

Abstract
Two identical single-ascending-dose studies evaluated the safety and pharmacokinetics (PK) of AVI-6002 and AVI-6003, two experimental combinations of phosphorodiamidate morpholino oligomers with positive charges (PMOplus) that target viral mRNA encoding Ebola virus and Marburg virus proteins, respectively. Both AVI-6002 and AVI-6003 were found to suppress disease in virus-infected nonhuman primates in previous studies. AVI-6002 (a combination of AVI-7537 and AVI-7539) or AVI-6003 (a combination of AVI-7287 and AVI-7288) were administered as sequential intravenous (i.v.) infusions of a 1:1 fixed dose ratio of the two subcomponents. In each study, 30 healthy male and female subjects between 18 and 50 years of age were enrolled in six-dose escalation cohorts of five subjects each and received a single i.v. infusion of active study drug (0.005, 0.05, 0.5, 1.5, 3, and 4.5 mg/kg per component) or placebo in a 4:1 ratio. Both AVI-6002 and AVI-6003 were safe and well tolerated at the doses studied. A maximum tolerated dose was not observed in either study. The four chemically similar PMOplus components exhibited generally similar PK profiles. The mean peak plasma concentration and area under the concentration-time curve values of the four components exhibited dose-proportional PK. The estimated plasma half-life of all four components was 2 to 5 h. The safety of the two combinations and the PK of the four components were similar, regardless of the target RNA sequence.
INTRODUCTION
Ebola virus and Marburg virus are filamentous, single-stranded, negative-sense RNA viruses of the family Filoviridae and causative agents of viral hemorrhagic fever (1). Clinically, filovirus infections are characterized by the acute onset of illness after a typical incubation period of 4 to 10 days, with symptoms initially consisting of fever, chills, myalgia, and malaise (2). Disease features may evolve to encompass anorexia, nausea, vomiting, abdominal pain, diarrhea, respiratory complaints, conjunctival injection, hypotension, edema, prostration, confusion, and coma. Hemorrhagic manifestations, coagulopathy, maculopapular rash, cytopenias, and increased transaminase levels may also be observed (2–4). Infections caused by Ebola and Marburg viruses are associated with a very high mortality rate (1, 2). Ebola virus is associated with case fatality rates of 47 to 89% (5), and of the 2,387 known cases of Ebola virus reported up to April 2014, 1,590 (66.6%) have been fatal (6). Death rates in Marburg hemorrhagic fever outbreaks have ranged from 24 to 88%, and of the 571 cases of Marburg hemorrhagic fever reported to date, 82.3% have been fatal (7).
Both viruses may be transmitted to the host by exposure of mucosal surfaces or abraded skin to infected body fluids or through parenteral inoculation; the contribution of aerosol or respiratory droplet transmission in the setting of natural epidemics is unknown (8). Because these viruses are highly lethal and readily transmitted from person to person, both Ebola virus and Marburg virus have been classified by the U.S. Centers for Disease Control and Prevention as potential “category A” agents of bioterrorism and deemed of high priority for focused preparedness efforts, including the development of effective counter measures to infection (9). Other than supportive care, no vaccine or established effective therapy is currently available to treat Ebola and Marburg virus infections.
AVI-6002 and AVI-6003 are two combination drugs under evaluation for postexposure prophylaxis of Ebola virus and Marburg virus, respectively. Both drugs were developed in 1:1 combinations: AVI-6002 consists of AVI-7537 and AVI-7539 (Fig. 1), and AVI-6003 consists of AVI-7287 and AVI-7288 (Fig. 2). Both drugs consist of phosphorodiamidate morpholino oligomers that have positive charges in the form of piperazine residues at defined locations along the backbone (PMOplus). The oligomers target specific mRNA sequences of viral proteins and physically block their translation. The addition of the piperazine residues impart positive charges to the antisense oligonucleotide and are thought to enhance binding to negatively charged viral RNA, possibly subverting the consequences of individual viral resistance mutations, should they evolve.
FIG 1.

The components of AVI-6002 are AVI-7537 and AVI-7539, each of which is a phosphorodiamidate morpholino oligomer in which the dimethylamine linkage sites have been replaced with a piperazine rings at defined locations along the backbone to confer a net positive charge.
FIG 2.

The components of AVI-6003 are AVI-7287 and AVI-7288, each of which is a phosphorodiamidate morpholino oligomer in which dimethylamine linkage sites have been replaced with a piperazine rings at defined locations along the backbone to confer a net positive charge.
Studies of the efficacy of AVI-6002 and AVI-6003 in the mouse, guinea pig, and nonhuman primate lethal challenge models were performed at the U.S. Army Medical Research Institute for Infectious Diseases (10, 11). In these studies, treatment with AVI-6002 resulted in high levels of survival in mice, guinea pigs, and rhesus monkeys after exposure to Ebola virus. Similarly, treatment with AVI-6003 resulted in high levels of survival in mice, guinea pigs, and cynomolgus macaques after exposure to Marburg virus. None of the untreated control animals and none of the animals that were treated with scrambled control survived in either the Ebola or Marburg virus nonhuman primate lethal challenge models. Dose-dependent survival was observed in both models, with high levels of survival observed on nonhuman primates treated with AVI-6002 or AVI-6003 at doses of 28 to 30 mg/kg. The human equivalent dose, based on scaling by body surface area, was approximately 9 mg/kg, or 4.5 mg/kg per component.
