

Abstract
Two open-label, single-dose, parallel-group studies were conducted to characterize the pharmacokinetics of the novel antibacterial tedizolid and the safety of tedizolid phosphate, its prodrug, in renally or hepatically impaired subjects. Tedizolid pharmacokinetics in subjects with severe renal impairment without dialysis support was compared with that of matched control subjects with normal renal function. Effects of hemodialysis on tedizolid pharmacokinetics were determined in a separate cohort of subjects undergoing long-term hemodialysis. Effects of hepatic impairment on tedizolid pharmacokinetics were determined in subjects with moderate or severe hepatic impairment and compared with those of matched control subjects with normal hepatic function. Each participant received a single oral (hepatic impairment) or intravenous (renal impairment) dose of tedizolid phosphate at 200 mg; hemodialysis subjects received two doses (separated by 7 days), before and after dialysis, in a crossover fashion. The pharmacokinetics of tedizolid was similar in subjects with severe renal impairment and controls (∼8% lower area under the concentration-time curve [AUC], with a nearly identical peak concentration) and in subjects undergoing hemodialysis before and after tedizolid phosphate administration (∼9% lower AUC, with a 15% higher peak concentration); <10% of the dose was removed during 4 h of hemodialysis. Tedizolid pharmacokinetics was only minimally altered in subjects with moderate or severe hepatic impairment; the AUC was increased approximately 22% and 34%, respectively, compared with that of subjects in the control group. Tedizolid phosphate was generally well tolerated in all participants. These results suggest that tedizolid phosphate dose adjustments are not necessary in patients with any degree of renal or hepatic impairment. (This study has been registered at ClinicalTrials.gov under registration numbers NCT01452828 [renal study] and NCT01431833 [hepatic study].)
INTRODUCTION
Management of infections resulting from Gram-positive pathogens, including strains resistant to older antibacterials, continues to pose challenges (1–3). Tedizolid phosphate is the prodrug of the active moiety tedizolid, a novel oxazolidinone antibacterial under investigation for use in the treatment of Gram-positive infections, including those caused by multidrug-resistant strains (4, 5). Tedizolid recently demonstrated noninferior efficacy to linezolid for the treatment of acute bacterial skin and skin structure infections (6–8). Tedizolid phosphate is administered once daily (200 mg) either orally or by intravenous infusion (9).
Many antibacterials necessitate adjustments to dose size, dose frequency, or both when patients with impaired renal or hepatic function are treated (10, 11) because chronic kidney disease and serious liver disease cause complex changes to the metabolism and elimination of many antibacterials (10, 11). These disorders are also associated with increased risk of serious infections, including those due to drug-resistant Gram-positive pathogens (10, 12, 13), and clinically significant adverse effects caused by antibacterial treatment itself (10, 14, 15). Because of aging populations, the prevalences of chronic kidney disease (16) and chronic liver disease (17) are increasing. Therefore, to achieve safe and successful treatment outcomes, it is necessary to understand the potential need for antibacterial dose adjustments in these special patient populations (10, 18, 19).
To elucidate this important point as it relates to the clinical use of tedizolid, two open-label, single-dose, parallel-group studies were undertaken to assess the pharmacokinetic properties of tedizolid and the safety of the prodrug tedizolid phosphate in subjects with moderately to severely impaired hepatic function or severely impaired renal function, including those requiring hemodialysis. These clinical trials are registered at www.ClinicalTrials.gov as NCT01452828 (renal study) and NCT01431833 (hepatic study).
MATERIALS AND METHODS
Study subjects.
Individuals with impaired renal or hepatic function and matched controls were enrolled in two open-label phase 1 trials to assess tedizolid pharmacokinetics. Subjects 18 to 75 years of age (renal study) and 18 to 70 years (hepatic study) with a body mass index (BMI) between 18.0 and 40.0 kg/m2 were eligible. Screening included assessing organ dysfunction by estimated glomerular filtration rate (renal study) and Child-Pugh classification (hepatic study). The presence of stable disease and the absence of confounding factors were determined by assessing patient medical history, physical examination results, laboratory findings, and electrocardiography results.
Subjects were excluded if they had received monoamine oxidase inhibitors or serotonergic agents within 14 days or sympathomimetic agents within 48 h of the first tedizolid phosphate dose. Lifestyle restrictions included avoidance of high-tyramine diets, alcohol, and strenuous exercise from the 48 h preceding tedizolid phosphate administration to the follow-up visit.
In the renal study, subjects with severely impaired renal function and not undergoing hemodialysis had an estimated glomerular filtration rate of <30 ml/min/1.73 m2 using the four-variable modification of the diet in renal disease formula (20), had stable hemoglobin and hematocrit values for the preceding 3 months, and had stable medication doses for the prior month. Subjects with end-stage renal disease necessitating long-term hemodialysis had a 3-month history of stable urea clearance during dialysis (≥1.2 times the total body water).
Additional exclusion criteria for the hepatic study included an alanine transaminase level ≥5 times the upper limit of normal for moderate disease and ≥8 times the upper limit of normal for severe disease, a hemoglobin concentration of <10 mg/dl for moderate disease and <9 mg/dl for severe disease, and a total bilirubin level of >5 mg/dl for moderate disease, with no limit for severe disease. Acute hepatic function deterioration within 8 weeks of screening, creatinine clearance of <50 ml/min, and electrocardiography abnormalities (including a corrected QT interval [a measure of the time between the start of the Q wave and the end of the T wave in the heart's electrical cycle] of >500 ms) were additional exclusion criteria.
