

Abstract
Eltrombopag is an orally bioavailable thrombopoietin receptor agonist approved for the treatment of thrombocytopenia associated with chronic immune (idiopathic) thrombocytopenic purpura and chronic hepatitis C virus (HCV) infection. This study evaluated the potential drug-drug interactions between eltrombopag and the HCV protease inhibitors boceprevir and telaprevir. In this open-label, 3-period, single-sequence, and crossover study, 56 healthy adult subjects were randomized 1:1 to cohort 1 (boceprevir) or 2 (telaprevir). The dosing was as follows: period 1, single 200-mg dose of eltrombopag; period 2, 800 mg boceprevir or 750 mg telaprevir every 8 hours (q8h) for 10 days; and period 3, single 200-mg dose of eltrombopag with either 800 mg boceprevir or 750 mg telaprevir q8h (3 doses). All doses were administered with food, and eltrombopag was administered specifically with low-calcium food. There was a 3-day washout between periods 1 and 2 and no washout between periods 2 and 3. Serial pharmacokinetic samples were collected for 72 h in periods 1 and 3 and for 8 h in period 2. The coadministration of eltrombopag increased the rate of boceprevir absorption, resulting in a 20% increase in the maximum concentration in plasma (Cmax), a 1-h-earlier time to Cmax (Tmax) for boceprevir, a 32% decrease in the concentration at the end of the dosing interval (Cτ), and no change in the area under the concentration-time curve over the dosing interval (AUC0-τ). The coadministration of eltrombopag did not alter telaprevir pharmacokinetics, and the coadministration of boceprevir or telaprevir did not alter eltrombopag pharmacokinetics. Dysgeusia, headache, and somnolence occurred in ≥2 subjects. One subject withdrew because of nausea, headache, dizziness, sinus pressure, and vomiting. There were no severe or serious adverse events. Dose adjustment is not required when eltrombopag is coadministered with boceprevir or telaprevir given the lack of clinically significant pharmacokinetic interaction.
INTRODUCTION
Eltrombopag is a thrombopoietin receptor agonist initially indicated for the treatment of thrombocytopenia in patients with chronic immune (idiopathic) thrombocytopenic purpura who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. In November 2012, eltrombopag was also approved for the treatment of thrombocytopenia in patients with chronic hepatitis C to allow the initiation and maintenance of interferon-based therapy (1, 2). Eltrombopag raises platelet counts to allow the initiation of interferon-based therapy and stabilizes platelet counts during therapy, minimizing the need for interferon dose reduction. Eltrombopag demonstrated improved sustained virologic response (SVR) compared with that of a placebo in patients with chronic hepatitis C who received pegylated interferon plus ribavirin as antiviral therapy (3). The eltrombopag pivotal clinical studies in patients with chronic hepatitis C were designed and conducted prior to the availability of the hepatitis C virus (HCV) protease inhibitors (PIs) boceprevir and telaprevir. The clinical pharmacology study described here was designed to evaluate the potential drug-drug interaction between eltrombopag and these HCV PIs, in anticipation that eltrombopag may be coadministered with HCV PIs in thrombocytopenic patients with chronic HCV infection.
MATERIALS AND METHODS
Study design.
This phase I, open-label, 3-period, single-sequence, and crossover study conducted in healthy adult subjects was designed to evaluate the potential drug-drug interaction between eltrombopag and the HCV PIs boceprevir and telaprevir. The subjects were randomized to 1 of 2 cohorts (Fig. 1). The subjects in cohort 1 received a single oral dose of 200 mg eltrombopag in period 1, 800 mg boceprevir every 8 h (q8h) for 10 days in period 2, and 800 mg boceprevir q8h for 1 day, with a single dose of 200 mg eltrombopag administered with the morning boceprevir dose in period 3. The subjects in cohort 2 received a single oral dose of 200 mg eltrombopag in period 1, 750 mg telaprevir q8h for 10 days in period 2, and 750 mg telaprevir q8h for 1 day, with a single dose of 200 mg eltrombopag administered with the morning telaprevir dose in period 3. There was a 3-day washout between periods 1 and 2 to allow for 72-h serial pharmacokinetic (PK) sampling. There was no washout between periods 2 and 3 to allow for an assessment of repeated-dose effects of boceprevir and telaprevir on eltrombopag PK. Boceprevir and telaprevir were administered with a moderate-fat (approximately 20 g fat) meal, and eltrombopag was administered with a moderate-fat low-calcium (approximately 50 mg calcium) meal. The subjects were admitted to the clinical research unit from the day prior to dosing (day −1) in period 1 and remained in the clinical research unit until the end of period 3, for a total of 18 days. The total duration of subject participation in the study was approximately 9 weeks, including screening within 28 days prior to enrollment, 3 treatment periods, and a final follow-up visit within 10 to 14 days after the last dose of the study drugs.
