

Abstract
Myxobacteria are Gram-negative soil-dwelling bacteria belonging to the phylum Proteobacteria. They are a rich source of promising compounds for clinical application, such as epothilones for cancer therapy and several new antibiotics. In the course of a bioactivity screening program of secondary metabolites produced by Sorangium cellulosum strains, the macrolide chlorotonil A was found to exhibit promising antimalarial activity. Subsequently, we evaluated chlorotonil A against Plasmodium falciparum laboratory strains and clinical isolates from Gabon. Chlorotonil A was highly active, with a 50% inhibitory concentration between 4 and 32 nM; additionally, no correlations between the activities of chlorotonil A and artesunate (rho, 0.208) or chloroquine (rho, −0.046) were observed. Per os treatment of Plasmodium berghei-infected mice with four doses of as little as 36 mg of chlorotonil A per kg of body weight led to the suppression of parasitemia with no obvious signs of toxicity. Chlorotonil A acts against all stages of intraerythrocytic parasite development, including ring-stage parasites and stage IV to V gametocytes, and it requires only a very short exposure to the parasite to exert its antimalarial action. Conclusively, chlorotonil A has an exceptional and unprecedented profile of action and represents an urgently required novel antimalarial chemical scaffold. Therefore, we propose it as a lead structure for further development as an antimalarial chemotherapeutic.
INTRODUCTION
Malaria is the most important parasitic disease worldwide, with an estimated 207 million cases causing 627,000 deaths in 2012 (1). Among Plasmodium spp. causing malaria in humans, Plasmodium falciparum is responsible for almost all severe and fatal cases and is the predominant species in sub-Saharan Africa. Even though the scaling up of malaria control programs has led to a reduction in its incidence and mortality rates (1) and first-generation vaccines show some efficacy against it (2, 3), chemotherapy remains the mainstay of treatment for all forms of malaria. A major threat for chemotherapy is the development of resistance, since resistant phenotypes have been reported for most registered antimalarials (4). In sub-Saharan Africa, resistance against the former first-line drugs chloroquine and sulfadoxine-pyrimethamine is widespread (5), and decreased activity of artemisinin derivatives is well documented in Southeast Asia (6). The loss of artemisinin activity is of particular concern, since all current efforts to control malaria are based on this class of compounds (7). To keep pace with the parasite's ability to develop resistance, researchers need to make a continuous effort to develop new drugs, especially in cases of severe malaria, for which only two alternative treatments (artesunate and quinine) are available (8).
Treatment and pharmacological requirements of drugs to treat uncomplicated or severe malaria differ fundamentally. Drugs for uncomplicated malaria should be available as an oral formulation, be easily administered (ideally once), and have a very good safety profile (e.g., wide therapeutic range; low toxicity, especially in children and pregnant or lactating women; and few unwanted side effects); ideally, they protect the individual for a prolonged time period and protract resistance development. In contrast, patients with severe and complicated malaria need fast-acting and usually parenterally administered drugs, which rapidly reduce the parasite burden and display activity against all parasite isolates within a narrow concentration range. Their pharmacokinetic profiles should allow parenteral administration in critically ill patients with organ failure. Since children are the most affected group, all antimalarials must be safe in this age group and must be available in pediatric formulations (9).
To control malaria on the epidemiological level and prevent the spread of resistant parasites, antimalarial drugs should act on gametocytes, the sexual stages of the parasite, to block transmission to mosquitoes. However, most current antimalarials do not act against gametocytes, and transmission of the parasite is not prevented (10). Nevertheless, gametocytocidal activity is highly desirable for new antimalarials to be used in elimination and eradication programs (11).
In the past, a particularly powerful way to find new chemotherapeutics against infectious diseases was by characterizing and developing natural compounds and their derivatizations (12). One reason for the success rate with natural products may be the long-lasting interaction of coevolving organisms, resulting in structural classes optimized for their respective targets. In malaria, most highly active compounds are plant derived and are thought to have a role in deterring herbivores (e.g., quinine) or to be potent herbicides (e.g., artemisinin) (13–16). Besides plants and fungi, bacteria, including the soil-dwelling myxobacteria (17, 18), are a rich source of biologically active compounds (19–23). We investigated the antimalarial properties of a chlorine-containing metabolite, chlorotonil A, whose antiplasmodial activity was primarily identified when screening a library of myxobacterial substances at the Swiss Tropical and Public Health Institute. Chlorotonil A, a tricyclic macrolide produced by Sorangium cellulosum, was first isolated and described by Gerth et al. (24). The total synthesis of this substance and its dehalogenated derivative was reported recently (Fig. 1) (25). In our study, we found chlorotonil A to exhibit potent in vitro and in vivo activity against P. falciparum and P. berghei, respectively. It acted against all erythrocytic stages of the parasite, showed a very rapid onset of action, and was active in vitro against the stages responsible for transmission.
FIG 1.
