

Abstract
Efflux is an important mechanism of bacterial multidrug resistance (MDR), and the inhibition of MDR pumps by efflux pump inhibitors (EPIs) could be a promising strategy to overcome MDR. 1-(1-Naphthylmethyl)-piperazine (NMP) and phenylalanine-arginine-β-naphthylamide (PAβN) are model EPIs with activity in various Gram-negative bacteria expressing AcrB, the major efflux pump of Escherichia coli, or similar homologous pumps of the resistance-nodulation-cell division class. The aim of the present study was to generate E. coli AcrB mutants resistant to the inhibitory action of the two model EPIs and to identify putative EPI target residues in order to better understand mechanisms of pump inhibition. Using an in vitro random mutagenesis approach focusing on the periplasmic domain of AcrB, we identified the double mutation G141D N282Y, which substantially compromised the synergistic activity of NMP with linezolid, was associated with similar intracellular linezolid concentrations in the presence and absence of NMP, and did not impair the intrinsic MICs of various pump substrates and dye accumulation. We propose that these mutations near the outer face of the distal substrate binding pocket reduce NMP trapping. Other residues found to be relevant for efflux inhibition by NMP were G288 and A279, but mutations at these sites also changed the susceptibility to several pump substrates. Unlike with NMP, we were unable to generate AcrB periplasmic domain mutants with resistance or partial resistance to the EPI activity of PAβN, which is consistent with the modes of action of PAβN differing from those of NMP.
INTRODUCTION
Multidrug resistance (MDR) has become an increasing problem in treating infectious diseases, particularly since the development of new antibiotics has stagnated dramatically. Efflux pumps are thought to contribute substantially to MDR of Gram-negative bacteria. In Pseudomonas aeruginosa, at least five efflux pumps of the resistance-nodulation-cell division (RND) class have been found to be involved in MDR phenotypes of clinical isolates (1, 2), and pumps of this class as well as of other classes are also found in clinical isolates of other Gram-negative pathogens. Efflux is supposed to facilitate the development of further resistance mechanisms by enabling the survival of pathogens in the presence of antibiotics that are pump substrates. Efflux also increases the level of resistance provided by target gene mutations alone. Inhibition of efflux therefore seems to be a promising strategy to reduce the level of resistance and to minimize its development.
The RND transporter superfamily provides major efflux systems in various Gram-negative bacteria. The best-investigated RND efflux pump is AcrB from Escherichia coli. It is part of the tripartite complex AcrAB-TolC that bridges the periplasmic space and the inner and outer membrane. Twelve transmembrane helices (TMs) anchor the AcrB pump within the inner membrane. A periplasmic domain is built from two large periplasmic loops comprising almost two-thirds of the protein (3, 4). Pump substrates are thought to be captured from the periplasm or the inner membrane lipid layer and to be extruded by a proton motive force-driven process through the outer membrane channel TolC (5, 6). A peristaltic-pump-like mechanism has been suggested as the mode of function, with asynchronic conformational changes of the AcrB protomers from an access to a binding and subsequently to an extrusion state (7–9).
AcrB and many other homologous RND efflux proteins are able to expel a wide variety of diverse compounds differing in size and chemical properties (10, 11). The determinants of differences in substrate specificity have been localized within the two large periplasmic loops between TM1 and TM2 and between TM7 and TM8 (12, 13). The promiscuity in substrate recognition is thought to be enabled by a large phenylalanine-rich deep (distal) binding pocket within the periplasmic pore domain of the protein (14, 15). Compounds are thought to bind within distinct areas of this flexible predominantly hydrophobic cavity (16, 17). In addition, recent studies have discovered an access (proximal) pocket separated from the distal binding pocket by a so-called “switch-loop.” This is supposed to be another important part of the drug pathway through the AcrB protomer (18, 19). The AcrAB-TolC complex is constitutively expressed in E. coli, and overexpression is inducible by drug exposure (20, 21), whereas the homologous E. coli transport systems AcrEF and YhiUV have been found to be expressed in vitro in AcrB-deficient E. coli strains only after several selection steps (22, 23).
A number of chemosensitizers inhibiting eukaryotic ABC transporters and overcoming drug resistance in cancer cells were in clinical development (24, 25). Although sensitizers that inhibit bacterial RND pump-mediated efflux so far have not been available for clinical use, such compounds have been described and include 1-(1-naphthylmethyl)-piperazine (NMP) and phenylalanine-arginine-β-naphthylamide (PAβN). Both NMP and PAβN are considered model efflux pump inhibitors (EPIs) with broad-spectrum efficacy for RND transporters. Interestingly, NMP appears to be most effective in E. coli in restoring the in vitro susceptibility to compounds usually active only in Gram-positive pathogens (such as linezolid and others), whereas PAβN was primarily studied in P. aeruginosa and described as being effective in restoring susceptibility to a number of fluoroquinolones (22, 26–28).