Two identical, first-in-human, single-ascending-dose studies of AVI-6002 and AVI-6003, respectively, were completed to assess the safety, tolerability, and pharmacokinetics (PK) of these compounds in healthy volunteers. The doses tested ranged from a very low dose, 0.005 mg/kg per component, a dose 100-fold lower than the human equivalent dose observed to have minimal adverse effects in toxicology studies, to 4.5 mg/kg per component, the human equivalent dose, as noted above.
MATERIALS AND METHODS
Study agents.
The chemical properties of the components of AVI-6002 (AVI-7537 and AVI-7539) and AVI-6003 (AVI-7287 and AVI-7288) are presented in Table 1. Each component is composed of morpholino analogs of A, C, G, T, and I nucleosides arranged on a phosphorodiamidate linkage platform in which five or six dimethylamine linkage sites have been replaced with a piperazine ring to confer a net 5+ positive charge to AVI-7537, AVI-7539, and AVI-7288 and a net 6+ charge to AVI-7287 (Fig. 1 and 2). The I (inosine) nucleoside was included because it pairs with A, C, and T, potentially maintaining target binding if a mutation emerges.
TABLE 1.
Study agents
| Parent compound | PMOplus component | Molecular mass (Da) | No. of nucleobases | Nucleobase composition (%) |
No. of piperazine rings | Target | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| A | C | G | I | T | ||||||
| AVI-6002 | AVI-7357 | 6,825.9 | 19 | 11 | 21 | 26 | 0 | 42 | 5 | Ebola viral protein 24, viral matrix protein that inhibits interferon signaling |
| AVI-7359 | 7,092.1 | 20 | 5 | 30 | 20 | 0 | 45 | 5 | Ebola viral protein 35, component of the RNA-dependent RNA polymerase that antagonizes the interferon pathway | |
| AVI-6003 | AVI-7287 | 7,491.4 | 21 | 14 | 24 | 14 | 10 | 38 | 6 | Marburg viral protein 24, viral matrix protein that inhibits interferon signaling |
| AVI-7288 | 8,179.0 | 23 | 43 | 17 | 13 | 4 | 22 | 5 | Marburg nucleoprotein, the major nucleoprotein involved in RNA encapsulation | |
Doses of both AVI-6002 and AVI-6003 of 0.01, 0.1, 1, 3, 6, and 9 mg/kg made up of equal parts of their respective individual components were administered as sequential intravenous (i.v.) infusions. Each component was diluted in 150 ml of normal saline prior to i.v. infusion over 30 min each. Placebo for both studies consisted of two sequential infusions of 150 ml of normal saline, administered i.v. over 30 min. Placebo and active drug preparations were indistinguishable.
Study design.
The designs of the two randomized, double-blind, placebo-controlled studies were identical. Each study protocol was reviewed and approved by the Institutional Review Board overseeing the research at the participating institution. In each study, a total of 30 subjects were enrolled into six cohorts of five subjects each. Within each cohort, subjects were randomized in a 4:1 ratio to active drug or placebo. The dose of active drug in the first cohort of each study was 0.005 mg/kg per component, and was escalated in each subsequent cohort to 0.05, 0.5, 1.5, 3, and 4.5 mg/kg per component, after cumulative blinded safety data were reviewed by an independent data safety monitoring board, prior to enrollment of subjects into the subsequent dose cohort.
Study subjects.
In both studies, subjects were males and females between the ages of 18 and 50 in good general health who were willing to use barrier methods of contraception or were of non-childbearing potential. Major exclusion criteria included pregnancy; breast-feeding; a body mass index (BMI) of <18 or >35 kg/m2; any clinically relevant abnormalities in physical examinations, vital signs, electrocardiograms (ECGs), clinical chemistry, hematology or urinalysis; an elevated serum creatinine level and/or a urine albumin-to-creatinine ratio (UACR) of >30 mg/g; glomerular filtration rate of <80 ml/min, based on Cockcroft-Gault calculation using lean (ideal) body weight; or a positive test for human immunodeficiency virus, hepatitis B, or hepatitis C or a known history of HIV infection.
Study procedures.
Study procedures were identical for both studies. After providing informed consent, subjects underwent screening procedures up to 21 days prior to study drug administration. Eligible subjects were admitted to the phase 1 unit the day prior to study drug administration and underwent day −1 assessments to confirm eligibility. Subjects received a single i.v. dose of AVI-6002, AVI-6003, or placebo. Subjects were observed in the study unit for 96 h after study drug administration for serial plasma and urine PK sampling. Safety was monitored through adverse event (AE) collection, telemetry, clinical laboratory assessments (hematology, coagulation, chemistry, urinalysis, and complement levels), oximetry, and ECGs. Subjects returned for follow-up visits 14, 21, and 28 days after study drug administration. AEs were coded by preferred term and system organ class using MedDRA version 14.1. The intensity of AEs and laboratory abnormalities was determined by using the criteria specified in the U.S. Food and Drug Administration's 2007 guidance document Guidance for Industry: Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventative Vaccine Clinical Trials (12).