Overall study design.
Screening visits were conducted within the 21 days (renal study) or 28 days (hepatic study) before the first tedizolid phosphate dose. Subjects entered the study center 1 day before the first dose. They remained at the center for routine blood and urine sample collection for 72 h (renal study) or 5 days (hepatic study) after the last dose. Additional details of sample collection are provided in the supplemental material. A final safety follow-up visit took place 7 days (±1 day) after the last dose.
In the renal study, the pharmacokinetic properties of a single 200-mg intravenous dose of tedizolid phosphate (in 250 ml saline infused over 1 h) were compared among healthy matched control subjects, subjects with severe renal impairment, and subjects with end-stage renal disease requiring long-term hemodialysis. In the hepatic study, controls and subjects with hepatic impairment were matched to compare the pharmacokinetic properties of a single oral 200-mg tedizolid phosphate dose. Serial plasma samples were collected from predose through 72 h postdose in the renal study and from predose through 96 h postdose in the hepatic study. In both studies, subjects were matched by sex, age (±10%), and BMI (±15%). Additional design elements for the individual studies are described in the supplemental material.
Ethical considerations.
Studies were conducted in accordance with current U.S. Food and Drug Administration regulations, International Conference on Harmonization Good Clinical Practice guidelines, and the Basic Principles of the Declaration of Helsinki.
Statistical analysis.
Standard noncompartmental analysis was conducted using the WinNonlin Professional edition (version 5.2; Pharsight Corporation, St. Louis, MO, USA). The following pharmacokinetic parameters were calculated for tedizolid and tedizolid phosphate when applicable: the peak plasma concentration (Cmax; μg/ml), the time to the peak plasma concentration (Tmax; hours), the area under the concentration-time curve from time zero to the time of the last collected sample (AUC0–t; μg · h/ml), the AUC from time zero to infinity (AUC0–∞; μg · h/ml), and the apparent terminal half-life (h). The geometric mean ratios for tedizolid Cmax, AUC0–t, and AUC0–∞ and corresponding 90% confidence intervals (CIs) were determined for each study group and their corresponding controls using analysis of variance models. AUC ratios and associated 90% CIs within a range of 0.5 to 2.0—boundaries that represent no clinically meaningful change for tedizolid plasma exposure—were prespecified, and studies were powered accordingly. For each comparison, the log-transformed pharmacokinetic parameter was the response variable, the group was the fixed factor, and the subject was the random effect. Plasma concentration-time profiles were generated for individuals receiving tedizolid phosphate, and median or mean plasma concentration-time profiles (linear and semilogarithmic scales) were generated for each treatment group.
RESULTS
Whether administered intravenously (to subjects with renal insufficiency) or orally (to subjects with hepatic insufficiency), tedizolid phosphate was rapidly and extensively converted to tedizolid. Therefore, presentation of study results focuses on tedizolid plasma kinetics.
Renal impairment study. (i) Pharmacokinetics.
Twenty-four subjects were enrolled in the renal impairment study, and 23 completed the study. One subject in the dialysis group completed only one dose of study drug and was later withdrawn for reasons other than an adverse event before completing the crossover dose administration. Groups were balanced in terms of age, sex, and BMI (see Table S1 in the supplemental material).
Tedizolid pharmacokinetics remained essentially unchanged in subjects with severe renal impairment, compared with healthy controls (Table 1; Fig. 1). Comparing the geometric mean pharmacokinetic exposure parameters between the severe renal impairment and control groups revealed no meaningful difference in Cmax or AUC (Cmax geometric mean ratio, 0.994; 90% CIs, 0.777 to 1.273; AUC0–∞, 0.925; 90% CIs, 0.698 to 1.227). When tedizolid pharmacokinetics was compared between infusions administered before and after hemodialysis, both Cmax and AUC were lower than observed in the control or severe renal impairment group (Table 1). However, there were no meaningful differences in the geometric mean values for Cmax or AUC when postdialysis and predialysis infusion data were compared (Cmax geometric mean ratio, 1.148; 90% CIs, 1.053 to 1.252; AUC0–∞ geometric mean ratio, 0.913; 90% CIs, 0.827 to 1.007). When samples were collected during high-flux hemodialysis from subjects who received infusion before dialysis, <10% of the administered tedizolid dose was removed by 4 h of hemodialysis (data not shown). Mean levels of tedizolid protein binding were similar (73.2% to 76.8%) across all groups.
TABLE 1.