FIG 1.

Study design. PK, pharmacokinetics; q8h, every 8 h.
Subjects.
All subjects provided written informed consent prior to enrollment in the study. The study was conducted in accordance with the standards of the site's institutional review board, the principles of good clinical practice, all applicable regulatory requirements, the Code of Federal Regulations, and the Declaration of Helsinki.
Healthy males and females were enrolled at Parexel (Baltimore, MD, USA). The subjects were between the ages of 18 and 64 years, inclusive, with a body mass index of 18.5 to 32.0 kg/m2 and no clinically significant abnormalities based on medical history, physical examination, clinical laboratory tests, and electrocardiogram. The subjects had to have tested negative for human immunodeficiency virus (HIV), hepatitis B virus, and HCV at the time of screening. The female subjects had a negative β-human chorionic gonadotropin pregnancy test. The male and female subjects who were not surgically sterile/postmenopausal agreed to use nonhormonal contraceptive methods, including abstinence, an intrauterine device, or 2 forms of barrier contraception.
Subjects were excluded from the study if they reported a history of deep vein thrombosis or other thromboembolic event(s), clotting factor abnormalities associated with hypercoagulability, or thrombocytopenia or bleeding due to abnormal platelet count/function. Elevated blood pressure (systolic, >140 mm Hg; diastolic, >90 mm Hg) or prolonged QT interval (corrected by Fredericia's formula) of >450 ms resulted in subject exclusion. Subjects with a history of cardiac abnormalities, such as atrial fibrillation, mitral valve prolapse, significant heart murmur, or vascular bruit, were excluded. Furthermore, potential subjects were excluded if they had a history of Gilbert's syndrome or alcohol dependency within 12 months, excessive alcohol consumption, or regular tobacco use within 6 months of screening. The anticipated use of prescription or nonprescription medication(s) during the study also excluded subjects from study participation, unless the investigator and sponsor felt that the medication(s) would not interfere with the study procedures or compromise the safety of the subject. Female subjects who were currently receiving hormonal contraception or hormone replacement therapy were also excluded.
Pharmacokinetic sampling and drug concentration assays.
Serial blood samples were collected via the forearm vein into EDTA anticoagulation tubes. Blood samples for determining plasma eltrombopag concentrations were collected predose and at 1, 2, 3, 4, 5, 6, 8, 12, 16, 24, 48, and 72 h following single-dose eltrombopag administration in periods 1 and 3. Blood samples for determining plasma boceprevir diastereomer concentrations were collected predose and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, and 8 h following repeated-dose administration on day 10 of period 2 and on day 1 of period 3. Blood samples for determining plasma telaprevir concentrations were collected predose and at 1, 2, 3, 4, 5, 6, and 8 h following repeated-dose administration on day 10 of period 2 and on day 1 of period 3. The samples were kept on wet ice until processed. The samples were processed and stored in a freezer (at −20°C) within 1 h of collection. The samples were centrifuged at 1,500 × g for 15 min in a refrigerated (4°C) centrifuge. Sample collection and storage tubes for boceprevir and telaprevir were prechilled, and 1.5 ml plasma and 75 μl of 85% phosphoric acid were mixed before storing in the freezer.
The plasma eltrombopag concentrations were determined by Pharmaceutical Product Development, Inc. (Richmond, VA, USA). Eltrombopag and internal standard SB-497115-[13C4] were isolated by protein precipitation using a 90:10 mixture of acetonitrile to 10 mM ammonium formate (pH 3.0). The samples were purified through an Ostro lipid removal 96-well plate. The final extract was analyzed by high-performance liquid chromatography-tandem mass spectrometry using negative ion electrospray. The assay was validated over the eltrombopag concentration range of 100 to 50,000 ng/ml. The eltrombopag assay imprecision (% coefficient of variation [CV]) was ≤4.70%, and inaccuracy (bias, % difference) was within 0.93% to 3.66%.