Pharmacokinetic evaluations.
Blood samples for pharmacokinetic determinations were drawn predose (within 30 min before dosing); at 5, 15, and 28 min after the start of the infusion of the first component of the study drug; at 5, 15, and 28 min after the start of the infusion of the second component of the study drug; and at 5, 10, 15, 30, and 45 min and 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48, 72, and 96 h after the completion of the study drug infusions. Urine was collected in aliquots for pharmacokinetic determinations at each of the following intervals: the start of dosing through the completion of the infusions, the completion of the infusions through 1 h, >1 h through 4 h, >4 h through 8 h, >8 h through 12 h, >12 h through 24 h, >24 h through 48 h, >48 h through 72 h, and >72 h through 96 h.
Plasma and urine concentrations were determined by using a validated capillary gel electrophoresis and fluorescent probe hybridization assay for each component (Helix Diagnostics, Inc., Madison, WI). Briefly, plasma and urine samples were prepared for hybridization using TRIzol LS extractions, followed by alcohol precipitation and then hybridized to a fluorescence-labeled oligonucleotide probe. The probe-analyte species were separated and detected by using capillary gel electrophoresis with laser-induced fluorescence.
The pharmacokinetics of the individual components of AVI-6002 and AVI-6003 were determined from blood and urine samples using a model-independent (noncompartmental) approach appropriate for a constant rate infusion (13). The pharmacokinetic parameters characterized included the maximum plasma concentration (Cmax), the plasma half-life, the time at which Cmax occurs (Tmax), the area under the plasma concentration-time curve from time zero to 24 h after the start of infusion of study drug (AUC0–24), the volume of distribution under steady-state conditions (Vss), plasma clearance (CLP), and renal (i.e., urinary) clearance (CLR). Pharmacokinetic and exploratory analyses were performed using WinNonlin v5.2 (Pharsight Corp., Mountain View, CA), SigmaPlot V 11.0 (Systat Software, Inc., San Jose, CA), GraphPad Prism v5.0 (La Jolla, CA), and Microsoft Excel 2003 (Microsoft Corp., Redmond, WA).
RESULTS
Study enrollment and disposition.
Demographics and baseline characteristics of the subjects who participated in the two studies are outlined in Table 2. Fifteen males and 15 females took part in the AVI-6002 study. The majority of the subjects were white (66.7%), eight were black or African-American (26.7%), and two were American Indian or Alaska Native (6.7%). The mean age was 26.4 years (range, 19 to 42 years) and was comparable across dose groups. The mean weight and BMI values were also similar across dose groups. All subjects were dosed, and all but two completed the 28-day study. One subject in the 1-mg/kg cohort withdrew consent on day 15, and one subject in the 6-mg/kg cohort was lost to follow-up after day 5; neither discontinuation was treatment related.
TABLE 2.
Demographic and baseline characteristics of study subjects
| Characteristic | AVI-6002 study |
AVI-6003 study |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo (n = 6) | Dose (mg/kg) |
Total (n = 30) | Placebo (n = 6) | Dose (mg/kg) |
Total (n = 30) | |||||||||||
| 0.01 (n = 4) | 0.1 (n = 4) | 1 (n = 4) | 3 (n = 4) | 6 (n = 4) | 9 (n = 4) | 0.01 (n = 4) | 0.1 (n = 4) | 1 (n = 4) | 3 (n = 4) | 6 (n = 4) | 9 (n = 4) | |||||
| No. of subjects (%) | ||||||||||||||||
| Gender | ||||||||||||||||
| Male | 2 (33) | 2 (50) | 2 (50) | 2 (50) | 2 (50) | 2 (50) | 3 (75) | 15 (50) | 5 (83) | 1 (25) | 2 (50) | 2 (50) | 2 (50) | 2 (50) | 2 (50) | 16 (53) |
| Female | 4 (67) | 2 (50) | 2 (50) | 2 (50) | 2 (50) | 2 (50) | 1 (25) | 15 (50) | 1 (17) | 3 (75) | 2 (50) | 2 (50) | 2 (50) | 2 (50) | 2 (50) | 14 (47) |
| Race | ||||||||||||||||
| White | 4 (67) | 2 (50) | 2 (50) | 3 (75) | 4 (100) | 3 (75) | 2 (50) | 20 (67) | 6 (100) | 2 (50) | 1 (25) | 1 (25) | 3 (75) | 3 (75) | 2 (50) | 18 (60) |
| Black or African-American | 1 (17) | 2 (50) | 1 (25) | 1 (25) | 0 | 1 (25) | 2 (50) | 8 (27) | 0 | 2 (50) | 2 (50) | 2 (50) | 0 | 1 (25) | 2 (50) | 9 (30) |
| Asian | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (25) | 1 (25) | 1 (25) | 0 | 0 | 3 (10) |
| American Indian or Alaska Native | 1 (17) | 0 | 1 (25) | 0 | 0 | 0 | 0 | 2 (7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mean (SD) | ||||||||||||||||
| Age (yr) | 28.0 (5.8) | 28.3 (5.1) | 30.3 (9.0) | 26.5 (6.0) | 23.5 (6.5) | 24.0 (5.7) | 23.3 (3.3) | 26.4 (6.0) | 33.2 (10.9) | 29.0 (8.9) | 35.0 (13.5) | 26.5 (5.3) | 32.8 (5.7) | 26.3 (7.5) | 33.8 (6.9) | 31.1 (8.7) |
