

Abstract
Mitochondrial thymidine kinase 2 (TK2) and deoxyguanosine kinase (dGK) catalyze the initial phosphorylation of deoxynucleosides in the synthesis of the DNA precursors required for mitochondrial DNA (mtDNA) replication and are essential for mitochondrial function. Antiviral nucleosides are known to cause toxic mitochondrial side effects. Here, we examined the effects of 3′-azido-2′,3′-dideoxythymidine (AZT) (zidovudine) on mitochondrial TK2 and dGK levels and found that AZT treatment led to downregulation of mitochondrial TK2 and dGK in U2OS cells, whereas cytosolic deoxycytidine kinase (dCK) and thymidine kinase 1 (TK1) levels were not affected. The AZT effects on mitochondrial TK2 and dGK were similar to those of oxidants (e.g., hydrogen peroxide); therefore, we examined the oxidative effects of AZT. We found a modest increase in cellular reactive oxygen species (ROS) levels in the AZT-treated cells. The addition of uridine to AZT-treated cells reduced ROS levels and protein oxidation and prevented the degradation of mitochondrial TK2 and dGK. In organello studies indicated that the degradation of mitochondrial TK2 and dGK is a mitochondrial event. These results suggest that downregulation of mitochondrial TK2 and dGK may lead to decreased mitochondrial DNA precursor pools and eventually mtDNA depletion, which has significant implications for the regulation of mitochondrial nucleotide biosynthesis and for antiviral therapy using nucleoside analogs.
INTRODUCTION
Nucleoside analogs are widely used as antiviral and anticancer agents, and today >50% of the FDA-approved antiviral and anticancer drugs are nucleoside or nucleobase analogs. These analogs are administered as prodrugs that need to be activated by cellular enzymes to exert their therapeutic potential. Deoxynucleoside kinases catalyze the initial phosphorylation of nucleoside analogs; once phosphorylated, these nucleotides are trapped inside the cell and are further metabolized to their active forms, which interact with the final targets. There are four deoxynucleoside kinases in mammalian cells, i.e., cytosolic thymidine kinase 1 (TK1) and deoxycytidine kinase (dCK) and mitochondrial thymidine kinase 2 (TK2) and deoxyguanosine kinase (dGK) (1, 2). Recent studies using TK1- and dCK-knockout mouse models indicate that cytosolic TK1 and dCK regulate hematopoiesis by linking salvage deoxynucleotide synthesis and replication stress (3, 4). The mitochondrial enzymes TK2 and dGK are vital for mitochondrial function, because deficiency in either causes severe mitochondrial DNA (mtDNA) depletion syndrome (MDS) in human patients and in TK2-knockout mouse models (5–8). TK2 has also been shown to play an important role in providing dTTP for nuclear DNA repair and thus genomic stability in quiescent cells (9).
The nucleoside analogs used in antiviral and anticancer therapy are often associated with mitochondrial toxicity. Mitochondrial myopathy, cardiomyopathy, and lipodystrophy associated with anti-HIV treatment using zidovudine (AZT) or 2′,3′-dideoxyinosine (ddI) (didanosine) were reported shortly after the introduction of these analogs in anti-HIV therapy in the 1990s. Furthermore, 1-β-arabinofuranosylcytosine (araC) (cytarabine) and 5-fluorouracil (Adrucil), which are used in anticancer therapy, are associated with cardiotoxicity and neurotoxicity (10, 11). The mechanism underlying the mitochondrial toxic effects of these types of nucleoside analogs has been postulated to occur via the inhibition of mitochondrial DNA (mtDNA) polymerase by the triphosphates of these analogs. However, recent studies using peripheral blood mononuclear cells from HIV-infected individuals treated with antiviral agents showed that depletion of both ribonucleotide and deoxynucleotide pools may be responsible for the observed mitochondrial toxicity independent of mtDNA polymerase inhibition (12). Other studies suggest that inhibition of mitochondrial TK2 and dGK may be a major contributing factor in nucleoside analog-induced mitochondrial toxicity, and the symptoms of AIDS patients treated with AZT or ddI resemble those reported for MDS patients (5, 6, 10, 11).
The nucleoside analogs used in antiviral therapy are often substrates or inhibitors of TK2 and dGK. Some of these analogs are also known to induce oxidative stress via increased reactive oxygen species (ROS) production (13–16). Recently, we showed that TK2 activity and protein levels were downregulated during oxidative stress (17) and that incubation with ddI led to reduced TK2 and dGK protein levels in U2OS cells (18). In the present study, we explored the effects of AZT on mitochondrial TK2 and dGK levels in cultured U2OS cells. We found that AZT treatment led to reductions in TK2 and dGK protein levels through oxidative stress mechanisms, which could be abrogated by uridine (Urd) supplementation. The mechanism of mitochondrial degradation of TK2 and dGK induced by AZT or oxidants was also investigated.
MATERIALS AND METHODS
Materials.
Uridine (Urd) and AZT were obtained from Sigma. Mouse monoclonal antibodies against α- and β-tubulin, cytochrome c oxidase subunit II (COX II), and cytochrome c oxidase subunit IV (COX IV) were purchased from Abcam. Polyclonal rabbit anti-human TK2 and anti-human dGK antibodies were produced using synthetic peptides of C-terminal sequences (TK2, NRDRILTPENRKHC; dGK, EEVTKQEDLMRVC) as the antigens (GenScript) and were purified from rabbit antisera by affinity chromatography on Sepharose resin (CNBr-activated Sepharose; GE Healthcare) coupled with the peptide antigen (17). A mouse monoclonal anti-human TK1 antibody (19) obtained from AroCell AB (Uppsala, Sweden) and a polyclonal rabbit anti-human dCK antibody (20) were used to detect the TK1 and dCK proteins, respectively. All antibodies were diluted in blocking buffer (0.05% Tween 20 and 5% nonfat milk powder in phosphate-buffered saline [PBS]). The protease inhibitor MG132 was purchased from UBPBio. Chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) was obtained from Life Technologies. The OxyBlot protein oxidation detection kit was purchased from Millipore.
Cell culture conditions.
