breuer W, Cabantchik I. 1997. Fluorescence analysis of the labile iron pool of mammalian cells. Anal. Biochem. 248:31–40. 10.1006/abio.1997.2126. [DOI] [PubMed] [Google Scholar]
- 30.Vermeire J, Naessens E, Vanderstraeten H, Landi A, Iannucci V, Van Nuffel A, Taghon T, Pizzato M, Verhasselt B. 2012. Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLoS One 7:e50859. 10.1371/journal.pone.0050859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quach P, Gutierrez E, Basha MT, Kalinowski DS, Sharpe PC, Lovejoy DB, Bernhardt PV, Jansson PJ, Richardson DR. 2012. Methemoglobin formation by triapine, di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone (Dp44mT), and other anticancer thiosemicarbazones: identification of novel thiosemicarbazones and therapeutics that prevent this effect. Mol. Pharmacol. 82:105–114. 10.1124/mol.112.078964. [DOI] [PubMed] [Google Scholar]
- 32.Simunek T, Boer C, Bouwman RA, Vlasblom R, Versteilen AM, Sterba M, Gersl V, Hrdina R, Ponka P, de Lange JJ, Paulus WJ, Musters RJ. 2005. SIH—a novel lipophilic iron chelator—protects H9c2 cardiomyoblasts from oxidative stress-induced mitochondrial injury and cell death. J. Mol. Cell. Cardiol. 39:345–354. 10.1016/j.yjmcc.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 33.Rouault TA. 2006. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2:406–414. 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 34.Lucas JJ, Szepesi A, Domenico J, Takase K, Tordai A, Terada N, Gelfand EW. 1995. Effects of iron-depletion on cell cycle progression in normal human T lymphocytes: selective inhibition of the appearance of the cyclin A-associated component of the p33cdk2 kinase. Blood 86:2268–2280. [PubMed] [Google Scholar]
- 35.Pahl PM, Yan XD, Hodges YK, Rosenthal EA, Horwitz MA, Horwitz LD. 2000. An exochelin of Mycobacterium tuberculosis reversibly arrests growth of human vascular smooth muscle cells in vitro. J. Biol. Chem. 275:17821–17826. 10.1074/jbc.M909918199. [DOI] [PubMed] [Google Scholar]
- 36.He J, Landau NR. 1995. Use of a novel human immunodeficiency virus type 1 reporter virus expressing human placental alkaline phosphatase to detect an alternative viral receptor. J. Virol. 69:4587–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuchipudi SV, Tellabati M, Nelli RK, White GA, Perez BB, Sebastian S, Slomka MJ, Brookes SM, Brown IH, Dunham SP, Chang KC. 2012. 18S rRNA is a reliable normalisation gene for real time PCR based on influenza virus infected cells. Virol. J. 9:230. 10.1186/1743-422X-9-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le NT, Richardson DR. 2003. Potent iron chelators increase the mRNA levels of the universal cyclin-dependent kinase inhibitor p21(CIP1/WAF1), but paradoxically inhibit its translation: a potential mechanism of cell cycle dysregulation. Carcinogenesis 24:1045–1058. 10.1093/carcin/bgg042. [DOI] [PubMed] [Google Scholar]
- 39.Klebanoff SJ, Mehlin C, Headley CM. 1997. Activation of the HIV type 1 long terminal repeat and viral replication by dimethylsulfoxide and related solvents. AIDS Res. Hum. Retroviruses 13:1221–1227. 10.1089/aid.1997.13.1221. [DOI] [PubMed] [Google Scholar]
- 40.Debebe Z, Nekhai S, Ashenafi M, Lovejoy DB, Kalinowski DS, Gordeuk VR, Byrnes WM, Richardson DR, Karla PK. 2012. Development of a sensitive HPLC method to measure in vitro permeability of E- and Z-isomeric forms of thiosemicarbazones in Caco-2 monolayers. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 906:25–32. 10.1016/j.jchromb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stefani C, Jansson PJ, Gutierrez E, Bernhardt PV, Richardson DR, Kalinowski DS. 2013. Alkyl substituted 2′-benzoylpyridine thiosemicarbazone chelators with potent and selective anti-neoplastic activity: novel ligands that limit methemoglobin formation. J. Med. Chem. 56:357–370. 10.1021/jm301691s. [DOI] [PubMed] [Google Scholar]
- 42.Moon SK, Jung SY, Choi YH, Lee YC, Patterson C, Kim CH. 2004. PDTC, metal chelating compound, induces G1 phase cell cycle arrest in vascular smooth muscle cells through inducing p21Cip1 expression: involvement of p38 mitogen activated protein kinase. J. Cell. Physiol. 198:310–323. 10.1002/jcp.10728. [DOI] [PubMed] [Google Scholar]