The development of improved bacterial EPIs requires a better understanding of substrate binding in RND pumps and its inhibition, including likely targets of diverse EPIs. Methods to evaluate substrate binding in AcrB in more detail have typically included cocrystallization studies with compounds (7, 29, 30), site-directed mutagenesis (14, 31), competition assays, and computer prediction of ligand binding (17, 19, 32). Unfortunately, few results are available concerning the mode of action of known EPIs. The effects of EPIs on the resistance phenotypes in several bacterial species are well known, and there is some evidence of their likely mode of binding at AcrB from computer simulation studies (16). Cocrystallization data are available for a novel pyridopyrimidine derivative with AcrB and the homologous P. aeruginosa RND transporter MexB and suggest a total inhibition of the functional rotation machinery by this relatively large molecule (33).
In vitro random mutagenesis, also often designated directed evolution, has become an important tool in studying structure-function relationship and engineering of enzymes (34). During the last decade, promising results with this method have been reported in studies of membrane proteins (35–37), including studies evaluating inhibition mechanisms of a eukaryotic neurotransmitter (38). In the present work we demonstrate the usefulness of such an in vitro random mutagenesis approach to study the possible mode of action of known RND-type EPIs. Here, we report the identification and characterization of E. coli mutants with partial resistance to the EPI NMP and define amino acids likely to be critical for NMP action.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and chemical compounds.
The bacterial strain used for the mutagenesis studies was the AcrAB-TolC-overexpressing E. coli strain 3-AG100 (gyrA marR mutant), a K-12 derivative described earlier (21). An AcrB-deficient E. coli strain, ΔAcrB(acrB::rpsLneo), was derived from 3-AG100 (see “In vitro random mutagenesis and library construction” below). Bacteria were cultivated in Luria-Bertani (LB) broth or on LB agar (1.5%) at 37°C supplemented with drug if needed and as indicated. Cells harboring the plasmid Red/ET (Gene Bridges, Dresden, Germany) were treated according to the manufacturer's instructions.
Chemicals were obtained from Sigma (Taufkirchen, Germany) with the following exceptions: NMP was purchased from Chess (Mannheim, Germany), linezolid (Zyvoxid; 2-mg/ml solution) from Pfizer (Berlin, Germany), silicon oils AR200 and AK100 from Wacker Chemicals (Burghausen, Germany), phosphate-buffered saline (PBS) from Lonza (Verviers, Belgium), and 1,2′-dinaphthylamine (1,2′-DINA) from TCI-Europe (Zwijndrecht, Belgium). The EPI NMP was used at a concentration of 100 μg/ml and PAβN at 25 μg/ml (the MIC of NMP is 512 μg/ml and that of PAβN is 1,024 μg/ml for E. coli strain 3-AG100), unless otherwise indicated.
Serial passage selection experiments.
E. coli 3-AG100 cells were cultivated overnight in LB broth with stepwise (2-fold)-increasing concentrations of linezolid, starting from 32 μg/ml in the presence of NMP. Subsequently, bacteria were plated on LB agar containing 64, 128, 256, and 512 μg/ml linezolid in combination with NMP. The susceptibility of saved clones was tested in the absence and presence of EPI. Similar experiments were performed with the combination of clarithromycin plus PAβN with an initial concentration of 4 μg clarithromycin per ml and PAβN, with subsequent plating on 8, 16, 32, and 64 μg/ml clarithromycin in the presence of PAβN. The initial drug and EPI concentrations used for the serial selections correspond to the starting MIC of the drug tested in combination with the EPI.
In vitro random mutagenesis and library construction.
MutazymII error-prone PCR (GeneMorphII random mutagenesis kit; Stratagene, La Jolla, CA, USA) was performed according to the manufacturer's instructions. The template DNA was prepared by amplifying the appropriate parts of acrB from extracted chromosomal DNA of E. coli 3-AG100 using a proofreading polymerase (PCR Extender system; 5PRIME, Hamburg, Germany). Primer pairs for template preparation were the same as for the Mutazym II reaction itself (see Table S1 in the supplemental material). An intermediate mutation frequency was adjusted using template amounts of 200 ng/kb. Concentration and desalting of error-prone PCR products were done by ethanol precipitation. Template DNA had been purified using a PCR purification kit (Qiagen, Hilden, Germany).
For the generation of 3-AG100 AcrB mutants, the appropriate part of the acrB gene was replaced directly within the bacterial chromosome by a two-step homologous recombination method using the Red/ET counterselection bacterial artificial chromosome (BAC) modification kit (Gene Bridges, Dresden, Germany). In the first step, the gene region of interest (bp 1 to 3150 for entire acrB, bp 85 to 990 for periplasmic loop 1, and bp 1681 to 2586 for periplasmic loop 2) was exchanged with an rpsL-neo cassette (template provided from the Red/ET kit; for oligonucleotides, see Table S1 in the supplemental material), yielding the AcrB-deficient strain ΔAcrB(acrB::rpsLneo), and in the second step, it was replaced by the MutazymII error-prone PCR products. Procedures were carried out according to Red/ET kit instructions with the following exceptions: modifications were realized directly within the bacterial chromosome, and thus, no additional drug for the maintenance of BACs was used; the rpsL-neo cassette was replaced by MutazymII error-prone PCR products instead of repair oligonucleotides; 100-bp homology arms downstream and upstream of the addressed region were used (generated by using appropriate primer pairs for the error-prone PCR [see Table S1 in the supplemental material]); and counterselection based on the replacement of the rpsL-neo cassette (streptomycin selection) was not necessary, since the repaired acrB served as resistance marker itself. Efficiency controls for the homologous recombination were performed by plating 50-μl aliquots from each transformation on 20 μg/ml linezolid (the linezolid MIC for the ΔAcrB strain was 16 μg/ml). The selection of AcrB mutants with potential inhibitor resistance was done by direct plating on drug-EPI combinations at the indicated concentrations (see Table 2).