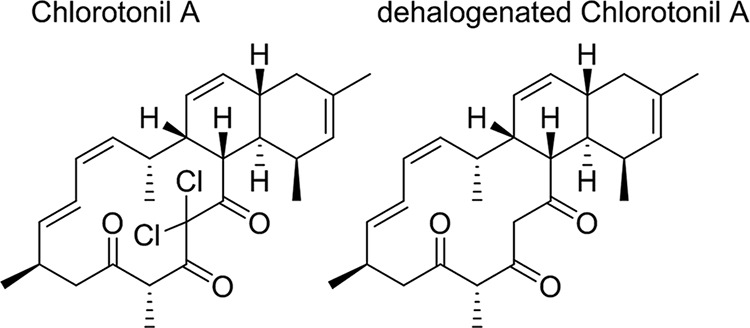
Chemical structures of chlorotonil A and its dehalogenated form.
MATERIALS AND METHODS
Parasite culture.
P. falciparum strains 3D7 (chloroquine sensitive) and Dd2 (chloroquine, sulfadoxine, and pyrimethamine resistant) were maintained in continuous culture as previously described (26). In brief, parasites were kept in complete culture medium [RPMI 1640, 25 mM 4-(2-hydroxyethyl) piperazine-N′-(4-butanesulfonic acid), 2 mM l-glutamine, 50 μg/ml gentamicin, and 0.5% (wt/vol) AlbuMax] at 37°C and in 5% CO2 and 5% oxygen at 5% hematocrit with daily changing of the medium. Synchronization was performed by sorbitol twice a week (27).
Chemicals.
Chlorotonil A (molecular weight [MW], 479.44) was isolated as described before (24), and a compound without chlorine atoms (MW, 410.54) was obtained by dehalogenation (see the structures in Fig. 1). Stock solutions of chlorotonil A and its dehalogenated form were prepared at 2.1 mM and 2.4 mM in tetrahydrofuran (THF). The comparator drug artesunate (Shin Poong Pharmaceutical Co.) (MW, 384.4) was prepared in 70% ethanol at 15 mM, chloroquine diphosphate (Sigma) (MW, 515.86) in double-distilled water at 9.7 mM, and epoxomicin (Sigma) (MW, 554.7) and dihydroartemisinin (Shin Poong Pharmaceutical Co.) (MW, 284.35) in dimethyl sulfoxide (DMSO) at 1 mM and 20 mM, respectively. Further dilutions of all drugs were done in complete culture medium. None of the solvent dilutions used interfered with parasite growth in the pilot experiments. All results given are in nM.
In vitro drug sensitivity assay of laboratory strains.
Drug sensitivity assays were performed according to standard procedures (28). In brief, 96-well plates were precoated with a 2-fold serial dilution of the respective drug. Ring-stage parasites were diluted to a parasitemia level of 0.05% with ≥0 erythrocytes and seeded at a hematocrit level of 1.5% in a total volume of 225 μl per well. After 3 days, the plates were freeze-thawed twice and analyzed by measuring histidine-rich protein 2 (HRP2) with an enzyme-linked immunosorbent assay (ELISA). To measure the delayed activity of the drug, we incubated parasites for 6 days (29); on days 2 and 4, the medium was changed (140 μl) without replacement of the drug. All experiments were done in duplicate and repeated at least three times.
In vitro drug sensitivity assay of clinical isolates.
We tested the activity of chlorotonil A compared to those of standard drugs (artesunate and chloroquine) against P. falciparum clinical isolates from patients with uncomplicated malaria in Lambaréné, Gabon, between February and May 2009. The investigations of the in vitro drug sensitivity of the clinical isolates were approved by the ethics committee of the International Foundation for the Albert Schweitzer Hospital in Lambaréné. Assent and informed consent were obtained from each child and his or her legal representative, respectively. A venous blood sample was taken into a lithium heparin tube and processed within 4 h. The assays were performed as for lab strains with minor modifications. Plates were precoated with a 3-fold dilution of each drug, and the parasites were incubated in a candle jar for 3 days at 37°C. Assays were performed only once, directly after the blood draw. Only samples in which the amount of detected HRP2 at least doubled within the 3 days were included in the analysis.
Stage-specific analysis.
Synchronized parasites at the ring, trophozoite, and schizont stages were incubated with the respective drug at a 1.5% hematocrit level and a parasitemia level between 1% and 4% in a 96-well plate for a total of 40 h. Samples were taken every 8 h to determine parasite development microscopically by Giemsa (Sigma)-stained thin blood smears. Artesunate (20 nM) was used as a positive control for stage-specific action against rings and trophozoites. For action against the schizont stage, epoxomicin (500 nM) and artesunate (500 nM) were used as controls. Chlorotonil A was used at a concentration of 40 nM for the determination of action against the ring and trophozoite stages and also at 500 nM against the schizont stage.
Evaluation of onset of action.
Assays were performed as for the in vitro standard drug sensitivity assay with laboratory strains but with removal of the drug after 1 h. In detail, synchronized ring-stage 3D7 parasites were seeded at 0.05% parasitemia level and 1.5% hematocrit level on precoated 96-well plates (chlorotonil A, artesunate, and chloroquine in a 3-fold serial dilution). After 1 h of incubation, the drugs were removed by washing with complete medium 3 times. A control plate, for which the washing step was omitted, was incubated in parallel to compare the results. Subsequently, incubation of the plates was continued for 3 days as for the standard assay before the HRP2 ELISA was performed. The 50% inhibition concentrations (IC50s) of the 1-h pulsed and standard plates of 3 independent experiments were compared. Differences in the IC50s are presented as mean fold change increase ± standard deviation with respect to the control plates. All experiments were performed in duplicate.