Mean tedizolid pharmacokinetics in the renal-impairment studya
| Study group | Cmax (μg/ml) | Tmax (h) | AUC0–t (μg · h/ml) | AUC0–∞ (μg · h/ml) | t1/2 (h) |
|---|---|---|---|---|---|
| Control (n = 8) | 3.11 (0.75) | 1.00 (1.00–2.50) | 32.02 (9.32) | 32.43 (9.53) | 12.25 (2.04) |
| Severe renal impairment (n = 8) | 3.12 (0.85) | 1.26 (1.00–2.00) | 29.69 (8.93) | 29.99 (8.97) | 12.85 (2.28) |
| Predialysis infusion (n = 7) | 2.53 (0.95) | 1.00 (0.50–1.50) | 22.97 (8.02) | 23.15 (8.10) | 11.41 (1.78) |
| Postdialysis infusion (n = 8) | 2.86 (1.01) | 1.50 (1.00–1.50) | 20.81 (4.65) | 21.01 (4.71) | 11.73 (2.33) |
AUC0–t, integrated area under the curve based on samples from time zero to the time of the last collected sample; AUC0–∞, area under the curve based on the terminal rate constant; Cmax, maximum concentration observed with a 200-mg dose; t1/2, tedizolid half-life; Tmax, time to reach the maximum concentration. Pharmacokinetic parameters are presented as means (standard deviations), except for Tmax values, which are presented as medians (ranges).
FIG 1.
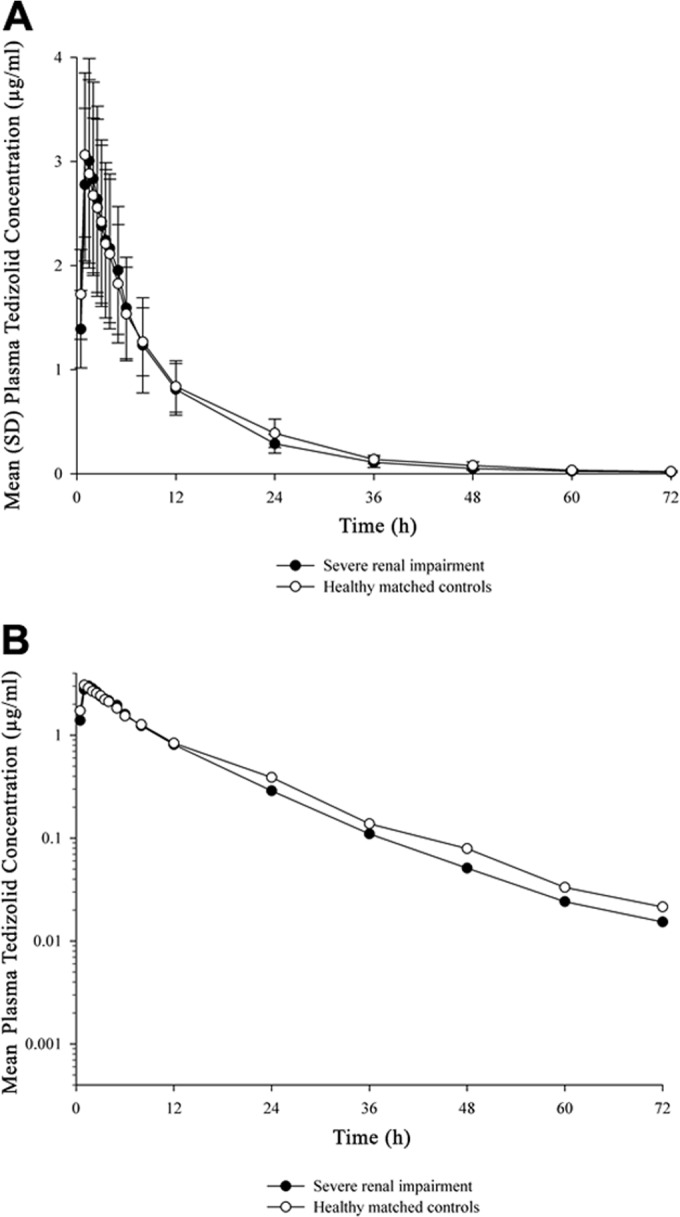
Plasma tedizolid concentrations over time in subjects with severe renal impairment and matched controls, shown on a linear scale (A) and on a semilogarithmic scale (B).
(ii) Tolerability and safety.
Tedizolid phosphate was generally well tolerated in subjects with severe renal impairment. Less than half the treated subjects experienced at least one treatment-emergent adverse event. This included three subjects in the control group, five in the nondialysis group, and three in the dialysis group. Headache was the only adverse event experienced by more than one subject per group. The severity of most treatment-emergent adverse events was mild or moderate; two severe treatment-emergent adverse events (nausea and vomiting) were reported for one subject with severe renal impairment. No serious adverse events were reported.
Clinically significant abnormal electrocardiography results were not observed in any participant. Five subjects in the end-stage renal disease group and four in the severe renal impairment group had abnormal electrocardiography results that were not clinically significant. None had a Bazett-corrected QT interval (QTcB) increase of ≥30 ms from predose. One subject in the end-stage renal disease group (underwent dialysis after infusion) had a postdose absolute QTcB interval of >500 ms, which was unchanged from baseline.
There were no substantial abnormalities in hematologic parameters beyond the diminished red blood cell levels associated with renal impairment and no significant coagulation panel changes. The majority of subjects in the renal-impairment groups had abnormal results for multiple chemistry laboratory tests at baseline and during follow-up, but these were deemed typical of this population. Creatinine and electrolyte level imbalances were particularly common.
Hepatic-impairment study. (i) Pharmacokinetics.
Thirty-two subjects were enrolled in the hepatic impairment group and completed the study. Groups were balanced in terms of age, sex, and BMI (see Table S2 in the supplemental material).