U2OS cells (U-2 OS, human osteosarcoma cell line; ATCC HTB-96) were maintained at 37°C in McCoy's 5A (modified) medium (Gibco Cell Culture) supplemented with 10% fetal bovine serum (FBS) (Gibco Cell Culture), 100 U/ml penicillin, and 0.1 mg/ml streptomycin, in a humid atmosphere with 5% CO2. The AZT and Urd used in cell cultures were dissolved in dimethyl sulfoxide (DMSO) as stock solutions and were diluted in the medium prior to use. The final DMSO concentration in complete medium was <0.05% (vol/vol) and thus should have no toxicity on cells.
Protein extraction and Western blot analysis.
Mitochondria were prepared by differential centrifugation, and the mitochondrial proteins were extracted as described previously (17). For total cellular protein extraction, approximately 1 × 106 cells were resuspended in 20 μl lysate buffer (50 mM Tris-HCl [pH 7.6], 150 mM KCl, 5 mM MgCl2, 5 mM dithiothreitol [DTT], 1× protease inhibitor cocktail [Roche], 0.5% NP-40, 0.3 M sucrose), and the cellular proteins were extracted by freezing and thawing three times, followed by sonication in an ice-water bath and centrifugation at 16,000 × g for 20 min at 4°C. The protein concentration was determined by the Bradford method (Bio-Rad protein assay), using bovine serum albumin (BSA) as a standard.
Mitochondrial proteins (30 μg/lane) or total cellular proteins (100 μg/lane) were subjected to 12% reducing SDS-PAGE and were transferred to Immobilon polyvinylidene difluoride (PVDF) membranes (Millipore) using a semidry transfer system. The membranes were incubated with blocking buffer at room temperature for 1 h and then were probed with the primary antibodies described above. Anti-mouse or anti-rabbit secondary antibodies conjugated with horseradish peroxidase (GE Healthcare) were applied to the membranes, and the target proteins were detected with an enhanced chemiluminescence detection system (ECL kit; GE Healthcare). The band intensity was quantified using Quantity One/Image Lab software (Bio-Rad).
Effects of AZT on the levels of mitochondrial TK2 and dGK.
Approximately 6 × 106 cells were seeded in 175-cm2 tissue culture flasks and incubated in the presence or absence of 20 μM AZT alone or in combination with 20 μM uridine in the medium for 3 days. The mitochondria were then isolated, and the levels of TK2 and dGK in the mitochondrial extracts were determined by Western blot analysis using polyclonal rabbit anti-human TK2 and dGK antibodies (17). To assess the effects of AZT in the absence or presence of Urd on cytosolic salvage enzymes (TK1 and dCK), total cell lysates (∼100 μg protein) were resolved by 12% SDS-PAGE, and the levels of TK1 and dGK were determined by Western blotting using antibodies against human TK1 (19) and dCK (20).
Total ROS level and protein oxidation measurements.
Triplicate samples of 0.3 × 106 U2OS cells were seeded into individual wells of 6-well flat-bottomed plates and incubated for 3 days in complete culture medium with various concentrations of AZT. To detect intracellular ROS production, the cells were treated with 5 μg/ml CM-H2DCFDA in PBS for 15 min at 37°C in the dark. The cells were then trypsinized and resuspended in PBS. The intensity of oxidized CM-H2DCFDA fluorescence was immediately measured using a FACScan flow cytometer with CellQuest software (Becton Dickinson). Data for 10,000 events were collected for each sample and analyzed with FlowJo software (TreeStar Inc.); the ROS levels are reported as the geometric mean florescence of the collected cells.
Protein oxidation (carbonylation) in both total cellular and mitochondrial extracts of U2OS cells was evaluated by OxyBlot assays, according to the manufacturer's instructions. In brief, 15 μg protein was reacted with 2,4-dinitrophenyl-hydrazine (DNPH) in 6% SDS (wt/vol) solution at room temperature for 15 min and then was subjected to Western blot analysis using an anti-dinitrophenyl (DNP) group antibody (1:150), to determine the contents of protein carbonyl groups. The blots were subsequently reprobed for α-tubulin or COX IV immunoreactivity, which was utilized as the loading control.
Effects of oxidant treatment on mitochondrial dGK and TK2 levels.
U2OS cells (initial culture of 6 × 106 cells/flask in 30 ml complete medium) were cultivated for 3 days in 175-cm2 flasks. Various concentrations of oxidants (hydrogen peroxide, tert-butylhydroperoxide [tBHP], or diamide) were added to the medium, and cells were incubated for an additional 30 min at 37°C. The cells were then harvested, and the mitochondria were isolated. The mitochondrial proteins (30 μg) were resolved by 12% SDS-PAGE and transferred to a PVDF membrane, and the endogenous dGK and TK2 protein levels were determined by Western blot analysis using the purified polyclonal rabbit anti-human TK2 and dGK antibodies, essentially as described previously (7).
In organello degradation of TK2 and dGK.
Mitochondria isolated from U2OS cells were further treated with 0.02% digitonin (wt/vol) for 3 min on ice, to remove the mitochondrial outer membrane, and the resulting mitoplasts were then washed twice with mitochondrial isolation buffer and resuspended in the same buffer at a protein concentration of 3 mg/ml. MG132 (10 μM) was added to the mitoplast suspension (200 μg protein), which was incubated at 37°C for 5 min before the addition of H2O2 (4 mM). The mixture was further incubated for 30 min at 37°C. At 0, 15, and 30 min, aliquots (equal to 40 μg protein) were withdrawn and centrifuged at 12,000 × g for 10 min at 4°C. The pellets were dissolved in 15 μl reducing SDS sample buffer and analyzed by SDS-PAGE and Western blotting. These experiments were repeated three times with similar results, and one representative Western blot is shown.
Statistical analysis.
Experimental data were analyzed using Student's t test performed with GraphPad Prism software. Differences with P values of <0.05 were considered significant.
RESULTS
AZT treatment led to downregulation of mitochondrial TK2 and dGK.