| Wt (kg) | 81.6 (10.9) | 77.8 (3.3) | 75.9 (14.8) | 65.1 (7.1) | 71.1 (10.5) | 73.0 (16.2) | 66.6 (7.7) | 73.6 (11.3) | 77.3 (14.0) | 75.7 (18.4) | 76.1 (12.9) | 73.7 (21.7) | 69.0 (4.5) | 69.8 (4.6) | 70.1 (5.0) | 73.4 (12.3) |
| BMIa (kg/m2) | 27.0 (3.0) | 27.1 (2.9) | 26.4 (3.0) | 22.6 (2.7) | 24.2 (3.1) | 26.3 (5.5) | 23.7 (1.7) | 25.5 (3.4) | 24.5 (4.4) | 26.3 (4.5) | 25.3 (3.3) | 24.0 (4.5) | 25.2 (3.3) | 23.8 (2.1) | 25.2 (2.2) | 24.9 (3.4) |
| Estimated CLCR (ml/min)b | 114.1 (28.4) | 107.8 (6.9) | 104.3 (21.0) | 108.2 (20.4) | 112.8 (16.2) | 103.2 (12.9) | 111.9 (16.8) | 109.2 (18.0) | 106.7 (14.5) | 110.7 (14.4) | 109.9 (29.4) | 118.0 (4.6) | 105.1 (8.3) | 118.4 (17.2) | 98.9 (14.5) | 109.5 (17.1) |
BMI, body mass index.
The estimated creatinine clearance (CLCR) was based on a Cockcroft-Gault calculation using the lean (ideal) body weight: (140 − age) × ideal body weight (in kg) × [0.85 if female]/[72 × serum creatinine (mg/dl)].
A total of 16 males and 14 females took part in the AVI-6003 study; 18 (60.0%) of the subjects were white, 9 (30.0%) were black or African-American, and 3 (10.0%) were Asian. The overall mean age was 31.1 years (range, 18 to 49 years) and was comparable across the dose groups. The mean weight and BMI values also were similar across dose groups. All subjects were dosed, and all completed the 28-day study except for one subject in the 0.1-mg/kg cohort who withdrew consent after experiencing a non-treatment-related serious adverse event (SAE).
Tolerability.
Both AVI-6002 and AVI-6003 were safe and well tolerated at all of the doses studied. Treatment-emergent adverse events (TEAEs) occurring two or more times in a given system organ class in either study are summarized in Table 3. In the AVI-6002 study, 12 (50%) of the AVI-6002-treated subjects experienced 20 TEAEs, 15 of which were considered treatment related. Overall, gastrointestinal disorders were most common, followed by nervous system disorders. No TEAEs related to abnormal laboratory results were reported. TEAEs occurring in more than one subject included headache (four AVI-6002 subjects; zero placebo subjects), nausea (three AVI-6002 subjects; zero placebo subjects), and sinus congestion (two AVI-6002 subjects; one placebo subject). No dose-dependent pattern was observed. All TEAEs were mild in severity, with the exception of four subjects who experienced one moderate episode each of headache, uveitis, contact dermatitis, and laceration. Nine AVI-6002-treated subjects experienced 15 treatment-related TEAEs, with headache and nausea being the only TEAEs occurring in more than one subject. One subject in the AVI-6002 9-mg/kg group developed moderate uveitis that was considered related to the drug. A retinal specialist suggested recurrent toxoplasmosis as the underlying cause of the uveitis based on the subject's country of origin (West Africa) and the presence of retinal scarring. The subject was treated, and the AE resolved on day 20. No SAEs, discontinuations due to AEs, or deaths were reported.
TABLE 3.
Treatment-emergent adverse events occurring more than once in a given system organ class in the two AVI studies
| TEAEa | No. (%) of subjects |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AVI-6002 study |
AVI-6003 study |
|||||||||||||||
| Placebo (n = 6) | Dose (mg/kg) |
AVI-6002 (n = 24) | Placebo (n = 6) | Dose (mg/kg) |
AVI-6003 (n = 24) | |||||||||||
| 0.01 (n = 4) | 0.1 (n = 4) | 1 (n = 4) | 3 (n = 4) | 6 (n = 4) | 9 (n = 4) | 0.01 (n = 4) | 0.1 (n = 4) | 1 (n = 4) | 3 (n = 4) | 6 (n = 4) | 9 (n = 4) | |||||
| Total | 5 (83) | 3 (75) | 0 | 2 (50) | 3 (75) | 3 (75) | 1 (25) | 12 (50) | 1 (17) | 2 (50) | 4 (100) | 1 (25) | 2 (50) | 2 (50) | 2 (50) | 13 (54) |
| Gastrointestinal disordersb | 1 (17) | 2 (50) | 0 | 0 | 2 (50) | 1 (25) | 0 | 5 (21) | 0 | 0 | 2 (50) | 0 | 1 (25) | 1 (25) | 1 (25) | 5 (21) |
| Nervous system disordersc | 0 | 3 (75) | 0 | 0 | 0 | 1 (25) | 0 | 4 (17) | 0 | 1 (25) | 2 (50) | 0 | 1 (25) | 1 (25) | 1 (25) | 6 (25) |
| Respiratory, thoracic, and mediastinal disordersd | 1 (17) | 1 (25) | 0 | 0 | 0 | 1 (25) | 0 | 2 (8) | 0 | 0 | 1 (25) | 0 | 0 | 0 | 1 (25) | 2 (8) |
| General disorders and administrative site conditionse | 3 (50) | 2 (50) | 0 | 0 | 0 | 0 | 0 | 2 (8) | 0 | 0 | 0 | 0 | 1 (25) | 0 | 0 | 1 (4) |
| Musculoskeletal and connective tissue disordersf | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 0 | 0 | 1 (25) | 1 (4) |
TEAE, treatment-emergent adverse event.