Overall, tedizolid pharmacokinetics (Table 2) and plasma concentration-time profiles (Fig. 2) after administration of 200-mg oral tedizolid phosphate were minimally different between subjects with moderate/severe hepatic impairment and matched controls. The largest pharmacokinetic differences between subjects with hepatic impairment and controls were seen in AUC0–∞, which were approximately 34% higher among those with severe impairment than in controls (geometric mean ratio, 1.341; 90% CIs, 0.927 to 1.939) and 22% higher among those with moderate impairment than in controls (geometric mean ratio, 1.216; 90% CIs, 0.862 to 1.716). Cmax values were relatively unchanged in patients with moderate or severe hepatic impairment compared with those in the control groups (moderate impairment geometric mean ratio, 1.093; 90% CIs, 0.849 to 1.408; severe impairment geometric mean ratio, 0.992; 90% CIs, 0.703 to 1.400).
TABLE 2.
Mean tedizolid pharmacokinetic parameters of the hepatic-impairment groupa
| Study group | Cmax (μg/ml) | Tmax (h) | AUC0–t (μg · h/ml) | AUC0–∞ (μg · h/ml) | t1/2 (h) |
|---|---|---|---|---|---|
| Moderate impairment (n = 8) | 2.08 (0.74) | 1.75 (0.50–3.00) | 29.89 (16.76) | 30.47 (17.50) | 14.94 (3.49) |
| Matched controls (n = 8) | 1.85 (0.49) | 2.00 (1.00–4.00) | 22.80 (5.63) | 23.00 (5.70) | 13.42 (3.93) |
| Severe impairment (n = 8) | 2.20 (1.07) | 2.00 (0.50–3.00) | 34.80 (20.72) | 35.23 (21.13) | 14.19 (2.92) |
| Matched controls (n = 8) | 2.12 (0.80) | 3.00 (1.00–8.00) | 24.37 (8.03) | 24.56 (8.05) | 13.68 (3.71) |
AUC0–t, integrated area under the curve based on samples from time zero to the time of the last collected sample; AUC0–∞, area under the curve based on the terminal rate constant; Cmax, maximum concentration observed with a 200-mg dose; t1/2, tedizolid half-life; Tmax, time to reach the maximum concentration. Pharmacokinetic parameters are presented as means (standard deviations), except for Tmax values, which are presented as medians (ranges).
FIG 2.
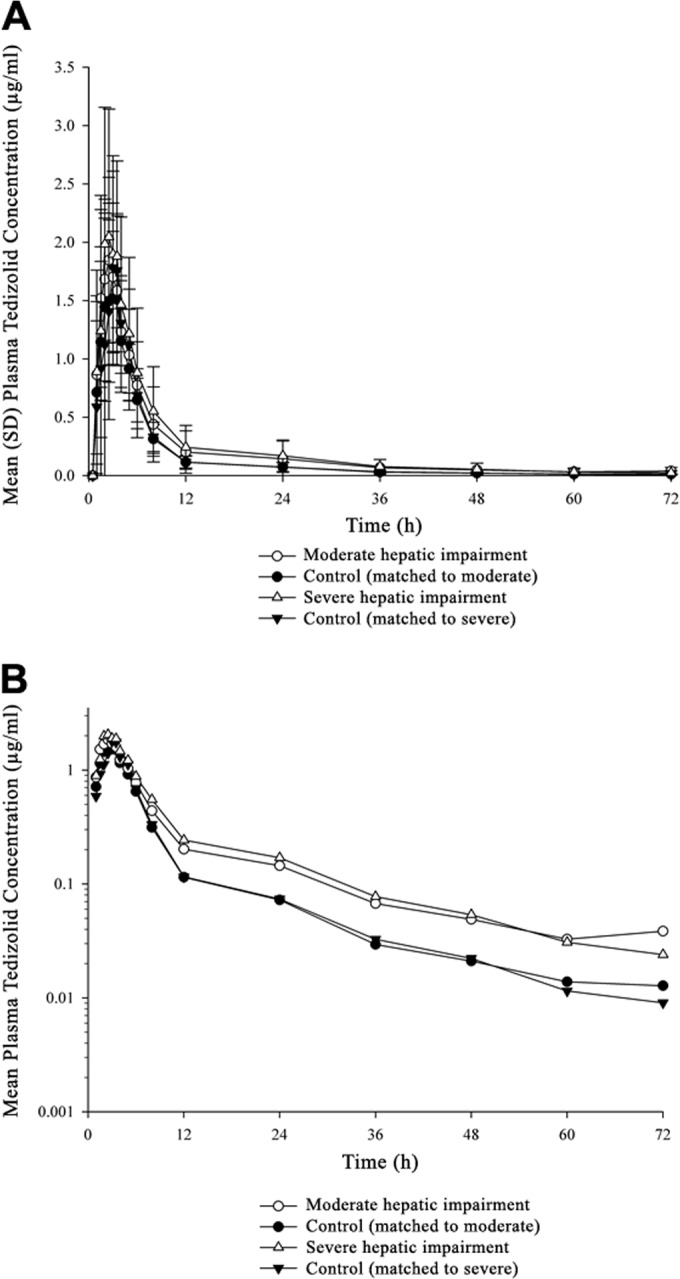
Plasma tedizolid concentrations over time in subjects with impaired hepatic function and matched controls, shown on a linear scale (A) and on a semilogarithmic scale (B).