In vivo efficacy.
The in vivo antimalarial activity of chlorotonil A was assessed against that of the rodent malaria strain P. berghei ANKA (provided by David Walliker, University of Edinburgh, United Kingdom) in BALB/c and Swiss CD1 mice in the 4-day suppression test (30, 31). In brief, 4 BALB/c mice and 5 Swiss CD1 mice were inoculated intravenously with 2 × 107 parasitized erythrocytes (diluted in phosphate-buffered saline [PBS]) obtained from a donor mouse. After being infected, the mice were treated at 2 h, 24 h, 48 h, and 72 h postinfection with different amounts of chlorotonil A powder (BALB/c, 36, 68, and 110 mg/kg of body weight [1 control]; Swiss CD1, 3 mice received 100 mg/kg [2 controls]) in 100 mg of peanut butter (Barney's Best) or placebo control (peanut butter only). Peanut butter with or without chlorotonil A was given to each mouse individually out of a syringe. Thin blood smears from tail blood were taken daily from days 1 to 5 and stained with 20% Giemsa stain (Sigma). A minimum of 1,000 erythrocytes per slide was counted microscopically to assess parasitemia. The activity is expressed as percent reduction of parasitemia in comparison to the control group according to the following equation: activity = 100 − (mean parasitemia treated/mean parasitemia control) × 100.
Activity was analyzed on day 4; additionally, we analyzed activity on day 5, which is recommended for slow-acting drugs (31). The mouse experiments were approved by the competent authority for animal experiments in Tübingen (no. T1/08) and performed according to German legislation.
Gametocytocidal activity.
The gametocytocidal activity of chlorotonil A was evaluated by an ATP bioluminescence assay as described previously (32). As comparators, artesunate, epoxomicin, and dihydroartemisinin were tested. In brief, gametocyte culture was initiated from synchronized 3D7 parasites with an increased concentration (0.75% [wt/vol]) of AlbuMax II solution, starting with a 10% hematocrit level and a 0.5% parasitemia level. Culture medium was changed daily without parasite dilution throughout the entire process. When the parasitemia level reached 5%, the volume of the medium was doubled, and the concentration of AlbuMax II was reduced to 0.5% (wt/vol). Between days 11 and 14, the cultures were treated with 50 mM N-acetyl-d-glucosamine (MP Biomedicals GmbH) to remove the asexual stages, and on day 15, the culture was purified by a NycoPrep 1.077 cushion density gradient and magnetic column separation in order to remove erythrocytes and enrich the gametocyte population.
The compounds were precoated in a 3-fold dilution in 96-well plates before 50,000 gametocytes were added to each well in a final volume of 100 μl and incubated at 37°C in 5% CO2 and 5% oxygen. After 48 h, ATP production of the gametocytes was measured by the BacTiterGlo assay (Promega) according to the manufacturer's protocol and recorded by a Victor3 V multilabel reader (PerkinElmer, Inc.). Each experiment was repeated at least three times.
Statistics.
Individual inhibitory concentrations were determined by nonlinear regression analysis of log-concentration-response curves using the drc-package v0.9.0 (33) of R v2.3.1 (34). The mean 50% and 90% inhibition concentrations and standard deviations are presented for each drug assayed in the laboratory strains. For clinical isolates, the median 50% and 90% inhibition concentrations and their ranges are given. Correlations between the IC50s of clinical isolates of the three different drugs were calculated with Spearman's (nonparametric) test for paired samples in JMP v5.0.1.2 software.
RESULTS
Chlorotonil A acts against laboratory and clinical P. falciparum isolates in vitro.
Chlorotonil A potently inhibits the growth of chloroquine-sensitive (3D7) and chloroquine-resistant (Dd2) P. falciparum parasite strains in vitro (Table 1). As reference compounds, chloroquine and artesunate were analyzed. To assess if chlorotonil A shows signs of hysteresis (also called delayed-death phenomenon) similar to those in some other antimalarial antibiotics, we performed a 6-day assay covering two intraerythrocytic cycles of parasite replication. The IC50s obtained in the 6-day assay were 10.6 ± 4.1 nM (3D7) and 23.5 ± 9.4 nM (Dd2), and the IC90s were 13.8 ± 6.4 nM (3D7) and 32.4 ± 13 nM (Dd2), indicating that chlorotonil A acts directly upon first contact with the parasite. The dehalogenated form of chlorotonil A showed no measurable activity against Dd2 and 3D7, even at the highest concentration tested (540 nM).
TABLE 1.
Inhibitory concentrations of tested compounds against P. falciparum strains 3D7 and Dd2a
| Compound | IC (mean ±SD) (nM) of indicated compound for strain: |
|||
|---|---|---|---|---|
| 3D7 |
Dd2 |
|||
| IC50 | IC90 | IC50 | IC90 | |
| Chlorotonil A | 9.1 ± 3 | 13.34 ± 3.2 | 18.1 ± 8.6 | 28.5 ± 8.5 |
| Chloroquine | 5.2 ± 1.2 | 8.2 ± 2.3 | 160.4 ± 62.1 | 228.6 ± 66.4 |
| Artesunate | 2.4 ± 1.3 | 7.5 ± 4.6 | 1.3 ± 0.2 | 1.8 ± 0.2 |
Each value is the mean inhibitory concentration of at least 3 independent experiments performed in duplicate.