Gastrointestinal disorders included nausea in three subjects and abdominal discomfort, upper abdominal pain, diarrhea, and vomiting in one subject each in the AVI-6002 study and abdominal distension, abdominal pain, gastroesophageal reflux disease, anorectal discomfort, aphthous stomatitis, nausea, and vomiting in one subject each in the AVI-6003 study.
Nervous system disorders included headache in four subjects and somnolence in one subject in the AVI-6002 study and headache in three subjects, dizziness in two subjects, and cataplexy, dysgeusia, and tremor in one subject each in the AVI-6003 study.
Respiratory, thoracic, and mediastinal disorders included sinus congestion in three subjects in the AVI-6002 study and nasal congestion and rhinorrhea in one subject each in the AVI-6003 study.
General disorders and administrative site conditions included chest discomfort, fatigue, feeling cold, and infusion site erythema in one one subject each in the AVI-6002 study and fatigue in one subject in the AVI-6003 study.
Musculoskeletal and connective tissue disorders included arthralgia and muscle tightness in one subject each in the AVI-6003 study.
Similarly, AVI-6003 was safe and well tolerated at all of the doses studied. Thirteen (54.2%) AVI-6003-treated subjects experienced 27 TEAEs (Table 3), 6 of which were considered treatment related. Overall, nervous system disorders were most common, followed by gastrointestinal disorders. No TEAEs related to abnormal laboratory results were reported. TEAEs occurring in more than one subject included headache (two AVI-6003 subjects; zero placebo subjects) and dizziness (two AVI-6003 subjects; zero placebo subjects). No dose-dependent pattern was observed. All TEAEs were mild in severity, except for anxiety and cataplexy of moderate severity and exacerbation of schizophrenia of severe intensity, all of which were reported in one subject, who had not disclosed his preexisting diagnosis of schizophrenia at the time of screening. The exacerbation of schizophrenia was considered an SAE, was assessed as severe, and was unrelated to study drug but was considered related to the study procedure (i.e., confinement to the study site) and led the subject to withdraw consent on day 21. Five (20.8%) AVI-6003 subjects experienced six treatment-related TEAEs, with headache being the only TEAE occurring in more than one subject. No deaths or discontinuations due to study treatment were reported.
No clinically significant or dose-dependent effects of AVI-6002 or AVI-6003 were reported on any of the safety endpoints evaluated, including clinical laboratory assessments, vital signs, ECGs, physical examinations, pulse oximetry, and cardiac telemetry, nor were any gross abnormalities in fluid status, nephrotoxicity, changes in renal function, or changes in the biomarkers of renal dysfunction observed. Specifically, no subjects in either study developed a confirmed increase in serum creatinine level of ≥0.3 mg/dl from the baseline or had a confirmed percentage increase of ≥50% from the baseline in the serum creatinine level, where the baseline was defined as the mean of the screening and day −1 values, and no subjects in either study developed a consistent or durable >3-fold increase in UACR from the predose on day 1 and to an absolute value of >30 mg/g, which were the individual stopping rules based on renal function for each study. Mild (grade 1) increases in alanine aminotransferase (ALT) or aspartate aminotransferase (AST) to 1.1- to 2.5-fold of the upper limit of normal were observed in two subjects in the AVI-6002 study and two subjects in the AVI-6003 study, but no dose-dependent pattern was observed. Mild (grade 1) increases in amylase to 1.1- to 1.5-fold the upper limit of normal were observed in two subjects in the AVI-6002 study and eight subjects in the AVI-6003 study, but no dose-dependent pattern was observed. No grade 2 or greater increases in AST, ALT, or amylase were observed in either study.
Pharmacokinetics.
The four PMOplus components displayed similar plasma PK properties, including similar time-concentration curves (Fig. 3), and absolute values of Cmax (Fig. 4) and AUC (Fig. 5) over the range of dosing from 0.005 to 4.5 mg/kg. These parameters changed in a dose proportional manner. Other PK parameters (e.g., Tmax, half-life, clearance, and volume of distribution) were nearly identical, particularly at the higher dose levels (Table 4).
FIG 3.

Time-concentration curves of the components of AVI-6002 (AVI-7537 [A] and AVI-7539 [B]) and the components of AVI-6003 (AVI-7287 [C] and AVI-7288 [D]).
FIG 4.