The long-term use of nucleoside analogs, e.g., AZT in anti-HIV therapy, is associated with mitochondrial toxicity, including myopathy, neuropathy, and lipodystrophy (21). To investigate whether AZT has any effect on mitochondrial TK2 and dGK protein levels, we incubated U2OS cells in the presence or absence of AZT for 3 days and then isolated mitochondria. Mitochondrial proteins were separated by SDS-PAGE, and the TK2 and dGK protein levels were determined by Western blot analysis. As shown in Fig. 1, both TK2 and dGK protein levels were significantly decreased, by 30% and 60%, respectively, in AZT-treated cells. The levels of cytochrome c oxidase subunit II (COX II), encoded by mitochondrial DNA, were decreased by ∼10% in the presence of AZT, whereas the levels of COX IV, encoded by nuclear DNA, were unchanged. Since AZT and its phosphorylated metabolites should have little effect on mitochondrial protein synthesis (22), the decreased COX II levels indicate that other mitochondrial factors in mtDNA replication, transcription, and/or translation are also altered. These results suggest that mitochondrial TK2 and dGK are specifically targeted by AZT treatment and that the TK2 and dGK deficiencies caused by AZT may have an impact on mtDNA synthesis, causing reductions in the levels of mtDNA-encoded proteins (e.g., COX II).
FIG 1.
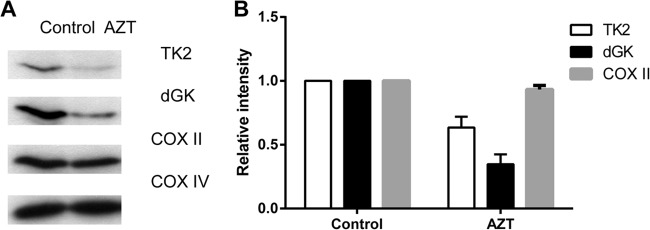
TK2 and dGK protein levels in cells treated with AZT. (A) U2OS cells were incubated in the presence or absence of 20 μM AZT for 3 days, and then mitochondria were isolated and used to determine the levels of TK2 and dGK by Western blot analysis using the corresponding antibodies. (B) The band intensities were quantified and are shown as TK2, dGK, and COX II levels relative to the controls after normalization to the level of COX IV.
AZT induced reactive oxygen species production.
Oxidative stress is a condition in which the cellular production of reactive oxygen species (ROS) exceeds the antioxidant defense capacity. In cultured cells, animal models, and HIV-infected patients treated with AZT, it has been shown that oxidative stress is correlated with AZT-induced adverse effects (13, 15). Therefore, we investigated the oxidative effects of AZT in U2OS cells treated with various concentrations of AZT for 3 days. The ROS levels were measured using CM-H2DCFDA, a cell membrane-permeable dye that reacts with superoxide. The fluorescence of oxidized CM-H2DCFDA was measured by flow cytometry, and the relative ROS levels, in comparison with untreated cells, are shown in Fig. 2. Reductions in cell numbers were observed for the cells treated with >50 μM AZT. Compared with untreated cells, ROS production was increased by approximately 16% in cells treated with 50 or 100 μM AZT. Incubation with higher concentrations of AZT elevated the intracellular superoxide levels by 40% (Fig. 2). These results demonstrated that AZT increased ROS production in U2OS cells, even with short-term exposure.
FIG 2.

AZT promotion of ROS production in U2OS cells after 3 days of exposure. Cells were prepared as described in Materials and Methods. (A) Representative flow cytometric offset histogram of the fluorescence of oxidized products of CM-H2DCFDA in U2OS cells exposed to various concentrations of AZT (from bottom to top, the curves depict treatment with 0, 50, 100, 500, and 1000 μM AZT). (B) Relative intracellular ROS levels in cells treated with AZT. The results are expressed as the average percentage ± standard error of the mean (SEM) of the control value (n = 3). *, P < 0.05, compared with the control; **, P < 0.01, compared with the control; ns, not significant. FL1-H represents the green fluorescence intensity resulting from CM-H2DCFDA oxidation.
Oxidant treatment led to downregulation of mitochondrial TK2 and dGK.
We previously showed that mitochondrial TK2 was selectively degraded in U2OS cells during oxidative stress, via a mechanism involving oxidative modifications (17). In mitochondria, dGK is a complementing enzyme for TK2 and is responsible for the phosphorylation of deoxyguanosine and deoxyadenosine. Similar to TK2, incubation of recombinant dGK with oxidized glutathione (GSSG) led to S-glutathionylation of dGK, presumably at the conserved cysteine residue (Cys-199) (see Fig. S1 in the supplemental material). In the mitochondria of H2O2-treated cells, we observed a significant increase in protein carbonylation, a marker of oxidative protein damage (see Fig. S2A in the supplemental material). Therefore, we determined the levels of dGK in H2O2-treated cells. A concentration-dependent decrease in mitochondrial dGK protein levels in the H2O2-treated cells was found, whereas the control protein COX IV was not affected (Fig. 3A). The effects of other cell membrane-permeable oxidizing agents (e.g., tBHP and diamide) on mitochondrial TK2 and dGK levels were also investigated. Incubation with 1 mM tBHP resulted in 80% and 65% reductions in TK2 and dGK protein levels, respectively. Similarly, incubation with 2 mM diamide led to ∼50% decreases in TK2 and dGK levels. No significant changes were observed for COX IV (Fig. 3B).
FIG 3.
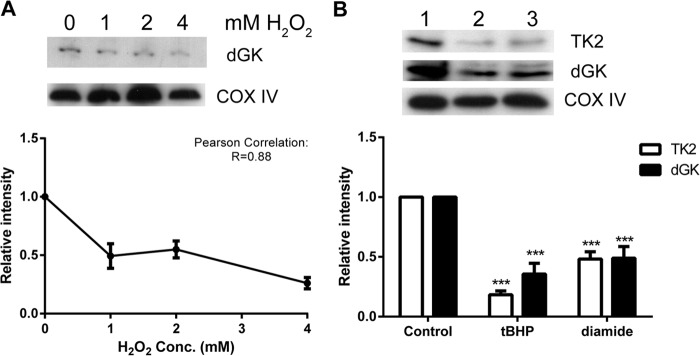
Effects of oxidants on mitochondrial dGK and TK2 protein levels. Approximately equal numbers of U2OS cells were incubated with various concentrations of H2O2 (A) or 1 mM tBHP (lane 2), 2 mM diamide (lane 3), or no treatment (lane 1) (control) (B) for 30 min at 37°C. Mitochondrial extracts were prepared, and 30 μg protein was subjected to 12% reducing SDS-PAGE. The endogenous TK2 protein levels with the tBHP and diamide treatments and the dGK protein levels in all groups were measured by Western blot analysis. The COX IV levels were used as the mitochondrial loading control. The experiments were repeated at least three times, and one representative blot is shown.