The concentrations of the boceprevir diastereomers SCH-534128 and SCH-534129 were determined by Pharmaceutical Product Development, Inc. (Middleton, WI, USA). SCH-534128 and SCH-534129 and internal standards 503034-d9 and 629144-d9 were isolated by solid-phase extraction and eluted from the solid-phase extraction plate. The extracts were dried and reconstituted. The final extract was analyzed by high-performance liquid chromatography-tandem mass spectrometry using positive ion atmospheric pressure chemical ionization. The assay was validated over the SCH-534128 concentration range of 5.20 to 5,200 ng/ml and over the SCH-534129 concentration range of 4.80 to 4,800 ng/ml. SCH-534128 assay imprecision (% CV) was ≤12.1%, and inaccuracy (bias, % difference) was within −7.12% to 3.59%. SCH-534129 assay imprecision (% CV) was ≤10.3%, and inaccuracy (bias, % difference) was within −7.84% to 4.12%.
Plasma telaprevir [(S)-telaprevir] concentrations were determined by Pharmaceutical Product Development, Inc. (Middleton, WI, USA). Telaprevir and the internal standard telaprevir-d5 were isolated through supported liquid extraction using 200 μl Biotage Isolute SLE+ (Biotage AB, Uppsala, Sweden), fixed-well plates, and eluted with 700 μl dichloromethane-methyl tertiary butyl ether (1:2 [vol/vol]). The eluate was evaporated under nitrogen steam at approximately 45°C, and the remaining residue was reconstituted with 800 μl methanol-water (50:50 [vol/vol]). The final extract was analyzed by high-performance liquid chromatography-tandem mass spectrometry using positive ion electrospray. The assay was validated over the telaprevir concentration range of 0.100 to 20.0 μg/ml. Telaprevir assay imprecision (% CV) was ≤6.02%, and inaccuracy (bias, % difference) was within −4.76% to 1.45%.
Safety assessments.
Safety was assessed throughout the study by the collection of adverse events (AEs) and the measurement of vital signs, electrocardiogram, and clinical laboratory tests. The frequency, severity, and relationship to treatment of AEs were evaluated.
Pharmacokinetic analysis.
Plasma boceprevir concentrations were calculated as the sum of the 2 diastereomer concentrations at each time point. The PK calculations were based on the actual sample collection times recorded. The PK parameters were calculated for each subject using standard noncompartmental methods (Phoenix WinNonlin version 6.2; Pharsight Corporation, St. Louis, MO, USA). The eltrombopag PK parameters, including the area under the plasma concentration-time curve from time zero extrapolated to infinity (AUC0-∞), the maximum concentration of drug in plasma (Cmax), time to Cmax (Tmax), and half-life (t1/2) were determined from single-dose plasma concentration-time data. The boceprevir and telaprevir PK parameters, Cmax, Tmax, AUC over the dosing interval (AUC0-τ), and concentration at the end of the dosing interval (Cτ) were determined from repeated-dose plasma concentration-time data.
Statistical analysis.
The study was powered for a 90% confidence interval (CI) for the ratio of combination versus single-drug PK parameters to be within 0.82 to 1.22 for boceprevir, 0.86 to 1.16 for telaprevir, and 0.79 to 1.27 for eltrombopag, for a point estimate of 1.0. Statistical analyses were performed using the SAS/STAT module of SAS, version 9.2 (SAS Institute, Cary, NC, USA). The plasma eltrombopag, boceprevir, and telaprevir PK parameters were natural log transformed before analysis by a mixed-effect analysis of variance model, fitting the terms for treatment as a fixed effect and subject as a random effect. Point estimates and 90% CI for the differences between eltrombopag in combination with boceprevir or telaprevir (period 3 [test]) and eltrombopag alone (period 1 [reference]) and between boceprevir or telaprevir in combination with eltrombopag (period 3 [test]) and boceprevir or telaprevir alone (period 2 [reference]) were constructed using the appropriate error terms. The point and 90% CI estimates on the log scale were back transformed to provide point and 90% CI estimates for the ratios of the geometric mean of the test treatment to the geometric mean of the reference treatment.