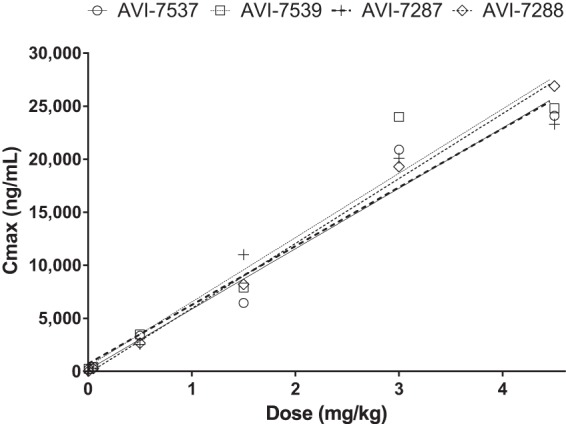
Mean Cmax by dose of components of AVI-6002 (AVI-7537 [○] and AVI-7539 [□]) and AVI-6003 (AVI-7287 [+] and AVI-7288 [◇]). Linear regression analysis yielded R2 values of 0.96 for AVI-7537, 0.94 for AVI-7539, 0.96 for AVI-7287, and 1.00 for AVI-7288. The Cmax (ng/ml) slopes per mg/kg dose are 5,701 ± 568 for AVI-7537, 6,069 ± 780 for AVI-7539, 5,545 ± 534 for AVI-7287, and 6,122 ± 182 for AVI-7288. An individual linear regression line is plotted for each agent.
FIG 5.
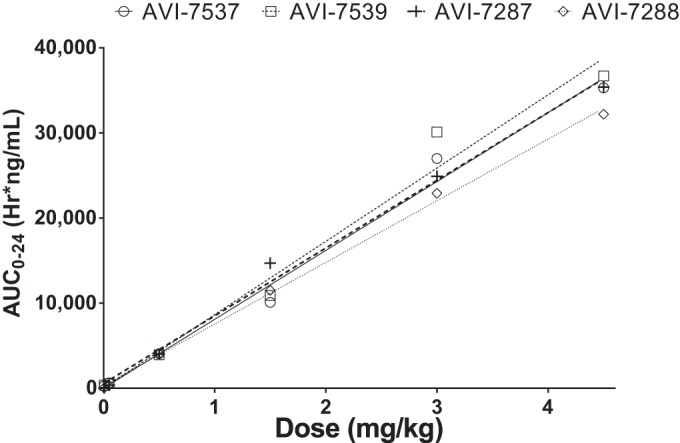
Mean AUC0–24 by dose of components of AVI-6002 (AVI-7537 [○] and AVI-7539 [□]) and AVI-6003 (AVI-7287 [+] and AVI-7288 [◇]). Linear regression analysis yielded R2 values of 0.99 for AVI-7537, 0.98 for AVI-7539, 0.99 for AVI-7287, and 1.00 for AVI-7288. The AUC0–24 (h·ng/ml) slopes per mg/kg dose are 8,095 ± 442 for AVI-7537, 8,612 ± 635 for AVI-7539, 7,961 ± 325 for AVI-7287, and 7,244 ± 154 for AVI-7288. An individual linear regression line is plotted for each agent.
TABLE 4.
PK parameters of components of AVI-6002 (AVI-7537 and AVI-7539) and AVI-6003 (AVI-7287 and AVI-7288)
| Component and dose (mg/kg) | Mean (SD) |
|||||
|---|---|---|---|---|---|---|
| Cmax (ng/ml) | AUC0–24 (h·ng/ml) | Tmax (h) | Plasma half-life (h) | CLP (ml/h/kg) | Vss (ml/kg) | |
| AVI-7537 | ||||||
| 0.005 | 185 (108) | 285 (173) | 0.5 (0.08) | 2.0 (0.30) | 31 (32.6) | 65 (68.2) |
| 0.05 | 425 (99) | 617 (237) | 0.5 (0.00) | 2.0 (0.57) | 89 (29.1) | 183 (23.8) |
| 0.5 | 3,360 (974) | 4,060 (462) | 0.5 (0.00) | 3.6 (0.25) | 122 (11.5) | 382 (139.0) |
| 1.5 | 6,460 (2,370) | 10,100 (2,140) | 0.5 (0.00) | 2.8 (0.81) | 152 (32.3) | 406 (124.0) |
| 3.0 | 20,900 (5,220) | 27,000 (6,520) | 0.5 (0.07) | 4.6 (1.17) | 114 (22.9) | 334 (42.6) |
| 4.5 | 24,100 (3,440) | 35,300 (3,700) | 0.5 (0.08) | 4.0 (0.71) | 126 (12.6) | 453 (70.8) |
| AVI-7539 | ||||||
| 0.005 | 242 (67) | 338 (72) | 0.5 (0.07) | 2.1 (0.09) | 15 (2.7) | 48 (40.3) |
| 0.05 | 399 (107) | 519 (150) | 0.5 (0.09) | 1.9 (0.63) | 103 (28.5) | 201 (13.8) |
| 0.5 | 3,480 (1,130) | 3,950 (658) | 0.5 (0.06) | 2.9 (0.80) | 127 (18.9) | 387 (125.0) |
| 1.5 | 7,910 (1,440) | 10,900 (1,950) | 0.5 (0.07) | 2.5 (0.92) | 140 (24.4) | 329 (91.2) |
| 3.0 | 24,000 (4,330) | 30,100 (6,250) | 0.5 (0.00) | 4.6 (1.21) | 101 (19.4) | 273 (38.6) |
| 4.5 | 24,800 (2,090) | 36,700 (2,420) | 0.6 (0.00) | 3.9 (1.01) | 121 (7.9) | 401 (35.1) |
| AVI-7287 | ||||||
| 0.005 | 34 (8) | 48 (11) | 0.6 (0.14) | 1.6 (0.92) | 94 (15.8) | 189 (99.8) |
| 0.05 | 313 (89) | 338 (65) | 0.5 (0.08) | 1.8 (0.26) | 151 (31.0) | 299 (65.3) |
| 0.5 | 2,550 (486) | 4,000 (729) | 0.5 (0.14) | 1.9 (0.46) | 129 (27.0) | 278 (33.9) |
| 1.5 | 11,000 (1,530) | 14,700 (3,470) | 0.5 (0.0) | 2.2 (0.33) | 105 (24.7) | 267 (50.4) |
| 3.0 | 20,100 (4,290) | 24,900 (3,200) | 0.5 (0.0) | 5.0 (2.04) | 120 (17.0) | 354 (88.9) |