In vitro recombinant TK2 and dGK were shown to be sensitive to FeII-catalyzed oxidation (see Fig. S2B in the supplemental material). Together, these results indicate that mitochondrial TK2 and dGK are sensitive to oxidative stress, and further studies may reveal the direct mechanisms involved in this oxidative stress-induced TK2 and dGK degradation.
Time-dependent in organello degradation of TK2 and dGK was observed.
To study the mechanism of mitochondrial TK2 and dGK downregulation during oxidative stress and to exclude the possibility of cytosolic protease involvement, we isolated mitoplasts from untreated U2OS cells and then treated the mitoplasts with H2O2 for 30 min in the presence or absence of MG132, a specific and reversible proteasome and mitochondrial Lon AAA+ protease inhibitor. At each time point, aliquots were withdrawn, and the mitoplasts were collected by centrifugation; the total proteins were dissolved in sample buffer and analyzed by SDS-PAGE, and the levels of TK2 and dGK proteins were determined. As shown in Fig. 4A, mitochondrial TK2 levels declined 40% in mitoplasts exposed to H2O2 for 30 min. In contrast, mitochondrial TK2 levels were unchanged when MG132 was present during the H2O2 treatment. Mitochondrial dGK levels were decreased by 30% at 30 min after exposure to H2O2, and this decrease in dGK protein levels was prevented by MG132. The mitochondrial loading control, COX IV, was present at similar levels at all time points (Fig. 4B). These results strongly indicate that the downregulation of mitochondrial TK2 and dGK levels during oxidative stress is due to selective proteolytic degradation in mitochondria.
FIG 4.

In organello degradation of mitochondrial TK2 and dGK. (A) Western blot analysis of dGK and TK2. Mitoplasts prepared from untreated U2OS cells were incubated with 10 μM MG132 at 37°C for 5 min, 4 mM H2O2 was added, and cells were incubated for an additional 30 min. At the time points indicated, equal amounts (∼40 μg mitochondrial protein) of mitoplast suspension were withdrawn and analyzed by Western blotting with the corresponding antibody. (B) Relative levels of TK2 and dGK. The mitochondrial TK2 and dGK band intensities were quantified and normalized to the COX IV levels; the values are represented as intensity relative to the zero time point.
Uridine supplementation reversed the effects of AZT on mitochondrial TK2 and dGK.
Uridine supplementation for AIDS patients treated with pyrimidine analogs has been shown to reduce toxic side effects (23). Therefore, it was interesting to test whether the addition of uridine to AZT-treated cells would have any effect on the levels of TK2 and dGK. After 3 days of treatment, cells were harvested, and mitochondria were isolated. Total mitochondrial proteins were used to determine the levels of TK2 and dGK proteins. Again, AZT caused pronounced reductions in both TK2 and dGK protein levels, and a reduction in COX II levels was also observed in the AZT-treated cells (Fig. 5A). Uridine supplementation resulted in the restoration of TK2 and dGK to levels similar to those of untreated cells. The COX II protein was also returned to normal levels in the presence of uridine, whereas COX IV levels were unchanged (Fig. 5A). These results demonstrate that treatment with AZT causes severe TK2 and dGK deficiencies and the addition of uridine protects mitochondria from these AZT-induced effects.
FIG 5.

Effects of uridine supplementation. (A) Uridine supplementation restored the levels of TK2 and dGK in cells treated with AZT. U2OS cells were treated with 20 μM AZT alone or in combination with 20 μM uridine for 3 days. (Left) The levels of mitochondrial TK2 and dGK were determined by Western blotting (lane 1, control; lane 2, AZT; lane 3, AZT plus Urd). (Right) The protein bands were quantified, the levels of TK2, dGK, and COX II were normalized to the levels of COX IV, and results are presented as levels relative to the control. (B) Uridine abrogated AZT-induced ROS production. (Left) The ROS levels were assessed by CM-H2DCFDA staining and flow cytometry (from bottom to top, the curves depict control, 20 μM AZT, 20 μM AZT plus 20 μM Urd, and 20 μM Urd). The offset histogram plot shown is representative of three independent experiments. (Right) The total cellular ROS levels are reported as the geometric mean fluorescence of 10,000 events, expressed as a percentage of the control value. (C) Uridine had no effect on protein oxidation. (Left) Representative Western blot analysis of protein carbonylation measured by using an OxyBlot kit. (Right) Relative levels of protein carbonyl contents for treated versus untreated U2OS cells. The levels of α-tubulin were used as the loading control. The data are presented as the mean ± SEM (n = 3). **, P < 0.01 versus the control; *, P < 0.05 versus the control; ns, not significant.
Uridine abrogated the effects of AZT on ROS production and protein oxidation.
Because uridine supplementation reversed the effects of AZT on TK2 and dGK levels, we investigated whether it also had effects on ROS production and protein oxidation. As shown in Fig. 5B, treatment with 20 μM AZT caused increases in ROS levels and the presence of uridine led to reduced ROS production (Fig. 5B). Compared to the controls, the addition of uridine to AZT-treated cells led to reduction of the protein carbonylation levels (Fig. 5C). Uridine alone had no effect on either ROS levels or protein oxidation (Fig. 5B and C).
Cytosolic TK1 and dCK protein levels were not affected by AZT.