TABLE 2.
Number of putative mutants derived from in vitro random mutagenesis of the periplasmic domain of AcrB
| Targeted AcrB region | Selection drug and concn (μg/ml)a | No. of mutants |
||||
|---|---|---|---|---|---|---|
| Plated | Selected overnight | MIC tested | Showing ≥4-fold decreased EPI efficacyb | Sequenced | ||
| Periplasmic loop 1 (residues 29–330) | NMP 100 + LZD 90 | 1.2 × 105 | 139 | 139 | 124 | 38c |
| NMP 100 + LZD 128 | 4 × 104 | |||||
| NMP 100 + CHL 8 | 5 × 104 | 24 | 21 | 5 | 5 | |
| PAβN 25 + NOV 16 | 6 × 104 | |||||
| PAβN 25 + CLR 16 | 8 × 104 | 34 | 34 | |||
| Periplasmic loop 2 (residues 561–862) | NMP 100 + LZD 90 | 1.1 × 105 | 2 | 2 | 2 | 2 |
| NMP 100 + CHL 8 | 6 × 104 | |||||
| PAβN 25 + NOV 16 | 1.1 × 105 | |||||
| PAβN 25 + CLR 16 | 3 × 105 | 450 | 183d | |||
LZD, linezolid; CHL, chloramphenicol; NOV, novobiocin; CLR, clarithromycin.
With at least one drug.
Mutants revealing NMP efficacy reduced ≥4-fold with at least 3 drugs or ≥8-fold with at least one drug.
A strain was selected for MIC testing if large colonies on CLR 16/PAβN 25 plates and/or colonies growing on 0.25 μg/ml rifampin in the presence of PAβN were observed.
Site-directed mutagenesis.
Mutations of interest were reconstructed by chromosomal site-directed mutagenesis using the Red/ET counterselection BAC modification kit. Oligonucleotides for cassette constructions are listed in Table S1 and repair oligonucleotides are listed in Table S2 in the supplemental material.
Standard genetic techniques.
Taq PCR amplification, DNA agarose gel electrophoresis, DNA extraction, and DNA purification and sequencing were carried out following standard procedures.
Susceptibility and synergy testing.
The MICs of drugs and compounds were determined by a standard microdilution assay in 96-well custom plates (Merlin, Bornheim-Hersel, Germany) using LB medium and inocula of 1.5 × 105 CFU/ml. In order to evaluate the efficiency of EPIs, the assays were performed in the absence and presence of NMP and PAβN. All assays were done at least in triplicate. Gentamicin was used as a control drug (no AcrB substrate).
A gradient MIC test was performed to measure more precisely the NMP efficacy in highly linezolid-resistant strains (MIC, 1,024 μg/ml) using stepwise (100 μg/ml per step) increasing drug concentrations in the absence of NMP and 10-μg/ml steps in the presence of NMP. Tests were also done at least in triplicate.
The fractional inhibitory concentration (FIC) index of the EPIs in combination with drug was investigated using a modified checkerboard method. Briefly, the MICs of NMP and PAβN in the presence of different concentrations of linezolid (4 up to 16 μg/ml) and in the presence of chloramphenicol (0.5 up to 1 μg/ml) were determined at least in duplicate, and the FIC index was calculated as described elsewhere (39).
Drug and dye uptake and efflux assays.
Bacteria were grown to an optical density at 600 nm (OD600) of 0.5 to 0.6 and washed twice with PBS. Cells were resuspended in the same buffer supplemented with 0.4% d-glucose to an OD600 of 0.4. After an incubation of 10 min at 37°C with shaking, linezolid or levofloxacin was added at a final concentration of 20 μg/ml or 10 μg/ml, respectively. Drug uptake was determined in the absence and presence of NMP. Aliquots of 1 ml were taken every 10 min and centrifuged through double silicon-oil layers (335 μl AR200, 165 μl AK100) to separate cells from the drug-containing medium. Pellets were lysed in 500 μl 0.1 M glycine-HCl, pH 3 (300 μl for the levofloxacin assay), at room temperature overnight. Linezolid determination in supernatants was carried out by a high-performance liquid chromatography (HPLC) method described earlier (40, 41). Standard curves with linezolid were prepared from 0.1 to 5 μg/ml. Levofloxacin was measured using a fluorescence plate reader (Tecan Safire) at an excitation wavelength of 292 nm and an emission wavelength of 496 nm. Standard curves were prepared from 0.003 to 10 μg/ml levofloxacin in the absence and presence of NMP. With HPLC of the cell lysates from the linezolid uptake assay, NMP peaks were detected at 282 nm, and NMP amounts were calculated using an NMP standard curve prepared from 5 to 100 μg/ml.