| 4.5 | 23,300 (2,770) | 35,400 (3,720) | 0.4 (0.11) | 3.7 (1.46) | 123 (12.3) | 537 (169.0) |
| AVI-7288 | ||||||
| 0.005 | 36 (7) | 45 (7) | 0.4 (0.12) | 1.2 (0.48) | 104 (17.6) | 158 (21.0) |
| 0.05 | 272 (97) | 322 (105) | 0.5 (0.06) | 1.6 (0.19) | 171 (67.9) | 303 (142.0) |
| 0.5 | 2,660 (271) | 4,060 (638) | 0.5 (0.01) | 1.7 (0.46) | 126 (23.4) | 241 (30.5) |
| 1.5 | 8,210 (460) | 11,500 (1,680) | 0.5 (0.01) | 2.4 (0.53) | 133 (19.2) | 277 (38.4) |
| 3.0 | 19,300 (290) | 22,900 (1,170) | 0.5 (0.01) | 4.1 (1.95) | 130 (7.2) | 315 (131.0) |
| 4.5 | 26,900 (5,040) | 32,200 (3,480) | 0.5 (0.06) | 5.5 (2.51) | 136 (11.6) | 569 (298.0) |
For all four components, most of the drug was excreted in the urine within the first 24 h. The AVI-7537 CLR was not measurable for the two lower doses, but the CLR/CLP values averaged 1.7, 21.6, 26.8, and 44.0% for the 0.5-, 1.5-, 3-, and 4.5-mg/kg dose levels, respectively. Similarly, the AVI-7539 CLR was not measurable for the two lower doses, but the CLR/CLP values averaged 1.3, 20.0, 30.7, and 27.1% for the 0.5-, 1.5-, 3-, and 4.5-mg/kg dose levels, respectively. The AVI-7287 CLR was not measurable for the three lower doses, but the CLR/CLP values averaged 11.0, 38.7, and 37.7% for the 1.5-, 3-, and 4.5-mg/kg dose levels. The AVI-7288 CLR was not measurable for the two lower doses (0.005 and 0.05 mg/kg), but the CLR/CLP values averaged 2.2, 25.4, 51.5, and 49.5% for the 0.5-, 1.5-, 3-, and 4.5-mg/kg dose levels. A consistent trend of increasing renal clearance with increasing dose was observed in the CLR/CLP values with averages of 1.7% at 0.5 mg/kg, 19.5% at 1.5 mg/kg, 31.9% at 3.0 mg/kg, and 39.6% at 4.5 mg/kg, which is likely to be due to low-affinity interactions between the four different PMOplus agents and plasma proteins.
DISCUSSION
In demographically similar study subjects, the safety of the two combinations (AVI-6002 or AVI-6003) and the PK of the four components (AVI-7537, AVI-7539, AVI-7287, or AVI-7288) in identical single-ascending-dose studies were similar. In both studies, all subjects received one dose of active study drug or placebo as planned.
Both AVI-6002 and AVI-6003 were shown to be safe and well tolerated at doses of 0.005, 0.05, 0.5, 1.5, 3, and 4.5 mg/kg per component. A maximum tolerated dose was not observed in either study. More subjects in the AVI-6002 study group experienced TEAEs than in the AVI-6003 study group (50% versus 21%), but no dose dependence was observed. Of note, more subjects in the placebo group of the AVI-6002 study experienced AEs than in the placebo group of the AVI-6003 study (83% versus 17%). In the AVI-6002 study, headache and nausea were reported more frequently with AVI-6002 than with placebo. However, these results were reported across the cohorts who received AVI-6002 with no apparent relationship to dose.