Total proteins were extracted from cells treated with AZT, alone or in the presence of uridine, and were analyzed by SDS-PAGE. The TK1 and dCK levels were determined by Western blotting using anti-human TK1 and dCK antibodies. As shown in Fig. 6, there were no significant alternations in the levels of TK1, dCK, or the control proteins (i.e., COX IV and β-tubulin). These results indicated that the effect of AZT is apparently mitochondrion specific and occurs via oxidative stress localized in the mitochondria.
FIG 6.
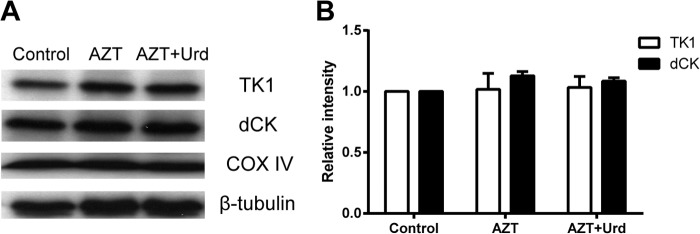
Cytosolic TK1 and dCK protein levels in cells treated with 20 μM AZT in the absence or presence of 20 μM uridine. (A) The levels of TK1 and dCK in total cellular lysates were determined by Western blotting (lane 1, control; lane 2, AZT; lane 3, AZT plus Urd). (B) The bands were quantified. The levels of COX II and COX IV were used as mitochondrial and nuclear DNA-encoded protein controls, respectively, and β-tubulin was used as the loading control. Data are the mean ± SEM of four independent experiments.
DISCUSSION
As the first FDA-approved, effective, HIV reverse transcriptase inhibitor, AZT is still used in combination with other anti-HIV drugs in highly active antiviral therapy (HAART) and has significantly increased the life expectancy of HIV-infected individuals. However, long-term treatment with AZT and/or other nucleoside analogs is often associated with toxic mitochondrial side effects, e.g., cardiomyopathy, neuropathy, and lipodystrophy (11, 24). Initially, the mechanism of mitochondrial toxicity induced by AZT and other nucleoside analogs was attributed to the inhibition of mtDNA polymerase by the triphosphate (TP) forms of the analogs, such as AZT-TP, causing mtDNA depletion and mitochondrial malfunction (25). This was supported by extensive studies of the interactions between human mtDNA polymerase and the nucleoside analog triphosphates, including AZT-TP (26–28). However, recent evidence suggested that inhibition of mitochondrial TK2 activity is likely to play an important role in the AZT-mediated mitochondrial toxicities, since in mitochondria AZT competitively inhibited thymidine phosphorylation by TK2 but deoxycytidine phosphorylation was stimulated by the same enzyme, leading eventually to imbalanced dTTP and dCTP pools. In addition, the concentrations of AZT-TP are unlikely to be high enough to inhibit mtDNA polymerase (22, 29, 30). Mitochondrial oxidative stress induced by nucleoside analogs may also contribute to mitochondrial dysfunction, since it is known that mtDNA polymerase is subject to extensive oxidative damage (31, 32). Recently, TK2 has been shown to be sensitive to oxidative stress induced by oxidants, and both TK2 and dGK are downregulated in cells treated with ddI (17, 18) (Fig. 7).
FIG 7.

Illustration of the consequences of AZT- or H2O2-induced oxidative damage. H2O2 or AZT treatment increases cellular and mitochondrial ROS levels and oxidative modifications and damage of mitochondrial proteins, lipids, and mtDNA. Oxidative modifications of TK2 and dGK lead to proteolytic degradation, e.g., downregulation. Deficiencies in TK2 or dGK result in imbalanced mitochondrial dNTP pools and eventually mtDNA depletion. Upon entering mitochondria, AZT can also inhibit TK2 activity, leading to imbalanced dTTP and dCTP pools that stall mtDNA synthesis. mtDNA depletion, in turn, leads to decreased synthesis and abundance of mtDNA-encoded proteins that are essential components of the mitochondrial electron transport chain complexes, thus producing mitochondrial dysfunction. dN, deoxynucleoside; dNMP, deoxynucleoside monophosphate; AZTMP, AZT monophosphate; AZTTP, AZT triphosphate; pol-γ, polymerase γ.
We showed here that treatment with AZT caused profound depletion of both mitochondrial TK2 and dGK proteins in U2OS cell but had no effect on the levels of cytosolic TK1 and dCK proteins or other control proteins (e.g., COX IV, α-tubulin, and β-tubulin). A decrease in the levels of COX II, an mtDNA-encoded protein, in AZT-treated cells was also observed. Since AZT and its metabolites have little effect on mitochondrial protein synthesis (22), the decreased COX II levels suggested that the downregulation of mitochondrial TK2 and dGK and/or other unknown factors affected mtDNA synthesis, transcription, and translation and thus mtDNA-encoded protein abundance.
Interestingly, the negative effects of AZT treatment on mitochondrial TK2 and dGK levels were similar to those of oxidant treatments, as shown in our previous study (17) and the data presented here. During oxidative stress, mitochondrial TK2 was shown to be S-glutathionylated at Cys-189, which resulted in the proteolytic degradation of TK2. The TK2 Cys-189 residue is conserved in the dGK (Cys-199) and dCK (Cys-185) sequences but not in the Drosophila melanogaster deoxynucleoside kinase sequence (17). Similar to TK2, dGK and dCK but not D. melanogaster deoxynucleoside kinase were S-glutathionylated in the presence of GSSG, which suggests that the conserved cysteine, e.g., Cys-199 in dGK, is responsible for S-glutathionylation. We also found that S-glutathionylation resulted in reduced dGK activity (see Fig. S1 in the supplemental material). Furthermore, TK2 and dGK are sensitive to transition metal-catalyzed protein oxidation. These results demonstrate that TK2 and dGK are sensitive to redox regulation under oxidative stress.