(ii) Tolerability and safety.
Eight subjects with hepatic impairment experienced a total of five treatment-emergent adverse events related to tedizolid phosphate: diarrhea (n = 2), flatulence, transient flushing, and fine downy hair growth on the scalp. No serious or severe adverse events or deaths related to the drug were reported during the study, and most events were mild. There were no serious electrocardiography changes. After tedizolid phosphate administration, no subject experienced a QTcB increase of >30 ms, compared with predose values, or an absolute QTcB interval of >500 ms.
Four subjects in the severe impairment group, three in the moderate impairment group, and none in the control group had substantially abnormal postbaseline values for hematologic parameters (seven had abnormal platelet values, and one had an abnormal absolute neutrophil count). All values were abnormal at baseline and did not worsen with tedizolid phosphate administration. The majority of subjects with moderate and severe impairment had baseline abnormalities for multiple chemistry laboratory test results, but no parameters worsened after tedizolid phosphate administration. The hematologic and chemistry laboratory abnormalities in the subjects with hepatic impairment were considered typical of such individuals. No subject had a clinically significant abnormal coagulation panel or urinalysis result.
DISCUSSION
Tedizolid elimination is primarily nonrenal; only ∼18% of the administered dose is eliminated in urine (∼1% as unchanged tedizolid, the remainder as metabolites) (21). However, because renal impairment can also affect nonrenal drug clearance (22), an empirical study was necessary to assess whether tedizolid dose adjustments are needed in patients with renal impairment. For instance, uremia can alter several aspects of nonrenal drug clearance (including membrane transport functions, but not first-pass effects) (19, 23, 24). For this reason, tedizolid phosphate was administered intravenously in the current study to permit better assessment of tedizolid metabolism and excretion. Under these sensitive assessment conditions, tedizolid pharmacokinetic parameters were comparable between controls and subjects with severe renal impairment.
By comparison, approximately 80% of the administered dose of linezolid is eliminated renally (including 30% as linezolid, 40% as the metabolite PNU-142586, and 10% as the metabolite PNU-142300) (25). Linezolid-associated thrombocytopenia rates are higher in patients with severe renal impairment than in subjects with normal renal function (26–28) and might be related to drug or metabolite accumulation, because renal insufficiency is also associated with significant increases in linezolid plasma metabolite levels (29–31). Some authors have suggested therapeutic trough monitoring during linezolid use to improve efficacy and safety outcomes in patients with renal insufficiency (32). However, dose adjustments and drug monitoring further complicate the treatment of critically ill patients, and antibacterial agents that can be safely and conveniently administered without dose adjustments are therefore preferable in this population.
Because of the fast clearance of small-molecular-weight compounds during dialysis, additional dosing considerations come into play when hemodialysis support is necessary (11, 18). Tedizolid exhibits high protein binding (∼80%) compared with the low (∼30%) protein binding of linezolid (33). Because dialysis clearance is associated with the free-drug fraction (18, 19), it is no surprise that tedizolid clearance during hemodialysis (i.e., ∼10% of the administered dose) is less than the ∼30% clearance for linezolid during dialysis (33). While linezolid should be administered only after dialysis (33), tedizolid phosphate may allow for more-flexible timing of dose administration in hemodialysis patients.
In the current study, mean AUC values in end-stage renal disease subjects were ∼25% lower than in subjects with severe renal impairment not undergoing dialysis or their matched controls. However, these differences (which were well within the prespecified no-effect boundaries) were not in the direction of change generally associated with impaired function of a clearance organ (19, 34) and therefore likely reflect minor variability between small groups of subjects rather than a physiological effect. Other available data suggest that even if the observed lower tedizolid AUC in subjects requiring hemodialysis had represented a true pharmacokinetic difference, a decrease this small in magnitude would have minimal impact on efficacy. Based on a target attainment study reported in the companion paper to this article, pharmacokinetic/pharmacodynamic changes that are analogous to an AUC decrease of one-third would still result in more than 95% of patients achieving the tedizolid pharmacokinetic/pharmacodynamic target for clinical efficacy, compared with 98% at the reference AUC; decreases in AUC of about 50% were required before probability fell below 90% (35).
Hepatic excretion via bile accounts for the majority of tedizolid elimination; ∼80% of the administered dose is eliminated in feces, primarily as a sulfate conjugate (with ∼2% as unchanged tedizolid) (21). Unlike with renal filtration, which correlates reasonably well with creatinine clearance, there is no widely accepted marker for hepatic function to predict the pharmacokinetics or pharmacodynamics of a given drug, and pharmacokinetic studies must be conducted for compounds with significant hepatic elimination (36).
The present study was conducted using oral tedizolid phosphate, because first-pass metabolism can be meaningfully decreased in patients with impaired liver function before significant changes in systemic clearance are evident (37). Given the potential impact of insufficient liver function on tedizolid metabolism, the study was originally designed to evaluate patients with moderate and mild hepatic impairment. When it was found that tedizolid AUC increases were small in moderately impaired subjects relative to those in matched controls, it was assumed that any alterations as a result of mild impairment would also be minimal, and the protocol was amended to study subjects with severe hepatic impairment as the best way to characterize the effects of hepatic insufficiency on tedizolid exposure.