RESULTS
Disposition and demographics.
This study enrolled 56 healthy subjects, with 28 in each cohort. Fifty-three subjects completed the study, including 26 subjects in cohort 1 and 27 subjects in cohort 2. One subject in cohort 2 was omitted from the eltrombopag PK analysis because of a lack of exposure following the administration of a single dose of eltrombopag with telaprevir in period 3; the subject had 3 quantifiable concentrations at the lower limit of quantification.
The majority of subjects were male (41 of 56 subjects [73%]) and African-American (40 of 56 subjects [71%]); 14 of 56 subjects (25%) were white, and 1 subject each was Asian and Native Hawaiian/Pacific Islander. The mean (range) age was 40 years (20 to 62 years), weight was 77.5 kg (52.2 to 109 kg), and body mass index was 25.9 kg/m2 (20.6 to 31.4 kg/m2). The demographics were similar between the 2 cohorts.
Pharmacokinetics.
The coadministration of a single dose of 200 mg eltrombopag with 800 mg boceprevir q8h (cohort 1) did not alter the plasma boceprevir AUC0-τ (Table 1). The plasma boceprevir Tmax occurred approximately 1 h earlier when eltrombopag was coadministered, Cmax increased approximately 20%, and Cτ decreased 32% (Table 1 and Fig. 2).
TABLE 1.
Summary of plasma boceprevir and telaprevir pharmacokinetic parameters and drug-drug interaction results
| Parametera | Data for drug(s) administeredb: |
GLS mean ratio (90% CI)c | |
|---|---|---|---|
| Boceprevir (cohort 1) or telaprevir (cohort 2) alone | Eltrombopag with boceprevir (cohort 1) or telaprevir (cohort 2) | ||
| Cohort 1 (n = 26) | |||
| Cmax (μg/ml) | 1.63 (1.43, 1.85) (32.1) | 1.95 (1.78, 2.14) (23.4) | 1.197 (1.110, 1.291) |
| AUC0-τ (μg · h/ml) | 5.16 (4.67, 5.70) (25.1) | 5.38 (4.88, 5.92) (24.4) | 1.043 (1.008, 1.079) |
| Cτ (μg/ml) | 0.100 (0.082, 0.123) (53.0) | 0.068 (0.056, 0.082) (50.4) | 0.677 (0.583, 0.786) |
| Tmax (h) | 3.00 (1.48, 5.02) | 2.00 (1.00, 3.05) | |
| Cohort 2 (n = 27) | |||
| Cmax (μg/ml) | 3.04 (2.69, 3.42) (31.2) | 2.94 (2.62, 3.30) (29.6) | 0.969 (0.913, 1.029) |
| AUC0-τ (μg · h/ml) | 18.8 (16.8, 21.1) (30.0) | 18.5 (16.6, 20.5) (27.0) | 0.981 (0.939, 1.025) |
| Cτ (μg/ml) | 2.00 (1.80, 2.22) (27.0) | 1.89 (1.71, 2.10) (25.9) | 0.948 (0.898, 0.999) |
| Tmax (h) | 4.00 (2.00, 7.88) | 4.00 (2.00, 5.03) | |
Cmax, maximum concentration of drug in plasma; AUC0-τ, area under the plasma concentration-time curve over the dosing interval; Cτ, concentration at the end of the dosing interval; Tmax, time to Cmax.
Eltrombopag was given as a 200-mg single dose, boceprevir at 800 mg q8h, and telaprevir at 750 mg q8h. The summary statistics are presented as geometric mean values (95% CIs) (% coefficient of variation), except Tmax, whose summary statistics are presented as the median (minimum, maximum).
GLS, geometric least squares.
FIG 2.
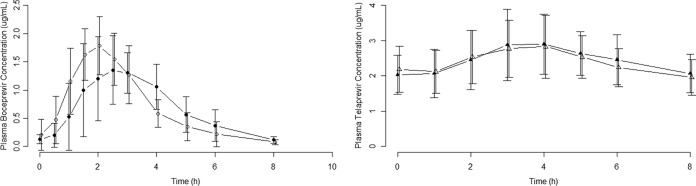
Plasma boceprevir and telaprevir concentration-time profiles. Values are means ± standard deviations (SD) (error bars). Closed circles, boceprevir; open circles, boceprevir plus eltrombopag; closed triangle, telaprevir; open triangles, telaprevir plus eltrombopag.