AZT treatment induces oxidative stress and mitochondrial damage, and increased ROS production causes oxidative modifications on mtDNA, proteins, and lipids, as observed in endothelial cells, fibroblasts, and adipocytes from AIDS patients and in animal model studies (13, 15, 33–38). The oxidation of protein side chains of proline, arginine, lysine, and threonine residues results in the generation and accumulation of carbonyl groups (i.e., aldehydes and ketones), which are regarded as biochemical markers of oxidative stress. Elevated levels of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG) in serum or urine have been used as markers of oxidative damage to DNA. In HIV patients and in mice, AZT treatment led to greater urinary excretion of 8-oxo-dG, which could be prevented by antioxidants (36). In mouse heart, AZT treatment caused marked increases in ROS production, mitochondrial glutathione oxidation, and more than 120% increases in 8-oxo-dG levels in heart mtDNA (37). Short-term AZT treatment of rats induced large increases in ROS, which led to single-stranded DNA breaks, lipid peroxidation, and protein carbonylation (38). In this study, 30-min H2O2 treatment resulted in significant increases in mitochondrial protein oxidation. Mitochondria contain high levels of iron(II), which is required for incorporation into cytochromes and other iron-containing proteins, including iron-sulfur-containing enzymes. ROS cause the release of FeII from iron-sulfur proteins, resulting in protein oxidation via the Fenton reaction. TK2 and dGK are readily oxidized in the presence of FeII in vitro and AZT treatment may also lead to TK2 and dGK oxidation in vivo, but further investigations are required to define the role of oxidative modifications in the mitochondrial toxicities of antiviral nucleoside analogs.
The AZT concentration used in this study is high, in comparison with clinical situations, and we think that AZT is still able to cause oxidative stress at low, clinically relevant concentrations, possibly at lower levels. For example, 20 μM AZT induced increased ROS production in macrophages, as measured by using more advanced techniques (13), and also in animal models mentioned above (36–38). These results suggest that the effects of AZT on TK2 and dGK may be cumulative effects of low-level ROS induced by AZT over 3 days.
Uridine supplementation during antiviral therapy has been shown to reduce peripheral neuropathy and encephalopathy in AIDS patients and in animal models, but the mechanism of this protective effect is not known (23, 39). After uptake in cells, uridine can be metabolized to various nucleoside and nucleotide intermediates, e.g., uridine, cytidine, deoxycytidine, and thymidine nucleotides such as UTP, CTP, dCTP, and dTTP. The latter can be derived from uridine in proliferating cells, leading to elevated dCTP and dTTP levels, which may contribute to increased mtDNA synthesis. This could protect mitochondria from mtDNA depletion and mitochondrial malfunction. We observed that uridine supplementation reduced the levels of ROS and the associated protein carbonylation, abrogating the effects of AZT on TK2 and dGK protein levels in U2OS cells. The COX II levels were also normalized after the addition of uridine, indicating that the addition of uridine released blocked mtDNA synthesis in the cells treated with AZT, possibly by increasing the dCTP and dTTP levels through restoration of TK2 and/or dGK activity and/or uridine metabolites. Although the mechanism of these protective effects is still not clear, the protective effects of uridine on TK2 and dGK levels may explain in part the uridine effects observed in HIV patients and in animal models in connection with antiviral treatment with nucleoside analogs (23, 39, 40).
Increases in oxidative stress, e.g., increases in ROS levels produced by antiviral nucleoside analogs or other agents, will result in oxidative modifications of mitochondrial TK2 and dGK and subsequent proteolytic degradation by mitochondrial proteases. Decreased TK2 and dGK levels will lead to impaired mitochondrial synthesis of deoxynucleoside triphosphates (dNTPs) and eventually depletion of mtDNA. Depletion of mtDNA results in decreased synthesis of mtDNA-encoded proteins that are essential components of the mitochondrial electron transport chain. Dysfunctional mitochondrial energy metabolism leads to increased mitochondrial ROS production and ultimately mitochondrial malfunction. Therefore, deficiencies in TK2 or dGK activity are likely responsible for the observed mtDNA depletion in these HIV patients, independent of mtDNA polymerase inhibition (Fig. 7), in line with a recent report showing that antiviral therapy caused depletion of both ribonucleotide and deoxynucleotide pools in HIV patients (12).
Recently, it was shown that antiviral ribonucleotides are substrates for the human mitochondrial RNA polymerase, thus adding an additional target for the antiviral agents (41). ddI, another anti-HIV nucleoside analog, also induced downregulation of TK2 and dGK in U2OS cells without concurrent induction of ROS or protein oxidation (18). Thus, the mechanism of nucleoside analog-induced mitochondrial toxicity is multifactorial, encompassing transport across cell and mitochondrial membranes, metabolic activation by cytosolic and mitochondrial kinases, and inhibition of enzymes in mtDNA precursor synthesis and mtDNA replication, transcription, and translation, in addition to the oxidative stress-caused damage to mitochondrial proteins and DNA described here (Fig. 7).
The selective sensitivity of mitochondrial TK2 and dGK to AZT-induced oxidative stress strongly suggests that downregulation of these two enzymes may be a major factor in mitochondrial replicative stress associated with AZT treatment. Furthermore, not only AZT but also many other nucleoside analogs used in antiviral therapy cause mtDNA depletion, including ddI (didanosine), stavudine, and lamivudine (42). Therefore, further studies of the effects of AZT and other nucleoside analogs should help to clarify the mechanisms underlying the adverse effects of these prodrugs.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by a grant from the Swedish Research Council.
Footnotes
Published ahead of print 2 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03613-14.