In the hepatic impairment study, the greatest increases in tedizolid AUCs were observed among subjects with severe impairment. However, the overall average AUC increase was only 34%, with CIs remaining well within the preestablished no-effect boundaries. Despite this small increase, tedizolid exposure was well tolerated. This is consistent with results from other single-dose studies (with about 50 subjects, using doses up to 1,200 mg, i.e., 6-fold higher) (9, 38) and from a phase 2 study (with 188 treated patients) that showed no safety differences between the 200-mg dose and a 300- or 400-mg tedizolid dose (7). Therefore, these small relative increases in exposure are unlikely to be clinically relevant. Pharmacokinetic variability was greater in subjects with moderate or severe hepatic dysfunction than in their respective control groups, possibly reflecting the heterogeneity of these populations. No appreciable changes in Cmax were observed with moderate or severe hepatic impairment.
For patients treated with linezolid, chronic liver disease and impaired liver function are risk factors for thrombocytopenia (26) and for isolated cases of delayed but rapid-onset lactic acidosis (39, 40). Linezolid pharmacokinetic changes have not been formally evaluated in subjects with severe hepatic impairment, but an increase in linezolid AUC of approximately1.3-fold was observed in subjects with mild to moderate hepatic impairment (33). This effect size is similar to the increase seen with tedizolid in subjects with even greater (i.e., severe) hepatic impairment, suggesting that larger increases in linezolid AUC might be expected in subjects with severe hepatic impairment.
Tedizolid is one of the only approved antibacterials active against S. aureus (including methicillin-resistant S. aureus) that has been studied in subjects with severe hepatic impairment. Furthermore, tedizolid is unique among antibacterials in that it lacks significant renal elimination. The current studies suggest that the pharmacokinetics of tedizolid is not meaningfully altered by impaired renal or hepatic function and that tedizolid was generally well tolerated in patients with such conditions. Therefore, tedizolid dose adjustments or adjustment of dose timing for hemodialysis patients is not necessary for patients with severe renal or hepatic impairment. This might be a practical advantage of tedizolid over other antibacterials that necessitate therapeutic monitoring or dose adjustments or have not been studied in patients with renal or hepatic impairment.
Supplementary Material
ACKNOWLEDGMENTS
These studies were funded by Cubist. S. Flanagan, E. Fang, and P. Prokocimer are employees of Cubist. S. L. Minassian is a consultant to Cubist and was compensated for supporting this research. D. Morris (Covance), R. Ponnuraj (Covance), T. C. Marbury (Orlando Clinical Research Center), and H. W. Alcorn (DaVita Clinical Research) conducted this research in the course of their employment, and their respective employers were compensated by Cubist for conducting this study. Medical writing support was provided by Bill Jacobs (Strategic HealthCOM, Somerville, NJ), funded by Cubist, and by Dominik Wolf, an employee of Cubist.
In collaboration with the authors, employees of the study sponsor were involved in study design, data analysis, interpretation of the results, and review/writing of the manuscript. The studies were conducted and data were collected at Covance (Madison, WI, USA), Orlando Clinical Research Center (Orlando, FL, USA), and DaVita Clinical Research (Minneapolis, MN, USA). All authors had full access to the data. The authors had final responsibility for the decision to submit for publication.
Footnotes
Published ahead of print 18 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03431-14.
REFERENCES
- 1.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48:1–12. 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Gould IM, David MZ, Esposito S, Garau J, Lina G, Mazzei T, Peters G. 2012. New insights into methicillin-resistant Staphylococcus aureus (MRSA) pathogenesis, treatment and resistance. Int. J. Antimicrob. Agents 39:96–104. 10.1016/j.ijantimicag.2011.09.028. [DOI] [PubMed] [Google Scholar]
- 3.Moran GJ, Abrahamian FM, Lovecchio F, Talan DA. 2013. Acute bacterial skin infections: developments since the 2005 Infectious Diseases Society of America (IDSA) guidelines. J. Emerg. Med. 44:e397–e412. 10.1016/j.jemermed.2012.11.050. [DOI] [PubMed] [Google Scholar]
- 4.Kanafani ZA, Corey GR. 2012. Tedizolid (TR-701): a new oxazolidinone with enhanced potency. Expert Opin. Invest. Drugs 21:515–522. 10.1517/13543784.2012.660250. [DOI] [PubMed] [Google Scholar]