To assess the variability of the activity in clinical isolates of P. falciparum, we measured the activity of chlorotonil A against parasites freshly isolated from patients with uncomplicated malaria in Lambaréné, Gabon. Of 28 collected clinical isolates, 25 (chlorotonil A and chloroquine) and 26 (artesunate) were successfully grown in culture and analyzed in the in vitro assay. Chlorotonil A was active against clinical isolates, with IC50s comparable to those obtained against laboratory strains; in addition, the determined IC50s for the clinical isolates showed narrow ranges (Table 2). No correlations between the activities of chlorotonil A and artesunate (rho, 0.208; n = 25) or chloroquine (rho, −0.046; n = 25) were observed.
TABLE 2.
Median inhibitory concentrations of 25 (chlorotonil A and chloroquine) and 26 (artesunate) different clinical isolates against the indicated compoundsa
| Compound | IC50 (median [range]) (nM) | IC90 (median [range]) (nM) |
|---|---|---|
| Chlorotonil A | 15.2 (3.7–32) | 37.1 (6.8–76.2) |
| Chloroquine | 47.2 (19.5–117) | 111.6 (44.2–191.1) |
| Artesunate | 0.6 (0.2–3.2) | 1.8 (0.6–4.7) |
Each experiment was performed once.
Chlorotonil A acts against all blood stages of the parasite.
To investigate the effects of chlorotonil A on the morphology of different stages, we incubated synchronous 3D7 ring, trophozoite, and schizont stages with the drug and stained thin blood smears by Giemsa stain after 8, 24, and 40 h. Chlorotonil A arrested parasite development in the ring and trophozoite stages. Morphologically, the parasites looked similar to artesunate-treated parasites, since their development stopped immediately after contact with the compound (Fig. 2A and B). When added to schizont-stage parasites, higher concentrations (>40 nM) were required. Incubation with 500 nM chlorotonil A led to an arrest in development, which is comparable to the activity of proteasome inhibitors (e.g., epoxomicin) that act against the schizont stage (Fig. 2C). Notably, artesunate added at the same concentration (500 nM) did not block the egress of parasites; furthermore, erythrocytes newly infected by merozoites were found (Fig. 2C, notice the presence of ring-stage parasites after 8 h). The chlorotonil A-induced arrest in the schizont stage was apparent after 8 h by thin blood smear. At later time points, arrested schizonts showed signs of degradation.
FIG 2.

Stage-specific inhibition of P. falciparum by chlorotonil A. Giemsa-stained thin blood smears of synchronous ring (A), trophozoite (B), and schizont (C) stages. Shown are P. falciparum 3D7 parasites after different incubation times (8, 24, and 40 h) with chlorotonil A and artesunate (ring and trophozoite stages) and epoxomicin and artesunate (schizont stage) at the indicated concentrations compared to those of the drug-free control.
Chlorotonil A acts quickly.
The standard in vitro susceptibility assay does not account for short drug pulses, but in vivo drug concentrations fluctuate, and high concentrations are maintained for limited time periods only. Therefore, we incubated ring-stage parasites with chlorotonil A or the comparator drugs (artesunate and chloroquine) for 1 h, removed the drug, and continued incubation as in the standard drug sensitivity assay. The IC50 of a 1-h pulse of chlorotonil A was only 1.3 ± 0.7-fold higher than that of the standard assay. In contrast, the IC50 of a 1-h pulse of artesunate was 15.6 ± 9.1-fold higher than that of the standard assay. As expected, chloroquine was not active when given as a short pulse at the ring stage, even at the highest concentration tested (160 μM).
Chlorotonil A is active in vivo.
Chlorotonil A is not soluble in most commonly used solvents. Nevertheless, a pilot experiment to assess the therapeutic efficacy of nonsolubilized orally administered chlorotonil A was performed in P. berghei ANKA-infected BALB/c mice and Swiss CD1 mice using the Peters 4-day suppression test (31). The activity levels of chlorotonil A on day 4 were 97% in BALB/c mice and 98% in Swiss CD1 mice compared to that of the control mice. On day 5, the antiplasmodial activity levels were 93% (BALB/c) and 85% (Swiss CD1). However, the cure was not complete with the standard protocol, as none of the mice were cleared of their parasitemia completely (Fig. 3). In BALB/c mice, all the doses (36, 68, and 110 mg/kg) resulted in a substantial reduction of parasitemia. This reduction showed some dose dependency; however, due to the small sample size, this was not analyzed formally. The mice did not show obvious signs of toxicity due to chlorotonil A treatment at either dose.
FIG 3.

Parasitemia levels of P. berghei in chlorotonil A-treated mice. Parasitemia levels in four P. berghei-infected BALB/c (balbc/1 to balbc/4) and five Swiss CD1 (swiss/1 to swiss/5) mice from day 1 (d1) to d5 in the 4-day suppression test. Mice indicated by black lines received either 36, 68, or 110 mg/kg (BALB/c) or 100 mg/kg (Swiss CD1) chlorotonil A, and mice indicated by the light-gray lines received placebo control.
Chlorotonil A is active against gametocytes.