For measurement of fluorescent-dye accumulation, bacterial cells freshly grown on LB agar plates were resuspended in PBS supplemented with 0.4% d-glucose to an OD600 of 1. Accumulation of dye was determined in the absence and presence of EPIs. Dye was added after 10 min of incubation with EPI at 34°C with shaking, and fluorescence was measured at 34°C over time with a fluorescence plate reader (Tecan Safire). Ethidium bromide was used at a final concentration of 25 μM (excitation, 518 nm; emission, 605 nm), Hoechst 33342 at 2.5 μM (excitation, 350 nm; emission, 461 nm), and berberine at 30 μg/ml (excitation, 355 nm; emission, 517 nm). Assays were done in triplicate with double measurement each, and the mean concentrations after 30 min of dye accumulation were determined by measuring the relative fluorescence. The 1,2′-DINA efflux assays were performed according to a protocol published earlier (42).
Visualization of amino acid residues in the three-dimensional structure of AcrB and prediction of NMP binding.
The residues identified from random mutagenesis were visualized, and the distances of side chains were measured within the binding state AcrB protomer (PDB code 2HRT; www.rcsb.org/pdb) using the molecular visualization program PyMOL (version 1.3, 2010; Schrodinger, LLC) (www.pymol.org). The putative orientation of NMP in correlation with the discovered residues was predicted due to its polarity, size, and availability of H donors for hydrogen bonds (the H bond cutoff center was set at 3.6 and the H bond cutoff edge was 3.2 in PyMOL). The three-dimensional structure of NMP is available from PubChem (http://pubchem.ncbi.nlm.nih.gov/).
RESULTS
Selection of EPI-resistant mutants from random mutagenesis experiments.
An initial approach using serial passages with drug-EPI combinations (in vivo mutagenesis) in order to select EPI-resistant mutants resulted in two mutants exhibiting decreased EPI susceptibility, but they also showed significant altered susceptibility to various pump substrates in the absence of EPIs, indicating functional changes that were not restricted to EPI activities (Table 1). Sequencing of acrB from the partially NMP-resistant strain 4B/15 revealed a single mutation causing a change of AcrB residue 288 from glycine to serine, whereas no mutations were detected in acrAB-tolC of the PAβN-resistant strain C5/1/17.
TABLE 1.
EPI efficacies detected with mutants derived from serial selection with drug/EPI compared to reference strains
| Strain | EPI efficacy witha: |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LVX |
CHL |
OXA |
LZD |
MIN |
NOV |
CLR |
RIF |
|||||||||
| NMP | PAβN | NMP | PAβN | NMP | PAβN | NMP | PAβN | NMP | PAβN | NMP | PAβN | NMP | PAβN | NMP | PAβN | |
| Reference strains | ||||||||||||||||
| AG100 | 2 | 2 (0.06) | 2 | 2 (2) | 4 | 4 (128) | 16 | 8 (256) | 8 | 4 (1) | 4 | 32 (64) | 4 | 64 (128) | 2 | 64 (8) |
| 3-AG100 | 8 | 8 (2) | 8 | 8 (16) | 8 | 8 (512) | 32 | 8 (1,024) | 8 | 16 (4) | 8 | 128 (1,024) | 2 | 32 (256) | 4 | 64 (16) |
| ΔAcrBb | 1 | 1 (0.06) | 2 | 1 (1) | 1 | 1 (0.5) | 1 | 2 (16) | 4 | 4 (0.25) | 8 | 8 (4) | 2 | 8 (4) | 2 | 128 (8) |
| Mutantsc | ||||||||||||||||
| 4B/15 | 4 | 8 (2) | 4 | 8 (32) | 2 | 4 (128) | 8 | 32 (>2,048) | 2 | 8 (4) | 4 | 16 (1,024) | 1 | 64 (128) | 2 | 128 (16) |
| C5/1/17 | 4 | 1 (0.25) | 4 | 2 (4) | 1 | 2 (128) | 16 | 4 (512) | 2 | 2 (1) | 4 | 16 (128) | 1 | 2 (2,048) | 1 | 8 (2) |
The relative EPI efficacy was defined as the ratio of the MICs in the absence and in the presence of the EPI (NMP or PAβN, respectively). MICs in µg/ml (without EPI) are given in parentheses; EPI efficacies and mutant MIC changes of ≥4-fold compared to values for the parental AcrB-overexpressing strain 3-AG100 are in bold. LVX, levofloxacin; CHL, chloramphenicol, OXA, oxacillin, LZD, linezolid, MIN, minocycline, NOV, novobiocin, CLR, clarithromycin; RIF, rifampin. Gentamicin was used in addition as a control drug (no AcrB substrate), showing no changes in relative NMP efficacy and MIC (data not shown) similar to rifampin (poor AcrB substrate).
AcrB knockout derived from the AcrB-overexpressing E. coli strain 3-AG100 (AG100 is the wild-type control strain).
Strain 4B/15 was from serial selection experiments with LZD/NMP (second-step mutant); C5/1/17 was selected with CLR/PAβN (third-step mutant).