- 5.Urbina O, Ferrández O, Espona M, Salas E, Ferrández I, Grau S. 2013. Potential role of tedizolid phosphate in the treatment of acute bacterial skin infections. Drug Des. Dev. Ther. 7:243–265. 10.2147/DDDT.S30728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prokocimer P, Bien P, Surber J, Mehra P, DeAnda C, Bulitta JB, Corey GR. 2011. Phase 2, randomized, double-blind, dose-ranging study evaluating the safety, tolerability, population pharmacokinetics, and efficacy of oral torezolid phosphate in patients with complicated skin and skin structure infections. Antimicrob. Agents Chemother. 55:583–592. 10.1128/AAC.00076-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prokocimer P, De Anda C, Fang E, Mehra P, Das A. 2013. Tedizolid phosphate vs linezolid for treatment of acute bacterial skin and skin structure infections: the ESTABLISH-1 randomized trial. JAMA 309:559–569. 10.1001/jama.2013.241. [DOI] [PubMed] [Google Scholar]
- 8.Moran GJ, Fang E, Corey GR, Das AF, De Anda C, Prokocimer P. 2014. Once-daily tedizolid for 6 days versus twice-daily linezolid for 10 days in acute bacterial skin and skin structure infections: results from a randomised controlled trial using an intravenous-to-oral switch strategy (ESTABLISH-2 study). Lancet Infect. Dis. 14:696–705. 10.1016/S1473-3099(14)70737-6. [DOI] [PubMed] [Google Scholar]
- 9.Flanagan SD, Bien PA, Muñoz KA, Minassian SL, Prokocimer PG. 2014. Pharmacokinetics of tedizolid following oral administration: single and multiple dose, effect of food, and comparison of two solid forms of the prodrug. Pharmacotherapy 34:240–250. 10.1002/phar.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergman SJ, Speil C, Short M, Koirala J. 2007. Pharmacokinetic and pharmacodynamic aspects of antibiotic use in high-risk populations. Infect. Dis. Clin. North Am. 21:821–846. 10.1016/j.idc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Heintz BH, Matzke GR, Dager WE. 2009. Antimicrobial dosing concepts and recommendations for critically ill adult subjects receiving continuous renal replacement therapy or intermittent hemodialysis. Pharmacotherapy 29:562–577. 10.1592/phco.29.5.562. [DOI] [PubMed] [Google Scholar]
- 12.Naqvi SB, Collins AJ. 2006. Infectious complications in chronic kidney disease. Adv. Chronic Kidney Dis. 13:199–204. 10.1053/j.ackd.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Canedo J, Ricciardi K, DaSilva G, Rosen L, Weiss EG, Wexner SD. 2013. Are postoperative complications more common following colon and rectal surgery in patients with chronic kidney disease? Colorectal Dis. 15:85–90. 10.1111/j.1463-1318.2012.03099.x. [DOI] [PubMed] [Google Scholar]
- 14.Rybak MJ, Lomaestro BM, Rotschafer JC, Moellering RC, Jr, Craig WA, Billeter M, Dalovisio JR, Levine DP. 2009. Therapeutic monitoring of vancomycin in adults: a consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am. J. Health Syst. Pharm. 66:82–98. 10.2146/ajhp080434. [DOI] [PubMed] [Google Scholar]
- 15.Ikuta S, Tanimura K, Yasui C, Aihara T, Yoshie H, Iida H, Beppu N, Kurimoto A, Yanagi H, Mitsunobu M, Yamanaka N. 2011. Chronic liver disease increases the risk of linezolid-related thrombocytopenia in methicillin-resistant Staphylococcus aureus-infected patients after digestive surgery. J. Infect. Chemother. 17:388–391. 10.1007/s10156-010-0188-8. [DOI] [PubMed] [Google Scholar]
- 16.Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, Van Lente F, Levey AS. 2007. Prevalence of chronic kidney disease in the United States. JAMA 298:2038–2047. 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- 17.Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, Srishord M. 2011. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol. 9:524–530. 10.1016/j.cgh.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 18.Matzke GR, Frye RF. 2008. Drug therapy individualization for subjects with renal insufficiency, p 833–844 In DiPiro J, Talbert RL, Yee G, Matzke G, Wells B, Posey LM. (ed), Pharmacotherapy: a pathophysiologic approach, 7th ed. McGraw-Hill Medical, New York, NY. [Google Scholar]
- 19.Nolin TD. 2008. Altered nonrenal drug clearance in ESRD. Curr. Opin. Nephrol. Hypertens. 17:555–559. 10.1097/MNH.0b013e3283136732. [DOI] [PubMed] [Google Scholar]
- 20.Levey AS, Coresh J, Greene T, Stevens LA, Zhang YL, Hendriksen S, Kusek JW, Van Lente F. 2006. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann. Intern. Med. 145:247–254. 10.7326/0003-4819-145-4-200608150-00004. [DOI] [PubMed] [Google Scholar]
- 21.Dreskin H, Boyea T, Barker J, Fang E, Prokocimer P. 2011. An evaluation of the absorption, metabolism, and excretion of orally administered [14C]-TR-701 FA in healthy subjects, poster A2-033. 51st Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC. [Google Scholar]
- 22.US Food and Drug Administration, Center for Drug Evaluation and Research. March 2010. Guidance for industry. Pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing and labeling. Draft guidance, revision 1 Center for Drug Evaluation and Research, US Food and Drug Administration, Silver Spring, MD: http://www.fda.gov/downloads/Drugs/./Guidances/UCM204959.pdf Accessed 15 April 2014. [Google Scholar]