To assess the activity of chlorotonil A against stage IV to V gametocytes, we used an in vitro bioluminescence assay (32). Chlorotonil A was active against late-stage gametocytes at concentrations similar to those against asexual blood stages. This is in contrast to artesunate and dihydroartemisinin, which act only when used in very high concentrations (Table 3). Epoxomicin served as an internal control for the assay, since it is known to be highly active against stage IV to V gametocytes.
TABLE 3.
Inhibitory concentrations of tested compounds against stage IV to V gametocytes of P. falciparum strain 3D7a
| Compound | IC50 (mean ± SD) (nM) | IC90 (mean ± SD) (nM) |
|---|---|---|
| Chlorotonil A | 29.6 ± 16.3 | 123.2 ± 36.9 |
| Artesunate | 8,917 ± 4,830 | 25,852 ± 19,563 |
| Dihydroartemisinin | 2,918 ± 964 | 16,603 ± 16,141 |
| Epoxomicin | 3.2 ± 2.6 | 14.3 ± 16.7 |
Each value is the mean inhibitory concentration of at least 3 independent experiments.
DISCUSSION
So far, continuous development of new drugs remains the only option for keeping pace with the potential of P. falciparum to adapt to manmade interventions. Myxobacteria, especially those in the genus Sorangium, have proven to be a valuable source for new chemotherapeutic compounds, such as soraphens, sorangicins, thuggacins, and epothilones (35–37), some of which are in advanced preclinical and clinical development. For example, ixabepilone (BMS-247550), an analogue of epothilone B, was approved by the Food and Drug Administration in 2007 for the treatment of breast cancer (38). Sorangium cellulosum strain So ce1525 was identified as our main producer of chlorotonil A and yields several natural products exhibiting different chemical scaffolds. New scaffolds are of particular interest for the development of new antimalarials, since most compounds in the development pipeline belong to a restricted number of drug classes.
Chlorotonil A shows several remarkable features which characterize it as a promising lead compound. Apart from being active against chloroquine-sensitive and chloroquine-resistant strains at low nanomolar concentrations, it acts against all blood stages of the malaria parasite and does not show a hysteresis effect, which is a characteristic of several antimalarial antibiotics (39, 40). Chlorotonil A acts against early in the ring and trophozoite stages of the parasite and is therefore able to reduce parasite biomass and the generation of parasite toxins immediately upon contact with the parasite (41). This is notable since it is one characteristic that needs to be fulfilled by drug candidates for the treatment of severe malaria; also, artemisinin derivatives have a similar profile of action. The mode of action of chlorotonil A is not known, but the loss of activity observed for the dehalogenated derivative points toward an important role of the chlorine-containing pharmacophore. It also acts against clinical isolates with a different genetic background, isolated from patients in Lambaréné, Gabon, an area of high-grade resistance against chloroquine and sulfadoxine-pyrimethamine (42, 43). The lack of correlation with its comparator drugs, chloroquine and artesunate, supports the assumption that chlorotonil A has a different mechanism of action than that of these two other drugs.
The first tests in the murine P. berghei model demonstrated that chlorotonil A is active in vivo, has low toxicity, and can be administered orally. Due to its poor solubility, it was given to the mice as a powder together with peanut butter because tetrahydrofuran, the solvent used for in vitro assays, is toxic for mice (44). Currently, we are exploring alternative formulations and chemical modifications to develop chlorotonil A from an early lead toward a drug development candidate. In addition, it would be interesting to test anthracimycin, a structural relative of chlorotonil A, and its dichloro derivative (45).
An exceptional property of chlorotonil A is its very rapid onset of action; 1 h of contact with the parasite is sufficient to exert its full activity. The two comparator drugs, artesunate and chloroquine, had strongly reduced activity levels when pulsed for only 1 h. Rapid onset, as seen for chlorotonil A, is especially important for drugs that are used to treat severe malaria, for which a rapid reduction of the parasitemia level is vital for the survival of the patient. In addition, rapid onset is important for drugs with a short half-life, like artesunate and dihydroartemisinin. These two drugs exhibit half-lives of <1 h (46), and thus, the time span of the therapeutic drug concentration above the antiparasitic threshold is short. This is one of the reasons why the cure rates of artemisinin derivatives are poor when given for <5 to 7 days as monotherapy (47). Its activity against late-stage gametocytes is an additional valuable feature of chlorotonil A. Late-stage gametocytes are especially difficult to target by drugs. Until now, primaquine has been the only licensed drug with known activity against them in vivo, but its hemolytic effect in individuals with glucose-6-phosphate dehydrogenase deficiency limits its use (48). Artemisinin derivatives decrease gametocyte carriage in vivo mainly by their rapid action, accompanied by the reduction of parasite biomass, but it has been shown that they cannot stop transmission completely (49, 50). The effect of artemisinin derivatives on gametocytes is still under discussion; they probably also act against early gametocytes, but only little activity against late-stage gametocytes has been reported (32, 51–53). This is also in concordance with our assay. Especially when compared side by side with chlorotonil A, the activity level of artemisinin is orders of magnitude lower. A new gametocytocidal drug would be a major step forward for the feasibility of elimination and resistance containment campaigns, especially in low-endemicity settings where transmission blocking is crucial for finally stopping the cycle.