REFERENCES
- 1.Eriksson S, Wang L. 1997. Substrate specificities, expression, and primary sequences of deoxynucleoside kinases: implications for chemotherapy. Nucleosides Nucleotides 16:653–659. 10.1080/07328319708002930. [DOI] [Google Scholar]
- 2.Arnér ESJ, Eriksson S. 1995. Mammalian deoxyribonucleoside kinases. Pharmacol. Ther. 67:155–186. 10.1016/0163-7258(95)00015-9. [DOI] [PubMed] [Google Scholar]
- 3.Choi O, Heathcote D, Ho K, Müller P, Ghani H, Lam E, Ashton-Rickardt P, Rutschmann S. 2012. A deficiency in nucleoside salvage impairs murine lymphocyte development, homeostasis, and survival. J. Immunol. 188:3920–3927. 10.4049/jimmunol.1102587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Austin W, Armijo A, Campbell D, Singh A, Hsieh T, Nathanson D, Herschman H, Phelps M, Witte O, Czernin J, Radu C. 2012. Nucleoside salvage pathway kinases regulate hematopoiesis by linking nucleotide metabolism with replication stress. J. Exp. Med. 209:2215–2228. 10.1084/jem.20121061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. 2001. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat. Genet. 29:342–344. 10.1038/ng751. [DOI] [PubMed] [Google Scholar]
- 6.Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, Anbinder Y, Berkowitz D, Hartman C, Barak M, Eriksson S, Cohen N. 2001. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat. Genet. 29:337–341. 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- 7.Akman H, Dorado B, López L, García-Cazorla A, Vilà M, Tanabe L, Dauer W, Bonilla E, Tanji K, Hirano M. 2008. Thymidine kinase 2 (H126N) knockin mice show the essential role of balanced deoxynucleotide pools for mitochondrial DNA maintenance. Hum. Mol. Genet. 17:2433–2440. 10.1093/hmg/ddn143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou X, Solaroli N, Bjerke M, Stewart J, Rozell B, Johansson M, Karlsson A. 2008. Progressive loss of mitochondrial DNA in thymidine kinase 2 deficient mice. Hum. Mol. Genet. 17:2329–2335. 10.1093/hmg/ddn133. [DOI] [PubMed] [Google Scholar]
- 9.Lee M-H, Wang L, Chang Z-F. 2014. The contribution of mitochondrial thymidylate synthesis in preventing the nuclear genome stress. Nucleic Acids Res. 42:4972–4984. 10.1093/nar/gku152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brinkman K, Smeitink J, Romijn J, Reiss P. 1999. Mitochondrial toxicity induced by nucleoside-analogue reverse-transcriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Lancet 354:1112–1115. 10.1016/S0140-6736(99)06102-4. [DOI] [PubMed] [Google Scholar]
- 11.Walker U, Setzer B, Venhoff N. 2002. Increased long-term mitochondrial toxicity in combinations of nucleoside analogue reverse-transcriptase inhibitors. AIDS 16:2165–2173. 10.1097/00002030-200211080-00009. [DOI] [PubMed] [Google Scholar]
- 12.Selvaraj S, Ghebremichael M, Li M, Foli Y, Langs-Barlow A, Ogbuagu A, Barakat L, Tubridy E, Edifor R, Lam W, Cheng Y-C, Paintsil E. 17 March 2014. Antiretroviral therapy-induced mitochondrial toxicity: potential mechanisms beyond polymerase-γ inhibition. Clin. Pharmacol. Ther. 10.1038/clpt.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amatore C, Arbault S, Jaouen G, Koh ACW, Leong WK, Top S, Valleron M-A, Woo CH. 2010. Pro-oxidant properties of AZT and other thymidine analogues in macrophages: implication of the azido moiety in oxidative stress. ChemMedChem 5:296–301. 10.1002/cmdc.200900464. [DOI] [PubMed] [Google Scholar]
- 14.Geller H, Cheng K, Goldsmith N, Romero A, Zhang A, Morris E, Grandison L. 2001. Oxidative stress mediates neuronal DNA damage and apoptosis in response to cytosine arabinoside. J. Neurochem. 78:265–275. 10.1046/j.1471-4159.2001.00395.x. [DOI] [PubMed] [Google Scholar]
- 15.Kline E, Bassit L, Hernandez-Santiago B, Detorio M, Liang B, Kleinhenz D, Walp E, Dikalov S, Jones D, Schinazi R, Sutliff R. 2009. Long-term exposure to AZT, but not D4T, increases endothelial cell oxidative stress and mitochondrial dysfunction. Cardiovasc. Toxicol. 9:1–12. 10.1007/s12012-008-9029-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hewish M, Martin S, Elliott R, Cunningham D, Lord C, Ashworth A. 2013. Cytosine-based nucleoside analogs are selectively lethal to DNA mismatch repair-deficient tumour cells by enhancing levels of intracellular oxidative stress. Br. J. Cancer 108:983–992. 10.1038/bjc.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun R, Eriksson S, Wang L. 2012. Oxidative stress induced S-glutathionylation and proteolytic degradation of mitochondrial thymidine kinase 2. J. Biol. Chem. 287:24304–24312. 10.1074/jbc.M112.381996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun R, Eriksson S, Wang L. 2014. Down-regulation of mitochondrial thymidine kinase 2 and deoxyguanosine kinase by didanosine: implications for mitochondrial toxicities of anti-HIV nucleoside analogs. Biochem. Biophys. Res. Commun. 450:1021–1026. 10.1016/j.bbrc.2014.06.098. [DOI] [PubMed] [Google Scholar]
- 19.Gasparri F, Wang N, Skog S, Galvani A, Eriksson S. 2009. Thymidine kinase 1 expression defines an activated G1 state of the cell cycle as revealed with site-specific antibodies and ArrayScan assays. Eur. J. Cell Biol. 88:779–785. 10.1016/j.ejcb.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Hatzis P, Al-Madhoon A, Jüllig M, Petrakis T, Eriksson S, Talianidis I. 1998. The intracellular localization of deoxycytidine kinase. J. Biol. Chem. 273:30239–30243. 10.1074/jbc.273.46.30239. [DOI] [PubMed] [Google Scholar]
- 21.Brinkman K, ter Hofstede HJ, Burger DM, Smeitink JA, Koopmans PP. 1998. Adverse effects of reverse transcriptase inhibitors: mitochondrial toxicity as common pathway. AIDS 12:1735–1744. 10.1097/00002030-199814000-00004. [DOI] [PubMed] [Google Scholar]