To avoid mutations unrelated to the direct EPI action and increasing the resistance to the substrate used in the selection experiments rather than to the EPI, we focused in further experiments on mutating acrB in vitro, with subsequent chromosomal replacement by homologous recombination. Taking into account the results of a preliminary study with a small library (1,360 mutants) of in vitro random mutagenesis variants of whole AcrB derived from parental strain 3-AG100 (see Table S3 in the supplemental material) and in order to further increase the efficiency of the approach, we decided to focus on the periplasmic domain of AcrB. The two large periplasmic loops, comprising about 300 amino acids each, were targeted separately, and large amounts of mutants were directly selected on agar with suitable antibiotic-EPI combinations. The preferred choices were linezolid in the presence of NMP and clarithromycin in the presence of PAβN. Each of these drugs appeared to be the optimal pump substrate most susceptible to the inhibitory action of the respective EPI. Further drug-EPI combinations were used (Table 2). The AcrB mutants obtained supposedly included those with reduced EPI efficacy as well as mutants with altered resistance to the antibiotic itself or both. Susceptibility testing in the absence and presence of EPI identified mutants with confirmed NMP resistance (≥4-fold-reduced EPI efficacy with at least one drug). Those exhibiting the most prominent reduced NMP efficacy (≥4-fold with at least 3 drugs or ≥8-fold with at least one drug) were analyzed for mutations within the periplasmic domain of AcrB (Tables 2 and 3). From the putative mutants selected with drug-PAβN combinations, no stable EPI-resistant phenotypes were detectable.
TABLE 3.
Amino acid changes detected in drug/NMP selected variants derived from in vitro random mutagenesis of the periplasmic domain of AcrB
| Group | Mutations in AcrB variantsa |
||
|---|---|---|---|
| Single | Double | Multiple | |
| A | G288S (9 variants) | T205I G288M | M20Ib R263C G288M |
| G288M | A26Tb G288S | V32M S128R G288C | |
| L270M G288S | A47V S82G G288S | ||
| M188L G288S | G271S G288S A311V | ||
| S82G A266T G288S A303V | |||
| B | A279T | A279V G296D | P40A V43I A279T |
| A209G A279T | D256E E269G A279T | ||
| E130V A279T | |||
| L219F A279T | |||
| A279T P331Tb,c | |||
| C | G141V (2 variants) | D59N G141N | Q63H G141D Q255E |
| G141D | Q112H G141D N282Y | ||
| G141Dc | L28Fb G141D K226T N282Y | ||
| D | V268M F332Lb | S608L F628L V773A | |
| N194D F332Lb,c | L75V L92M S135N | ||
| L117V L177Mc | Q63R A209V I277N | ||
| A42T D202N | |||
| F628L E645I | |||
The most frequently mutated residues and the two G141D N282Y mutations are in bold. Residues of the distal binding pocket are underlined. Frequency is indicated if it is >1.
Mutation outside the periplasmic domain that appeared incidentally due to the method used.
From mutants selected on chloramphenicol-NMP; others were selected on linezolid-NMP.
AcrB mutations among strains with significantly altered NMP efficacy.
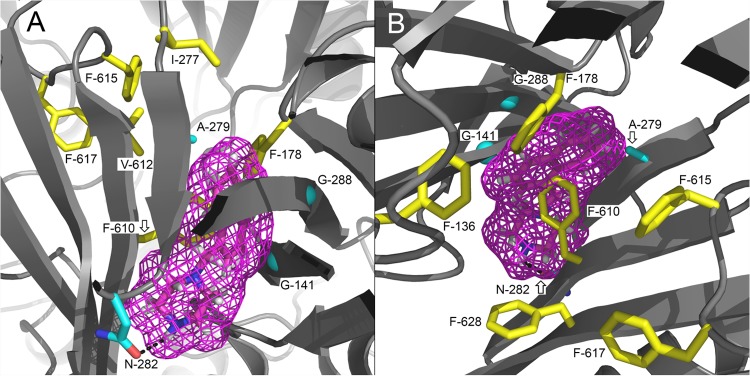
The by far most frequently mutated AcrB residue was glycine 288. This residue was altered in 44% of the sequenced mutants, most often to serine and less often to methionine (3 mutants) and to cysteine (1 mutant). G141 and A279 were found to be replaced in 19% of the sequenced mutants, predominantly by polar amino acids but also by valine (Table 3). Notably, the double mutation G141D N282Y was detected in two mutants (1D and LN-I-3) obtained from independently generated error-prone PCR products, and both showed a prominent reduced NMP efficacy with linezolid in MIC testing (Table 4). As shown in Fig. 1, the most frequently altered residues (G288S, A279T, and G141D N282Y) appear to be close to the phenylalanine-rich substrate-binding pocket.
TABLE 4.