The coadministration of a single dose of 200 mg eltrombopag with 750 mg telaprevir q8h (cohort 2) did not alter the plasma telaprevir PK (Table 1 and Fig. 2).
The coadministration of repeat doses of 800 mg boceprevir q8h (cohort 1) or 750 mg telaprevir q8h (cohort 2) with a single dose of 200 mg eltrombopag did not alter plasma eltrombopag exposure to a clinically significant extent, with mean changes in plasma eltrombopag AUC0-∞ and Cmax of 4% to 10% (Table 2 and Fig. 3).
TABLE 2.
Summary of plasma eltrombopag pharmacokinetic parameters and drug-drug interaction results
| Parametera | Eltrombopag administeredb: |
GLS mean ratio (90% CI)c | |
|---|---|---|---|
| Alone | With boceprevir (cohort 1) or telaprevir (cohort 2) | ||
| Cohort 1 | |||
| n | 28 | 26 | 26 |
| Cmax (μg/ml) | 22.0 (19.2, 25.1) (35.3) | 19.9 (18.1, 22.0) (24.5) | 0.923 (0.804, 1.060) |
| AUC0-∞ (μg · h/ml) | 313 (269, 363) (39.9) | 294 (260, 331) (30.4) | 0.962 (0.853, 1.085) |
| Tmax (h) | 3.98 (3.00, 8.00) | 4.00 (2.00, 6.05) | |
| t1/2 (h) | 23.0 (21.7, 24.5) (15.9) | 24.0 (22.5, 25.5) (15.4) | |
| Cohort 2 | |||
| n | 27 | 26 | 26 |
| Cmax (μg/ml) | 18.0 (15.0, 21.5) (47.9) | 16.1 (13.7, 18.9) (41.0) | 0.899 (0.794, 1.017) |
| AUC0-∞ (μg · h/ml) | 257 (209, 315) (55.6) | 238 (198, 287) (48.3) | 0.939 (0.853, 1.035) |
| Tmax (h) | 3.05 (2.00, 6.05) | 4.00 (3.00, 5.03) | |
| t1/2 (h) | 22.6 (21.0, 24.4) (19.3) | 19.0 (17.8, 20.3) (16.6) | |
Cmax, maximum concentration of drug in plasma; AUC0-∞, area under the plasma concentration-time curve from zero extrapolated to infinity; Tmax, time to Cmax; t1/2, half-life.
Eltrombopag was given as a 200-mg single dose, boceprevir at 800 mg q8h, and telaprevir at 750 mg q8h. The summary statistics are presented as geometric mean values (95% CIs) (% coefficient of variation), except Tmax, whose summary statistics are presented as the median (minimum, maximum).
GLS, geometric least squares.
FIG 3.
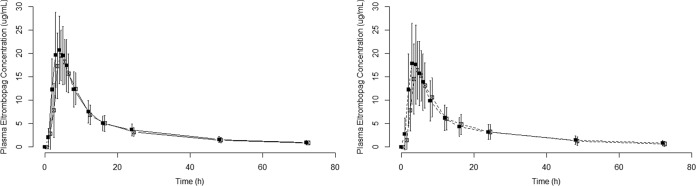
Plasma eltrombopag concentration-time profiles. Values are means ± standard deviations (SD) (error bars). Solid line and closed squares, eltrombopag; open squares, eltrombopag plus boceprevir; dashed line and closed squares, eltrombopag; open squares, eltrombopag plus telaprevir.
Safety.
No new safety signal was observed during the study. The AEs were of mild or moderate severity, and there were no severe AEs. Dysgeusia, headache, and somnolence occurred in ≥2 subjects, and these AEs occurred when eltrombopag was coadministered with boceprevir and telaprevir. Three subjects withdrew from the study; two subjects in cohort 1 withdrew consent during period 2 while receiving 800 mg boceprevir q8h, and 1 subject in cohort 2 withdrew due to AEs on day 6 of period 2 while receiving 750 mg telaprevir q8h. The AEs leading to subject withdrawal included nausea, headache, dizziness, sinus pressure, and vomiting and were considered to be related to the study drug.