The chemically similar components of AVI-6002 and AVI-6003 exhibited generally similar PK profiles. The mean Cmax and AUC values of the four components showed a dose-dependent increase suggestive of dose-proportional PK. The estimated plasma half-life of all four components was 2 to 5 h. Most of the drug that was excreted in the urine was excreted within the first 24 h. Renal clearance increased linearly with dose, and urinary excretion of intact drug accounted for no more than 44.0% of AVI-7537 total elimination, 30.7% of AVI-7539 total elimination, 38.7% of AVI-7287 total elimination, and 51.5% of AVI-7288 total elimination. The plasma protein binding of these components is likely to be weak, resulting in a greater filtered fraction in the kidney. The observed increase in renal excretion with dose may be reflected in the increase in Vss observed at higher doses.
As expected, the safety of the two combinations and the PK of the four components was similar, given their similar chemical properties. The molecular masses and nucleobase compositions of the four components differed, but these differences do not appear to affect the pharmacokinetic properties of the compounds.
Initially, AVI-6002 and AVI-6003 were developed as two drug combinations because the early mouse and guinea pig lethal challenge models suggested they were the best candidates for postexposure prophylaxis for Ebola and Marburg virus infections, respectively. However, subsequent studies in nonhuman primate lethal virus challenge models demonstrated that AVI-7537 for Ebola virus and AVI-7288 for Marburg virus were the active components of the parent combination compounds (10). Therefore, the single agents are now considered the lead clinical candidates. Additional studies in nonhuman primates and humans are under way to estimate the protective human dose, based on appropriate PK parameters such as AUC and CL.
Through these single-ascending-dose studies, four PMOplus compounds directed against Ebola and Marburg viruses demonstrate human safety profiles that are equivalent to placebo. One of the major advantages of the PMOplus platform technology is the ability to very rapidly respond to new or emerging infectious disease threats with safety demonstrated in the chemistry, while efficacy and specificity are based upon the sequence used for pathogen-specific mRNA translation inhibition.
ACKNOWLEDGMENTS
This study was conducted under contract with the Joint Product Management Office of BioDefense Therapeutics (BD-Tx). BD-Tx is a component of the Medical Countermeasure Systems Joint Project Management Office (JPM-MCS) within the U.S. Department of Defense's Joint Program Executive Office for Chemical and Biological Defense.
The views expressed here are those of the authors and do not necessarily represent the views or official position of the JPM-MCS or BD-Tx.
Editorial assistance was provided by Richard S. Perry and was supported by Sarepta Therapeutics, Cambridge, MA.
Footnotes
Published ahead of print 25 August 2014
REFERENCES
- 1.MacNeil A, Rollin PE. 2012. Ebola and Marburg hemorrhagic fevers: neglected tropical diseases? PLoS Negl. Trop. Dis. 6:e1546. 10.1371/journal.pntd.0001546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mehedi M, Groseth A, Feldmann H, Ebihara H. 2011. Clinical aspects of Marburg hemorrhagic fever. Future Virol. 6:1091–1106. 10.2217/fvl.11.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartman AL, Towner JS, Nichol ST. 2010. Ebola and Marburg hemorrhagic fever. Clin. Lab. Med. 30:161–177. 10.1016/j.cll.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Fauci A, Braunwald E, Kasper D, Hauser S, Longo D, Hameson J, Loscalzo J. 2008. Harrison's principles of internal medicine, 17th ed. McGraw-Hill Book Co, New York, NY. [Google Scholar]
- 5.Feldmann H, Geisbert TW. 2011. Ebola haemorrhagic fever. Lancet 377:849–862. 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.World Health Organization. 2014. Ebola virus disease: fact sheet 103, April 2014. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs103/en/. [Google Scholar]
- 7.World Health Organization. 2012. Marburg haemorrhagic fever: fact sheet, November 2012. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs_marburg/en/. [Google Scholar]
- 8.Nakayama E, Saijo M. 2013. Animal models for Ebola and Marburg virus infections. Front. Microbiol. 4:267. 10.3389/fmicb.2013.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Centers for Disease Control and Prevention. 2014. Emergency preparedness and response: bioterrorism agents/diseases. U.S. Centers for Disease Control and Prevention, Atlanta, GA: http://www.bt.cdc.gov/agent/agentlist-category.asp. [Google Scholar]
- 10.Iversen PL, Warren TK, Wells JB, Garza NL, Mourich DV, Welch LS, Panchal RG, Bavari S. 2012. Discovery and early development of AVI-7537 and AVI-7288 for the treatment of Ebola virus and Marburg virus infections. Viruses 4:2806–2830. 10.3390/v4112806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warren TK, Warfield KL, Wells J, Swenson DL, Donner KS, Van Tongeren SA, Garza NL, Dong L, Mourich DV, Crumley S, Nichols DK, Iversen PL, Bavari S. 2010. Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nat. Med. 16:991–994. 10.1038/nm.2202. [DOI] [PubMed] [Google Scholar]
- 12.U.S. Department of Health and Human Services/U.S. Food and Drug Administration Center for Biologics Evaluation and Research. 2007. Guidance for industry: toxicity grading scale for healthy adult and adolescent volunteers enrolled in preventive vaccine clinical trials. DHHS/FDA, Washington, DC. [Google Scholar]
- 13.Gibaldi M, Perrier D. 1982. Pharmacokinetics, 2nd ed. Marcel Dekker, New York, NY. [Google Scholar]