- 43.Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. 2004. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 23:1949–1956. 10.1038/sj.emboj.7600196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gartel AL. 2006. Inducer and inhibitor: “antagonistic duality” of p21 in differentiation. Leuk. Res. 30:1215–1216. 10.1016/j.leukres.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 45.Iordanskiy S, Waltke M, Feng Y, Wood C. 2010. Subtype-associated differences in HIV-1 reverse transcription affect the viral replication. Retrovirology 7:85. 10.1186/1742-4690-7-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nowak DE, Tian B, Jamaluddin M, Boldogh I, Vergara LA, Choudhary S, Brasier AR. 2008. RelA Ser276 phosphorylation is required for activation of a subset of NF-kappaB-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol. Cell. Biol. 28:3623–3638. 10.1128/MCB.01152-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanazawa S, Peterlin BM. 2001. Combinations of dominant-negative class II transactivator, p300 or CDK9 proteins block the expression of MHC II genes. Int. Immunol. 13:951–958. 10.1093/intimm/13.7.951. [DOI] [PubMed] [Google Scholar]
- 48.Charles S, Ammosova T, Cardenas J, Foster A, Rotimi J, Jerebtsova M, Ayodeji AA, Niu X, Ray PE, Gordeuk VR, Kashanchi F, Nekhai S. 2009. Regulation of HIV-1 transcription at 3% versus 21% oxygen concentration. J. Cell. Physiol. 221:469–479. 10.1002/jcp.21882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aboul-ela F, Karn J, Varani G. 1996. Structure of HIV-1 TAR RNA in the absence of ligands reveals a novel conformation of the trinucleotide bulge. Nucleic Acids Res. 24:3974–3981. 10.1093/nar/24.20.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen ZJ. 2005. Ubiquitin signalling in the NF-kappaB pathway. Nat. Cell Biol. 7:758–765. 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nekhai S, Kumari N, Dhawan S. 2013. Role of cellular iron and oxygen in the regulation of HIV-1 infection. Future Virol. 8:301–311. 10.2217/fvl.13.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsimberidou AM, Alvarado Y, Giles FJ. 2002. Evolving role of ribonucleoside reductase inhibitors in hematologic malignancies. Expert Rev. Anticancer Ther. 2:437–448. 10.1586/14737140.2.4.437. [DOI] [PubMed] [Google Scholar]
- 53.Allouch A, David A, Amie SM, Lahouassa H, Chartier L, Margottin-Goguet F, Barre-Sinoussi F, Kim B, Saez-Cirion A, Pancino G. 2013. p21-mediated RNR2 repression restricts HIV-1 replication in macrophages by inhibiting dNTP biosynthesis pathway. Proc. Natl. Acad. Sci. U. S. A. 110:E3997–E4006. 10.1073/pnas.1306719110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim YS, Kang KR, Wolff EC, Bell JK, McPhie P, Park MH. 2006. Deoxyhypusine hydroxylase is a Fe(II)-dependent, HEAT-repeat enzyme. Identification of amino acid residues critical for Fe(II) binding and catalysis [corrected]. J. Biol. Chem. 281:13217–13225. 10.1074/jbc.M601081200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoque M, Hanauske-Abel HM, Palumbo P, Saxena D, D'Alliessi Gandolfi D, Park MH, Pe'ery T, Mathews MB. 2009. Inhibition of HIV-1 gene expression by ciclopirox and deferiprone, drugs that prevent hypusination of eukaryotic initiation factor 5A. Retrovirology 6:90. 10.1186/1742-4690-6-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hanauske-Abel HM, Saxena D, Palumbo PE, Hanauske AR, Luchessi AD, Cambiaghi TD, Hoque M, Spino M, Gandolfi DD, Heller DS, Singh S, Park MH, Cracchiolo BM, Tricta F, Connelly J, Popowicz AM, Cone RA, Holland B, Pe'ery T, Mathews MB. 2013. Drug-induced reactivation of apoptosis abrogates HIV-1 infection. PLoS One 8:e74414. 10.1371/journal.pone.0074414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yuan S, Zhang X, Lu L, Xu C, Yang W, Ding J. 2004. Anticancer activity of methoxymorpholinyl doxorubicin (PNU 152243) on human hepatocellular carcinoma. Anticancer Drugs 15:641–646. 10.1097/01.cad.0000132238.23050.f5. [DOI] [PubMed] [Google Scholar]
- 58.Gordeuk VR, Delanghe JR, Langlois MR, Boelaert JR. 2001. Iron status and the outcome of HIV infection: an overview. J. Clin. Virol. 20:111–115. 10.1016/S1386-6532(00)00134-7. [DOI] [PubMed] [Google Scholar]