- 22.McKee EE, Bentley AT, Hatch M, Gingerich J, Susan-Resiga D. 2004. Phosphorylation of thymidine and AZT in heart mitochondria: elucidation of a novel mechanism of AZT cardiotoxicity. Cardiovasc. Toxicol. 4:155–167. 10.1385/CT:4:2:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venhoff N, Lebrecht D, Deveaud C, Beauvoit B, Bonnet J, Müller K, Kirschner J, Venhoff A, Walker U. 2010. Oral uridine supplementation antagonizes the peripheral neuropathy and encephalopathy induced by antiretroviral nucleoside analogues. AIDS 24:345–352. 10.1097/QAD.0b013e328335cdea. [DOI] [PubMed] [Google Scholar]
- 24.Smith RL, de Boer R, Brul S, Budovskaya Y, van der Spek H. 2013. Premature and accelerated aging: HIV or HAART? Front. Genet. 3:328. 10.3389/fgene.2012.00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewis W, Dalakas MC. 1995. Mitochondrial toxicity of antiviral drugs. Nat. Med. 1:417–422. [DOI] [PubMed] [Google Scholar]
- 26.Feng JY, Johnson AA, Johnson KA, Anderson KS. 2001. Insights into the molecular mechanism of mitochondrial toxicity by AIDS drugs. J. Biol. Chem. 276:23832–23837. 10.1074/jbc.M101156200. [DOI] [PubMed] [Google Scholar]
- 27.Johnson A, Ray A, Hanes J, Suo Z, Colacino J, Anderson KS, Johnson KA. 2001. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J. Biol. Chem. 276:40847–40856. 10.1074/jbc.M106743200. [DOI] [PubMed] [Google Scholar]
- 28.Lim SE, Copeland WC. 2001. Differential incorporation and removal of antiviral deoxynucleotides by human DNA polymerase gamma. J. Biol. Chem. 276:23616–23623. 10.1074/jbc.M101114200. [DOI] [PubMed] [Google Scholar]
- 29.Morris GW, Iams TA, Slepchenko KG, McKee EE. 2009. Origin of pyrimidine deoxynucleotide pools in perfused rat heart: implications for 3′-azido-3′-deoxythymidine-dependent cardiotoxicity. Biochem. J. 422:513–520. 10.1042/BJ20082427. [DOI] [PubMed] [Google Scholar]
- 30.Wang L, Sun R, Eriksson S. 2011. The kinetic effects on thymidine kinase 2 by enzyme bound dTTP may explain the mitochondrial side effects of antiviral nucleoside analogs. Antimicrob. Agents Chemother. 55:2552–2558. 10.1128/AAC.00109-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graziewicz M, Day B, Copeland WC. 2002. The mitochondrial DNA polymerase as a target of oxidative damage. Nucleic Acids Res. 30:2817–2824. 10.1093/nar/gkf392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis W, Day BJ, Copeland WC. 2003. Mitochondrial toxicity of NRTI antiviral drugs: an integrated cellular perspective. Nat. Rev. Drug Discov. 2:812–822. 10.1038/nrd1201. [DOI] [PubMed] [Google Scholar]
- 33.Caron M, Auclairt M, Vissian A, Vigouroux C, Capeau J. 2008. Contribution of mitochondrial dysfunction and oxidative stress to cellular premature senescence induced by antiretroviral thymidine analogues. Antivir. Ther. 13:27–38. [PubMed] [Google Scholar]
- 34.Mak I, Chmielinska J, Kramer J, Weglicki W. 2009. AZT-induced oxidative cardiovascular toxicity: attenuation by Mg-supplementation. Cardiovasc. Toxicol. 9:78–85. 10.1007/s12012-009-9040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stankov M, Panayotova-Dimitrova D, Leverkus M, Vondran F, Bauerfeind R, Binz A, Behrens G. 2012. Autophage inhibition due to thymidine analogues as novel mechanism leading to hepatocyte dysfunction and lipid accumulation. AIDS 26:1995–2006. 10.1097/QAD.0b013e32835804f9. [DOI] [PubMed] [Google Scholar]
- 36.de la Asuncion J, Del Olmo M, Sastre J, Millan A, Pellin A, Pallardo F. 1998. AZT treatment induces molecular and ultrastructural oxidative damage to muscle mitochondria. J. Clin. Invest. 102:4–9. 10.1172/JCI1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de la Asuncion J, Del Olmo M, Gomez-Cambronero L, Sastre J, Pallardo F, Vina J. 2004. AZT induces oxidative damage to cardiac mitochondria: protective effect of vitamins C and E. Life Sci. 76:47–56. 10.1016/j.lfs.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 38.Szabados E, Fischer G, Toth K, Csete B, Nemeti B, Trombitas K, Habon T, Endrei D, Sumegi B. 1999. Role of reactive oxygen species and poly-ADP-ribose polymerase in the development of AZT-induced cardiomyopathy in rat. Free Radic. Biol. Med. 26:309–317. 10.1016/S0891-5849(98)00199-3. [DOI] [PubMed] [Google Scholar]
- 39.Lebrecht D, Deveaud C, Beauvoit B, Bonnet J, Kirschner J, Walker U. 2008. Uridine supplementation antagonizes zidovudine-induced mitochondrial myopathy and hyperlactatemia in mice. Arthritis Rheum. 58:318–326. 10.1002/art.23235. [DOI] [PubMed] [Google Scholar]
- 40.Walker UA, Lebrecht D, Reichard W, Kirschner J, Bisse E, Iversen L, Venhoff AC, Venhoff N. 28 February 2014. Zidovudine induces visceral mitochondrial toxicity and intraabdominal fat gain in a rodent model of lipodystrophy. Antivir. Ther. 10.3851/IMP2758. [DOI] [PubMed] [Google Scholar]
- 41.Arnold J, Sharma S, Feng J, Ray A, Smidansky E, Kireeva M, Cho A, Perry J, Vela J, Park Y, Xu Y, Tian Y, Babusis D, Barauskus O, Peterson B, Gnatt A, Kashlev M, Zhong W, Cameron C. 2012. Sensitivity of mitochondrial transcription and resistance of RNA polymerase II dependent nuclear transcription to antiviral ribonucleotides. PLoS Pathog. 8:e1003030. 10.1371/journal.ppat.1003030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y, Song F, Gao Z, Ding W, Qiao L, Yang SY, Chen X, Jin R, Chen D. 2014. Long-term exposure of mice to nucleoside analogues disrupts mitochondrial DNA maintenance in cortical neurons. PLoS One 9:e85637. 10.1371/journal.pone.0085637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.