- 23.Vilay AM, Churchwell MD, Mueller BA. 2008. Clinical review: drug metabolism and nonrenal clearance in acute kidney injury. Crit. Care 12:235. 10.1186/cc7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun H, Frassetto LA, Huang Y, Benet LZ. 2010. Hepatic clearance, but not gut availability, of erythromycin is altered in patients with end-stage renal disease. Clin. Pharmacol. Ther. 87:465–472. 10.1038/clpt.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez F, Couthtrie M, Tukey RH. 2010. Drug metabolism, p 123–143 In Brunton L, Chambers B, Knollman B. (ed), Goodman & Gilman's the pharmacologic basis of therapeutics, 12th ed. McGraw-Hill Education, New York, NY. [Google Scholar]
- 26.Takahashi Y, Takesue Y, Nakajima K, Ichiki K, Tsuchida T, Tatsumi S, Ishihara M, Ikeuchi H, Uchino M. 2011. Risk factors associated with the development of thrombocytopenia in patients who received linezolid therapy. J. Infect. Chemother. 17:382–387. 10.1007/s10156-010-0182-1. [DOI] [PubMed] [Google Scholar]
- 27.Sasaki T, Takane H, Ogawa K, Isagawa S, Hirota T, Higuchi S, Horii T, Otsubo K, Ieiri I. 2011. Population pharmacokinetic and pharmacodynamic analysis of linezolid and a hematologic side effect, thrombocytopenia, in Japanese patients. Antimicrob. Agents Chemother. 55:1867–1873. 10.1128/AAC.01185-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsumoto K, Takeshita A, Ikawa K, Shigemi A, Yaji K, Shimodozono Y, Morikawa N, Takeda Y, Yamada K. 2010. Higher linezolid exposure and higher frequency of thrombocytopenia in patients with renal dysfunction. Int. J. Antimicrob. Agents 36:179–181. 10.1016/j.ijantimicag.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 29.Slatter JG, Stalker DJ, Feenstra KL, Welshman IR, Bruss JB, Sams JP, Johnson MG, Sanders PE, Hauer MJ, Fagerness PE, Stryd RP, Peng GW, Shobe EM. 2001. Pharmacokinetics, metabolism, and excretion of linezolid following an oral dose of [(14)C]linezolid to healthy human subjects. Drug Metab. Dispos. 29:1136–1145. [PubMed] [Google Scholar]
- 30.Brier ME, Stalker DJ, Aronoff GR, Batts DH, Ryan KK, O'Grady M, Hopkins NK, Jungbluth GL. 2003. Pharmacokinetics of linezolid in subjects with renal dysfunction. Antimicrob. Agents Chemother. 47:2775–2780. 10.1128/AAC.47.9.2775-2780.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfizer. 2013. Zyvox. US physician prescribing information. Pfizer, New York, NY. [Google Scholar]
- 32.Pea F, Viale P, Cojutti P, Del Pin B, Zamparini E, Furlanut M. 2012. Therapeutic drug monitoring may improve safety outcomes of long-term treatment with linezolid in adult patients. J. Antimicrob. Chemother. 67:2034–2042. 10.1093/jac/dks153. [DOI] [PubMed] [Google Scholar]
- 33.MacGowan AP. 2003. Pharmacokinetic and pharmacodynamic profile of linezolid in healthy volunteers and subjects with Gram-positive infections. J. Antimicrob. Chemother. 51(Suppl 2):ii17–ii25. 10.1093/jac/dkg248 [DOI] [PubMed] [Google Scholar]
- 34.Dreisbach AW, Lertora JJ. 2008. The effect of chronic renal failure on drug metabolism and transport. Expert Opin. Drug Metab. Toxicol. 4:1065–1074. 10.1517/17425255.4.8.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lodise TP, Drusano GL. 2014. Use of pharmacokinetic/pharmacodynamic systems analyses to inform dose selection of tedizolid phosphate. Clin. Infect. Dis. 58(Suppl 1):S28–S34. 10.1093/cid/cit615. [DOI] [PubMed] [Google Scholar]
- 36.Center for Biologics Evaluation and Research. 2003. Guidance for industry: pharmacokinetics in patients with impaired liver function: study design, data analysis, and impact on dosing and labeling. Center for Biologics Evaluation and Research, Food and Drug Administration, Rockville, MD: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072123.pdf Accessed 15 April 2014. [Google Scholar]
- 37.Reidenberg MM, Breckenridge A. 1998. Drugs and the liver. Clin. Pharmacol. Ther. 64:353–354. 10.1016/S0009-9236(98)90064-9. [DOI] [PubMed] [Google Scholar]
- 38.Das D, Tulkens PM, Mehra P, Fang E, Prokocimer P. 2014. Tedizolid phosphate for the management of acute bacterial skin and skin structure infections: safety summary. Clin. Infect. Dis. 58(Suppl 1):S51–S57. 10.1093/cid/cit618. [DOI] [PubMed] [Google Scholar]
- 39.Pea F, Scudeller L, Lugano M, Baccarani U, Pavan F, Tavio M, Furlanut M, Rocca GD, Bresadola F, Viale P. 2006. Hyperlactacidemia potentially due to linezolid overexposure in a liver transplant recipient. Clin. Infect. Dis. 42:434–435. 10.1086/499533. [DOI] [PubMed] [Google Scholar]
- 40.Su E, Crowley K, Carcillo JA, Michaels MG. 2011. Linezolid and lactic acidosis: a role for lactate monitoring with long-term linezolid use in children. Pediatr. Infect. Dis. J. 30:804–806. 10.1097/INF.0b013e3182186035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.