Chlorotonil A exhibits the promising characteristics of a potential new lead compound, namely, its low toxicity, oral availability, rapid onset of action, and activity against all erythrocytic stages of the parasite. In addition, it is active against the transmission stages of the parasite. Improved derivatives and dose regimens are required prior to clinical development, but at this stage, it is already evident that chlorotonil A has unique features that warrant its further assessment.
ACKNOWLEDGMENTS
We thank all the study participants, their families, and the teams in Lambaréné, Gabon, and Tübingen, Germany. We also thank Marcel Kaiser and Reto Brun at the Swiss Tropical and Public Health Institute for screening the myxobacterial library for in vitro antimalarial activity.
Blood products for parasite cultures were kindly provided by Torsten J. Schulze, Blood Donation Centre, Institute of Transfusion Medicine and Immunology, Mannheim, Germany.
Footnotes
Published ahead of print 11 August 2014
For this virtual institution, see http://www.dzif.de/en/.
REFERENCES
- 1.WHO. 2013. World malaria report, 2013. WHO, Geneva, Switzerland. [Google Scholar]
- 2.Greenwood BM, Targett GA. 2011. Malaria vaccines and the new malaria agenda. Clin. Microbiol. Infect. 17:1600–1607. 10.1111/j.1469-0691.2011.03612.x. [DOI] [PubMed] [Google Scholar]
- 3.The RTS,S Clinical Trials Partnership. 2012. A phase 3 trial of RTS,S/AS01 malaria vaccine in African infants. N. Engl. J. Med. 367:2284–2295. 10.1056/NEJMoa1208394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petersen I, Eastman R, Lanzer M. 2011. Drug-resistant malaria: molecular mechanisms and implications for public health. FEBS Lett. 585:1551–1562. 10.1016/j.febslet.2011.04.042. [DOI] [PubMed] [Google Scholar]
- 5.D'Alessandro U, Buttiens H. 2001. History and importance of antimalarial drug resistance. Trop. Med. Int. Health 6:845–848. 10.1046/j.1365-3156.2001.00819.x. [DOI] [PubMed] [Google Scholar]
- 6.Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM. 2008. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med. 359:2619–2620. 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 7.WHO. 2011. Guidelines for the treatment of malaria, 2nd ed. WHO, Geneva, Switzerland. [Google Scholar]
- 8.Tschan S, Kremsner PG, Mordmuller B. 2012. Emerging drugs for malaria. Expert Opin. Emerg. Drugs 17:319–333. 10.1517/14728214.2012.702754. [DOI] [PubMed] [Google Scholar]
- 9.Kremsner PG, Krishna S. 2004. Antimalarial combinations. Lancet 364:285–294. 10.1016/S0140-6736(04)16680-4. [DOI] [PubMed] [Google Scholar]
- 10.Dechy-Cabaret O, Benoit-Vical F. 2012. Effects of antimalarial molecules on the gametocyte stage of Plasmodium falciparum: the debate. J. Med. Chem. 55:10328–10344. 10.1021/jm3005898. [DOI] [PubMed] [Google Scholar]
- 11.Alonso PL, Brown G, Arevalo-Herrera M, Binka F, Chitnis C, Collins F, Doumbo OK, Greenwood B, Hall BF, Levine MM, Mendis K, Newman RD, Plowe CV, Rodriguez MH, Sinden R, Slutsker L, Tanner M. 2011. A research agenda to underpin malaria eradication. PLoS Med. 8:e1000406. 10.1371/journal.pmed.1000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newman DJ, Cragg GM. 2012. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 75:311–335. 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bagchi GD, Jain DC, Kumar S. 1997. Arteether: a potent plant growth inhibitor from Artemisia annua. Phytochemistry 45:1131–1133. 10.1016/S0031-9422(97)00126-X. [DOI] [Google Scholar]
- 14.Frey M, Chomet P, Glawischnig E, Stettner C, Grun S, Winklmair A, Eisenreich W, Bacher A, Meeley RB, Briggs SP, Simcox K, Gierl A. 1997. Analysis of a chemical plant defense mechanism in grasses. Science 277:696–699. 10.1126/science.277.5326.696. [DOI] [PubMed] [Google Scholar]
- 15.Rosenthal GA, Berenbaum MR. 1981. Herbivores: their interactions with secondary plant metabolites, volume 1: the chemical participants, 2nd ed. Academic Press, New York, NY. [Google Scholar]
- 16.Laska M, Rivas Bautista RM, Hernandez Salazar LT. 2009. Gustatory responsiveness to six bitter tastants in three species of nonhuman primates. J. Chem. Ecol. 35:560–571. 10.1007/s10886-009-9630-8. [DOI] [PubMed] [Google Scholar]
- 17.Sayers EW, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman DJ, Madden TL, Maglott DR, Miller V, Mizrachi I, Ostell J, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Yaschenko E, Ye J. 2009. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 37:D5–D15. 10.1093/nar/gkn741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. 2009. GenBank. Nucleic Acids Res. 37:D26–D31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerth K, Pradella S, Perlova O, Beyer S, Muller R. 2003. Myxobacteria: proficient producers of novel natural products with various biological activities—past and future biotechnological aspects with the focus on the genus Sorangium. J. Biotechnol. 106:233–253. 10.1016/j.jbiotec.2003.07.015. [DOI] [PubMed] [Google Scholar]