DISCUSSION
This study was designed to evaluate the drug-drug interaction between eltrombopag and boceprevir and between eltrombopag and telaprevir, in anticipation that eltrombopag may be coadministered with these HCV PIs in thrombocytopenic patients with chronic HCV infection.
Subjects with chronic HCV have higher plasma eltrombopag exposures than do healthy subjects and subjects with idiopathic thrombocytopenic purpura (ITP) for the same dose (2). A 200-mg dose of eltrombopag was chosen for the study to match the exposures observed in patients with chronic HCV infection. Plasma eltrombopag exposure following the administration of a single 200-mg dose to healthy adult subjects in this drug-drug interaction study was similar to the steady-state exposure achieved with 75 to 100 mg once-daily dosing in patients with chronic HCV infection (2).
The potential for a drug-drug interaction between eltrombopag and the HCV PIs boceprevir and telaprevir was considered low. Eltrombopag was not expected to significantly alter plasma boceprevir or telaprevir exposure because eltrombopag is not an inhibitor of CYP3A4 or P-glycoprotein, and eltrombopag is not an inducer. Eltrombopag had not been evaluated for aldo-keto reductase inhibition, the primary enzyme responsible for boceprevir metabolism. Boceprevir and telaprevir were not expected to significantly alter plasma eltrombopag exposure because eltrombopag is not a CYP3A4 substrate and it is metabolized through multiple pathways.
A lack of interaction between eltrombopag and telaprevir was confirmed, and boceprevir does not alter plasma eltrombopag PK, but changes in boceprevir PK were observed when eltrombopag was coadministered. During the reporting of this study, boceprevir was identified as a breast cancer resistance protein substrate (4). Eltrombopag is a breast cancer resistance protein inhibitor and increased the rate of boceprevir absorption, resulting in the observed changes in plasma boceprevir Tmax, Cmax, and Cτ. The coadministration of eltrombopag did not alter the extent of boceprevir absorption (no change in plasma boceprevir AUC0-τ), because the majority of a boceprevir dose is absorbed (5).
During the study, boceprevir was administered with a low-calcium meal when coadministered with eltrombopag and with a normal-calcium meal when administered alone. A contribution of the different meal type to the interaction cannot be ruled out; however, prior studies of the bioavailability of boceprevir under different meal conditions (high- versus low-fat meals) suggest that boceprevir is not very sensitive to meal type (6).
Phase II monotherapy and dual therapy (boceprevir plus interferon) dose-ranging studies suggest a relationship between plasma boceprevir Cτ and virologic response (6). A 50% effective concentration (EC50) of 98 ng/ml was identified in the phase II dual therapy exposure response analysis, similar to the in vitro 50% inhibitory concentration (IC50) of 100 ng/ml. However, when boceprevir was administered at the therapeutic dose of 800 mg 3 times daily in combination with both interferon and ribavirin (triple therapy) in phase III studies, no relationship between plasma boceprevir Cτ and SVR (primary efficacy endpoint) was observed (7). The reason(s) no relationship was identified between plasma boceprevir Cτ and SVR may have been the use of triple therapy in phase III as opposed to dual therapy in phase II, the administration of a single dose level in phase III as opposed to dose ranging in phase II, and the difference in the clinical endpoints between the phase III and phase II studies.
Moreover, no relationship between plasma boceprevir Cτ and SVR was observed in an analysis that included data from HIV/HCV-coinfected and HCV-monoinfected patients, despite an approximate 27% lower plasma boceprevir Cτ in HIV/HCV-coinfected patients (8). The lack of exposure response observed in the coinfected population supports that a 27% lower plasma boceprevir Cτ did not compromise efficacy (8). The 32% reduction in the plasma boceprevir Cτ observed with eltrombopag coadministration is not considered clinically significant because it is similar to the 27% lower Cτ identified in the HIV/HCV-coinfected population and the 26% reduction observed with raltegravir, for which no dose adjustment is required (5).
When patients use eltrombopag in combination with boceprevir or telaprevir, they must be compliant with food recommendations for the products. Boceprevir and telaprevir are dosed q8h and require dosing with food (5, 9). Eltrombopag is dosed once daily and must be administered ≥4 h apart from polyvalent metal cation-containing products, including dairy (1, 2). To enable coadministration, eltrombopag can be administered with boceprevir or telaprevir with a moderate-fat low-calcium meal, as done in this study, or eltrombopag can be administered separately from boceprevir and telaprevir to avoid polyvalent metal cations in standard meals.