Changes in NMP efficacies detected with a subset of mutants derived from in vitro random mutagenesis of the periplasmic domain of AcrB and of the corresponding site-directed generated variants
| Type and strain | Amino acid change(s) in AcrBa | NMP efficacy with drugb |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| LVX | CHL | OXA | LZD | MIN | NOV | CLR | RIF | ||
| Parent | |||||||||
| 3-AG100 | None | 8 (2) | 8 (16) | 8 (512) | 32c (1,024) | 8 (4) | 8 (1,024) | 2 (256) | 4 (16) |
| Mutants from random mutagenesis | |||||||||
| LN-I-46 | G288S | 2 (1) | 8 (32) | 2 (256) | 8 (1,024) | 4 (8) | 2 (512) | 1 (128) | 2 (16) |
| 2DD | G288M | 2 (1) | 8 (32) | 2 (256) | 8 (1,024) | 2 (4) | 2 (512) | 1 (128) | 4 (16) |
| LN-I-14 | A279T | 2 (2) | 2 (32) | 4 (512) | 4 (1,024) | 4 (8) | 2 (512) | 1 (256) | 4 (16) |
| CN7 | G141D | 2 (1) | 4 (16) | 2 (256) | 16 (1,024) | 4 (4) | 4 (1,024) | 1 (128) | 2 (16) |
| 1D | Q112H G141D N282Y | 4 (1) | 8 (16) | 2 (256) | 4 (1,024) | 4 (2) | 8 (1,024) | 1 (256) | 4 (16) |
| LN-I-3 | L28F G141D L226T N282Y | 4 (1) | 8 (16) | 4 (512) | 4 (1,024) | 16 (8) | 8 (1,024) | 2 (256) | 4 (32) |
| Mutants from site-directed mutagenesis | G288S | 2 (1) | 8 (16) | 4 (256) | 16c (1,024) | 4 (2) | 4 (256) | 1 (128) | 2 (8) |
| A279T | 8 (2) | 16 (32) | 8 (512) | 16c (1,024) | 4 (2) | 2 (256) | 1 (128) | 2 (8) | |
| A279T G288S | 4 (1) | 16 (32) | 8 (128) | 16c (1,024) | 8 (2) | 8 (64) | 4 (64) | 8 (8) | |
| N282Y | 8 (1) | 16 (16) | 2 (256) | 32 (1,024) | 8 (2) | 4 (512) | 2 (512) | 4 (16) | |
| G141D | 8 (1) | 8 (16) | 8 (256) | 32 (1,024) | ND | ND | 4 (256) | 4 (16) | |
| G141D N282Y | 4 (1) | 8 (16) | 4 (256) | 4c (1,024) | 8 (2) | 8 (1,024) | 2 (256) | 4 (16) | |
Boldface in this column indicates the occurrence of double mutation G141D N282Y.
Relative NMP efficacy was defined as the ratio of the MICs in the absence and in the presence of NMP. MICs in µg/ml (without NMP) are given in parentheses. NMP efficacies and mutant MIC changes of ≥4-fold compared to parental AcrB overexpressing strain 3-AG100 are in bold. ND, not detected; LVX, levofloxacin; CHL, chloramphenicol; OXA, oxacillin; LZD, linezolid; MIN, minocycline; NOV, novobiocin; CLR, clarithromycin; RIF, rifampin. Gentamicin was used in addition as a control drug (no AcrB substrate) showing no changes in relative NMP efficacy and MIC (data not shown) similar to rifampin (poor AcrB substrate).
See also NMP efficacies determined by gradient MIC testing (Fig. 2).
FIG 1.

Positions of the most frequently selected mutations from in vitro random mutagenesis of the periplasmic domain of AcrB (drug/NMP selection). Nonmutated (A and C) and mutated (B and D) side chains are shown as cyan sticks (glycines as cyan spheres), and distal binding pocket phenylalanine side chains are shown as yellow sticks. A side view from the periplasmic outer face (A and B) and a top view from the TolC docking domain (C and D) of the binding state AcrB protomer (PDB code 2HRT; www.rcsb.org/pdb) are shown. Dotted lines show distance (Å) between side chains of the double mutation. Images were created using PyMOL (www.pymol.org).
Susceptibility phenotypes of site-directed reconstructed mutants.
The impact of the AcrB double mutation G141D N282Y as well as of the single mutations G288S, A279T, G141D, and N282Y was evaluated with site-directed generated AcrB mutants derived from 3-AG100. Additionally, an A279T G288S mutant was constructed and investigated.
To exclude a single-nucleotide polymorphism (SNP) phenomenon of the most frequently occurring mutation, G288S, we also checked an alternative triplet, TCC instead of AGT (occurring from random mutagenesis). TCC was found to be the most common serine-coding triplet within AcrB. However, there were no detectable differences in MIC between the G288SAGT- and G288STCC-containing strains.
As shown in Table 4, (partial) NMP resistance in the single G141D mutant was not confirmed after site-directed reconstruction, and phenotypes of both reconstructed G141D and N282Y single mutants resembled the phenotype of the parental strain, 3-AG100. The combination of the two mutations, in contrast, was associated with much-diminished NMP efficacy predominantly limited to linezolid (the NMP inhibitory efficacy changed from a 32- to a 4-fold MIC reduction). Detailed gradient MIC testing (steps of 100 μg/ml for strains with an MIC of >500 μg/ml) confirmed the significant (partial) NMP resistance of the G141D N282Y mutant (Fig. 2), indicating that the combination of the two mutations was essential for significant loss of NMP efficacy with linezolid.
FIG 2.