- 20.Schneiker S, Perlova O, Kaiser O, Gerth K, Alici A, Altmeyer MO, Bartels D, Bekel T, Beyer S, Bode E, Bode HB, Bolten CJ, Choudhuri JV, Doss S, Elnakady YA, Frank B, Gaigalat L, Goesmann A, Groeger C, Gross F, Jelsbak L, Jelsbak L, Kalinowski J, Kegler C, Knauber T, Konietzny S, Kopp M, Krause L, Krug D, Linke B, Mahmud T, Martinez-Arias R, McHardy AC, Merai M, Meyer F, Mormann S, Munoz-Dorado J, Perez J, Pradella S, Rachid S, Raddatz G, Rosenau F, Ruckert C, Sasse F, Scharfe M, Schuster SC, Suen G, Treuner-Lange A, Velicer GJ, Vorholter FJ, Weissman KJ, Welch RD, Wenzel SC, Whitworth DE, Wilhelm S, Wittmann C, Blocker H, Puhler A, Muller R. 2007. Complete genome sequence of the myxobacterium Sorangium cellulosum. Nat. Biotechnol. 25:1281–1289. 10.1038/nbt1354. [DOI] [PubMed] [Google Scholar]
- 21.Diez J, Martinez JP, Mestres J, Sasse F, Frank R, Meyerhans A. 2012. Myxobacteria: natural pharmaceutical factories. Microb. Cell Fact. 11:52. 10.1186/1475-2859-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weissman KJ, Muller R. 2010. Myxobacterial secondary metabolites: bioactivities and modes-of-action. Nat. Prod. Rep. 27:1276–1295. 10.1039/c001260m. [DOI] [PubMed] [Google Scholar]
- 23.Wenzel SC, Müller R. 2009. The biosynthetic potential of myxobacteria and their impact in drug discovery. Curr. Opin. Drug Discov. Dev. 12:220–230. [PubMed] [Google Scholar]
- 24.Gerth K, Steinmetz H, Hofle G, Jansen R. 2008. Chlorotonil A, a macrolide with a unique gem-dichloro-1,3-dione functionality from Sorangium cellulosum, So ce1525. Angew. Chem. Int. Ed. Engl. 47:600–602. 10.1002/anie.200703993. [DOI] [PubMed] [Google Scholar]
- 25.Rahn N, Kalesse M. 2008. The total synthesis of chlorotonil A. Angew. Chem. Int. Ed. Engl. 47:597–599. 10.1002/anie.200703930. [DOI] [PubMed] [Google Scholar]
- 26.Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675. 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 27.Lambros C, Vanderberg JP. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65:418–420. 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- 28.Noedl H, Bronnert J, Yingyuen K, Attlmayr B, Kollaritsch H, Fukuda M. 2005. Simple histidine-rich protein 2 double-site sandwich enzyme-linked immunosorbent assay for use in malaria drug sensitivity testing. Antimicrob. Agents Chemother. 49:3575–3577. 10.1128/AAC.49.8.3575-3577.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Held J, Westerman R, Kremsner PG, Mordmuller B. 2010. In vitro activity of mirincamycin (U24729A) against Plasmodium falciparum isolates from Gabon. Antimicrob. Agents Chemother. 54:540–542. 10.1128/AAC.01090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peters W. 1970. The chemotherapy of rodent malaria. X. Dynamics of drug resistance. II. Acquisition and loss of chloroquine resistance in Plasmodium berghei observed by continuous bioassay. Ann. Trop. Med. Parasitol. 64:25–40. [PubMed] [Google Scholar]
- 31.Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S. 2004. Antimalarial drug discovery: efficacy models for compound screening. Nat. Rev. Drug Discov. 3:509–520. 10.1038/nrd1416. [DOI] [PubMed] [Google Scholar]
- 32.Lelièvre J, Almela MJ, Lozano S, Miguel C, Franco V, Leroy D, Herreros E. 2012. Activity of clinically relevant antimalarial drugs on Plasmodium falciparum mature gametocytes in an ATP bioluminescence “transmission blocking” assay. PLoS One 7:e35019. 10.1371/journal.pone.0035019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ritz C, Streibig JC. 2005. Bioassay analysis using R. J. Stat. Software 12:1–22. [Google Scholar]
- 34.R Development Core Team. 2009. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 35.Nicolaou KC, Winssinger N, Pastor J, Ninkovic S, Sarabia F, He Y, Vourloumis D, Yang Z, Li T, Giannakakou P, Hamel E. 1997. Synthesis of epothilones A and B in solid and solution phase. Nature 387:268–272. 10.1038/387268a0. [DOI] [PubMed] [Google Scholar]
- 36.Irschik H, Reichenbach H, Hofle G, Jansen R. 2007. The thuggacins, novel antibacterial macrolides from Sorangium cellulosum acting against selected Gram-positive bacteria. J. Antibiot. (Tokyo) 60:733–738. 10.1038/ja.2007.95. [DOI] [PubMed] [Google Scholar]