The AEs were consistent with the established safety profile of the individual study medications. No new safety signals were identified when eltrombopag was coadministered with either boceprevir or telaprevir. In conclusion, dose adjustment is not required when eltrombopag is coadministered with boceprevir or telaprevir given the lack of a clinically significant PK interaction.
ACKNOWLEDGMENTS
We thank all study participants and Jessika Weldon, Mary Alexander, John Zhang, and Parexel (Baltimore, MD, USA).
This research was sponsored by GlaxoSmithKline, Inc. M. B. Wire, J. Kleha, and D. Theodore are employees of GlaxoSmithKline.
Footnotes
Published ahead of print 25 August 2014
REFERENCES
- 1.GlaxoSmithKline. 2014. PROMACTA (eltrombopag) United States product information. GlaxoSmithKline, Research Triangle Park, NC. [Google Scholar]
- 2.Glaxo Operations UK. 2014. REVOLADE (eltrombopag) annex I: summary of product characteristics. Glaxo Operations UK, Hertfordshire, United Kingdom: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001110/WC500089964.pdf. [Google Scholar]
- 3.Afdhal NH, Dusheiko GM, Giannini EG, Chen PJ, Han KH, Mohsin A, Rodriguez-Torres M, Rugina S, Bakulin I, Lawitz E, Shiffman ML, Tayyab GU, Poordad F, Kamel YM, Brainsky A, Geib J, Vasey SY, Patwardhan R, Campbell FM, Theodore D. 2014. Eltrombopag increases platelet numbers in thrombocytopenic patients with HCV infection and cirrhosis, allowing for effective antiviral therapy. Gastroenterology 146:442–452.e1. 10.1053/j.gastro.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 4.Chu X, Cai X, Cui D, Tang C, Ghosal A, Chan G, Green MD, Kuo Y, Liang Y, Maciolek CM, Palamanda J, Evers R, Prueksaritanont T. 2013. In vitro assessment of drug-drug interaction potential of boceprevir associated with drug metabolizing enzymes and transporters. Drug Metab. Dispos. 41:668–681. 10.1124/dmd.112.049668. [DOI] [PubMed] [Google Scholar]
- 5.Merck Sharp & Dohme Corp. 2012. VICTRELIS (boceprevir) highlights of prescribing information. Merck Sharp & Dohme Corp., Whitehouse Station, NJ: http://www.merck.com/product/usa/pi_circulars/v/victrelis/victrelis_pi.pdf. [Google Scholar]
- 6.US Food and Drug Administration Center for Drug Evaluation and Research. 2011. Clinical pharmacology and biopharmaceutics review(s). Application no. 202258Orig1s000 US Food and Drug Administration, Silver Spring, MD: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202258orig1s000clinpharmr.pdf. [Google Scholar]
- 7.Stone JA, Wenning LA, Hang Y, Su J, Gupta S, Tsai K, Brass CA, O'Mara E. 2011. Assessment of boceprevir pharmacokinetic/pharmacodynamic relationships for sustained viral response and occurrence of anemia from phase 3 data, poster 1342. 62nd Annu. Meet. Am. Assoc. Study Liver Dis., San Francisco, CA, 4 to 8 November 2011. [Google Scholar]
- 8.Wenning LA, Flexner C, Liu R, Poland B, Tsai K, Ping Feng H, Stone JA, Wahl J, Sklar P, Greaves W, Sulkowski M. 2012. Assessment of boceprevir (VICTRELIS) pharmacokinetic/pharmacodynamic relationship with sustained viral response (SVR) and occurrence of anemia: results in HCV/HIV co-infected patients and in combined mono- and co-infected patients, poster 770. 63nd Annu. Meet. Am. Assoc. Study Liver Dis., Boston, MA, 9 to 13 November 2012. [Google Scholar]
- 9.Vertex Pharmaceuticals. 2013. INCIVEK (telaprevir) highlights of prescribing information. Vertex Pharmaceuticals, Inc., Cambridge, MA: http://pi.vrtx.com/files/uspi_telaprevir.pdf. [Google Scholar]