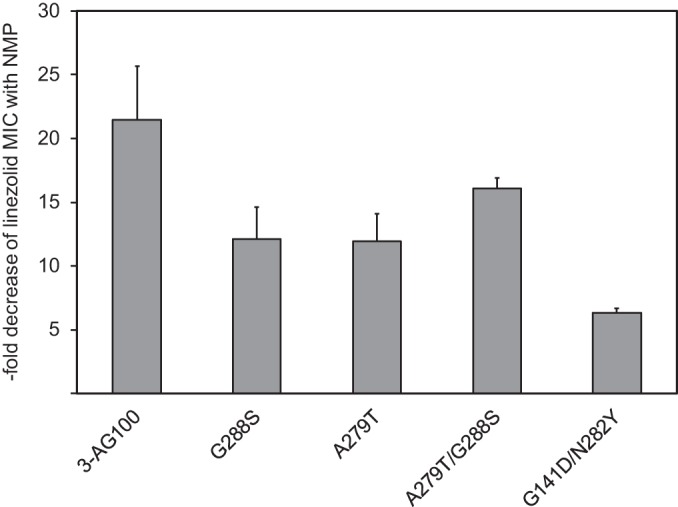
NMP efficacy with linezolid determined from gradient MIC testing (with the parental AcrB-overexpressing strain 3-AG100 and with site-directed-mutagenesis-generated AcrB mutants). Efficacy is reported as the ratio of the MICs in the absence and presence of NMP (values are means from at least four independent assays). Error bars indicate standard deviations (SD).
Table 4 shows that the NMP resistance phenotype of the initial G288S and A279T single mutants was less clear in reconstructed mutants and nonsignificant when tested by standard MIC assays in the presence of the EPI. However, gradient MIC testing confirmed partial NMP resistance with linezolid in the two site-directed reconstructed single mutants. The combination of the two mutations resulting in a constructed A279T G288S mutant failed to produce the enhanced NMP resistance that could have been expected (Fig. 2).
Synergy testing with FIC index calculation showed that the synergistic effect of NMP and linezolid seen with the wild-type AcrB strain 3-AG100 (FIC index, 0.28) was completely lost with the G141D N282Y-containing AcrB variant (FIC index, 1.25). In contrast, the FIC index with chloramphenicol-NMP remained unchanged in this mutant (0.63 in both wild-type and mutant cells). Linezolid-PAβN resulted in only subtle differences between 3-AG100 (FIC index, 0.38) and the G141D N282Y mutant (FIC index, 0.56), with both values being close to the breakpoint for synergy (<0.5 = synergy).
Drug and dye uptake phenotypes of site-directed reconstructed mutants.
An almost complete loss of the pump-inhibitory property of NMP was observed with the G141D N282Y mutant as well as with the AcrB variant harboring the mutation G288S when the uptake of linezolid was monitored over 30 min (Fig. 3). Similar results were obtained with the A279T mutant. We confirmed that the NMP concentration within the cells did not change over time in AcrB-overexpressing E. coli 3-AG100, the AcrB knockout strain, and the mutants (data not shown), and thus, NMP itself remained a poor substrate (or not a substrate) of AcrB in the mutants. Interestingly, in the absence of NMP, the mutants appeared to pump out the drug even more efficiently compared to the parental AcrB E. coli strain 3-AG100, showing that AcrB function with linezolid was not impaired (Fig. 3).
FIG 3.
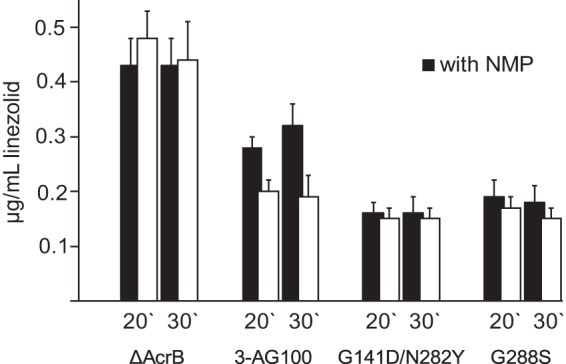
Linezolid uptake with an AcrB knockout strain, the parental AcrB-overexpressing strain 3-AG100, and site-directed-mutagenesis-generated AcrB mutants in the presence and absence of NMP determined after 20 and 30 min of drug exposure. Values are derived from at least 2 independent assays with 2 HPLC measurements each. Mutant AcrB-A279T had been included into one assay and behaved similarly to the AcrB-G288S variant (data not shown). Error bars indicate SD.
Using a levofloxacin uptake assay, we confirmed a diminished efflux-inhibitory efficacy of NMP for this substrate in both the double mutant (50% levofloxacin uptake after 30 min compared with the parental strain in the presence of NMP) and the G288S mutant (58%) (data not shown).