- 37.Reichenbach H, Höfle G. 2008. Discovery and development of the epothilones: a novel class of antineoplastic drugs. Drugs R. D. 9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cristofanilli M. 2012. Advancements in the treatment of metastatic breast cancer (MBC): the role of ixabepilone. J. Oncol. 2012:703858. 10.1155/2012/703858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goodman CD, Su V, McFadden GI. 2007. The effects of anti-bacterials on the malaria parasite Plasmodium falciparum. Mol. Biochem. Parasitol. 152:181–191. 10.1016/j.molbiopara.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 40.Dahl EL, Rosenthal PJ. 2007. Multiple antibiotics exert delayed effects against the Plasmodium falciparum apicoplast. Antimicrob. Agents Chemother. 51:3485–3490. 10.1128/AAC.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skinner TS, Manning LS, Johnston WA, Davis TM. 1996. In vitro stage-specific sensitivity of Plasmodium falciparum to quinine and artemisinin drugs. Int. J. Parasitol. 26:519–525. [DOI] [PubMed] [Google Scholar]
- 42.Mombo-Ngoma G, Oyakhirome S, Ord R, Gabor JJ, Greutelaers KC, Profanter K, Greutelaers B, Kurth F, Lell B, Kun JF, Issifou S, Roper C, Kremsner PG, Grobusch MP. 2011. High prevalence of dhfr triple mutant and correlation with high rates of sulphadoxine-pyrimethamine treatment failures in vivo in Gabonese children. Malar. J. 10:123. 10.1186/1475-2875-10-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borrmann S, Binder RK, Adegnika AA, Missinou MA, Issifou S, Ramharter M, Wernsdorfer WH, Kremsner PG. 2002. Reassessment of the resistance of Plasmodium falciparum to chloroquine in Gabon: implications for the validity of tests in vitro vs. in vivo. Trans. R. Soc. Trop. Med. Hyg. 96:660–663. 10.1016/S0035-9203(02)90345-7. [DOI] [PubMed] [Google Scholar]
- 44.Werawattanachai N, Towiwat P, Unchern S, Maher TJ. 2007. Neuropharmacological profile of tetrahydrofuran in mice. Life Sci. 80:1656–1663. 10.1016/j.lfs.2007.01.050. [DOI] [PubMed] [Google Scholar]
- 45.Jang KH, Nam SJ, Locke JB, Kauffman CA, Beatty DS, Paul LA, Fenical W. 2013. Anthracimycin, a potent anthrax antibiotic from a marine-derived actinomycete. Angew. Chem. Int. Ed. Engl. 52:7822–7824. 10.1002/anie.201302749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diem Thuy LT, Ngoc Hung L, Danh PT, Na-Bangchang K. 2008. Absence of time-dependent artesunate pharmacokinetics in healthy subjects during 5-day oral administration. Eur. J. Clin. Pharmacol. 64:993–998. 10.1007/s00228-008-0506-6. [DOI] [PubMed] [Google Scholar]
- 47.Borrmann S, Adegnika AA, Missinou MA, Binder RK, Issifou S, Schindler A, Matsiegui PB, Kun JF, Krishna S, Lell B, Kremsner PG. 2003. Short-course artesunate treatment of uncomplicated Plasmodium falciparum malaria in Gabon. Antimicrob. Agents Chemother. 47:901–904. 10.1128/AAC.47.3.901-904.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White NJ. 2013. Primaquine to prevent transmission of falciparum malaria. Lancet Infect. Dis. 13:175–181. 10.1016/S1473-3099(12)70198-6. [DOI] [PubMed] [Google Scholar]
- 49.Beshir KB, Sutherland CJ, Sawa P, Drakeley CJ, Okell L, Mweresa CK, Omar SA, Shekalaghe SA, Kaur H, Ndaro A, Chilongola J, Schallig HD, Sauerwein RW, Hallett RL, Bousema T. 2013. Residual Plasmodium falciparum parasitemia in Kenyan children after artemisinin-combination therapy is associated with increased transmission to mosquitoes and parasite recurrence. J. Infect. Dis. 208:2017–2024. 10.1093/infdis/jit431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Targett G, Drakeley C, Jawara M, von Seidlein L, Coleman R, Deen J, Pinder M, Doherty T, Sutherland C, Walraven G, Milligan P. 2001. Artesunate reduces but does not prevent posttreatment transmission of Plasmodium falciparum to Anopheles gambiae. J. Infect. Dis. 183:1254–1259. 10.1086/319689. [DOI] [PubMed] [Google Scholar]
- 51.D'Alessandro S, Silvestrini F, Dechering K, Corbett Y, Parapini S, Timmerman M, Galastri L, Basilico N, Sauerwein R, Alano P, Taramelli D. 2013. A Plasmodium falciparum screening assay for anti-gametocyte drugs based on parasite lactate dehydrogenase detection. J. Antimicrob. Chemother. 68:2048–2058. 10.1093/jac/dkt165. [DOI] [PubMed] [Google Scholar]
- 52.Dechy-Cabaret O, Benoit-Vical F. 2012. Effects of antimalarial molecules on the gametocyte stage of Plasmodium falciparum: the debate. J. Med. Chem. 55:10328–10344. 10.1021/jm3005898. [DOI] [PubMed] [Google Scholar]
- 53.Delves M, Plouffe D, Scheurer C, Meister S, Wittlin S, Winzeler EA, Sinden RE, Leroy D. 2012. The activities of current antimalarial drugs on the life cycle stages of Plasmodium: a comparative study with human and rodent parasites. PLoS Med. 9:e1001169. 10.1371/journal.pmed.1001169. [DOI] [PMC free article] [PubMed] [Google Scholar]