Finally, we tested AcrAB-TolC efflux pump function using dye accumulation and efflux assays. Again, the mutants were compared with the parental AcrB-overexpressing strain 3-AG100 and with the corresponding ΔAcrB strain serving as the control for maximum dye accumulation. The results of the 1,2′-DINA real-time efflux assay confirmed the maintained capacity of the mutants to efficiently expel this pump substrate. The accumulation of ethidium (−80%), Hoechst 33342 (−60%), and berberine (−30%) was found to be diminished in the G288S and A279T mutants in comparison with 3-AG100, likely indicating some improved efflux capacity. A similar phenomenon was seen in the case of the G141D N282Y mutant, which exhibited less ethidium (−40%) and berberine (−10%) accumulation but Hoechst 33342 accumulation similar to that of the parental strain. We observed a decreased NMP activity (partial EPI resistance) in combination with Hoechst 33342 for the G141D N282Y mutant (−25%), but not for the other mutants, and not with other dyes. No significant changes were seen in the dye accumulation experiments with regard to the efficacy of PAβN (data not shown).
DISCUSSION
We aimed to investigate in more detail the mode of function of the well-characterized model EPIs NMP and PAβN using a random-mutagenesis approach with EPI resistance as the readout. Since in vivo random mutagenesis of an E. coli strain by serial selection steps had predominantly induced adaptation by addressing targets other than the AcrAB-TolC efflux transporter itself, we decided to directly mutate AcrB in vitro. Taking into account the results from a preliminary study with the entire acrB gene that resulted in a number of mutations in the periplasmic domain of the pump, we finally focused directly on in vitro random mutagenesis of this region.
The major result of our study was the identification of a double AcrB mutation (G141D N282Y) that, in fact, could be confirmed in a mutant reconstructed by site-directed mutagenesis to provide a loss of NMP efficacy, primarily with linezolid as the pump substrate, but also a partial NMP resistance with Hoechst 33342 and levofloxacin when measured in accumulation assays. Unlike with most other mutants described above, the NMP resistance in combination with these compounds could not be explained by altered pump efficiency for the substrates themselves. Interestingly, the NMP or partial NMP resistance associated with this double mutation was absent or nonsignificant with other drugs known to be AcrB pump substrates, and the mutation was not associated with reduced intrinsic pump efficiency for the substrates included in our test panel. Also, there was no EPI cross-resistance between NMP and PAβN. This relatively high specificity of the mutation regarding the NMP-drug combination is remarkable.
Other frequent mutations that provided relative NMP resistance were the residues G288 and A279. Interestingly, mutations at both sites did not act synergistically in the sense of enhanced NMP resistance. Both as single mutations and as the double mutation A279T G288S, they were associated with much more altered pump function in the sense of diminished efficiency for (and higher susceptibility to) particular substrates, such as novobiocin and clarithromycin, as well as enhanced efficiency for other substrates, such as several dyes, and it was difficult to separate the NMP resistance from these diverse EPI-independent functional alterations. Major alterations in defined substrate efflux would be expected to affect the relative NMP inhibitory efficacy and make the interpretation of the results in the sense of EPI resistance difficult. In fact, when we applied our own strict definition of EPI resistance (≥4-fold decreased efficacy in MIC testing) and considered only reconstructed mutants with confirmed unaltered substrate susceptibility in MIC testing, the G141D N282Y double mutation was the only EPI resistance mutation observed.
Most mutations identified here were found to be at highly conserved residues when 15 homologous transporter proteins from the AcrB/AcrD/AcrF family were compared (43). They were derived from periplasmic loop 1, and the mutated residues are located near the outer face of the distal substrate binding pocket (Fig. 1) across the F615- and F617-containing flexible switch-loop (18). Previous molecular dynamics computer simulations proposed an interaction of NMP with this loop (17), and another study predicted NMP to be bound to a lower area (“cave”) of the distal binding pocket. The findings in the present study indicate that NMP binding might occur more closely to F610, a residue belonging to the backside of the so-called “groove,” an upper part of the large distal binding pocket (16).
An imaginary plane between G141 and N282 would be located close to F610. Considering the size of the NMP molecule, it might fit between residues G141 and N282 (Fig. 4). The distal binding pocket is known to be located at the interface of the subdomains PN2 and PC1 of the periplasmic pore domain (8). We suggest an interaction of the piperazine portion of the NMP molecule with the polar amino acid N282 on the outside of PC1 by a putative hydrogen bond. Thus, the hydrophobic aromatic naphthyl ring system gets close to two β-strands from subdomain PN2 containing G141 and G288. Furthermore, it would be stuck between these two β-strands and another β-strand belonging to subdomain PC1 harboring residue A279 (Fig. 4). It is conceivable that the alteration of G141 to the bulkier and polar aspartic acid in combination with the change of N282 to an aromatic tyrosine residue might impede proper NMP incorporation. With the suggested NMP orientation, an influence of G288 or A279 when mutated to polar or bulkier amino acids also seems plausible. A compensatory effect seen with the double mutation G288S A279T might be caused by repulsion of the two polar residues shifting them away from the putative binding site of NMP.
FIG 4.
Two views of suggested NMP binding within the AcrB binding state protomer (PDB code 2HRT; www.rcsb.org/pdb). Binding pocket side chains are in yellow, residues detected from random mutagenesis are cyan sticks, and glycines are cyan spheres. The NMP molecule is in magenta. The dotted black line indicates a putative hydrogen bond. Oxygen is in red, nitrogen is blue, and hydrogen is white. Images were created using PyMOL (www.pymol.org).
As we know from previous site-directed mutagenesis studies, replacement of F610 by alanine affects the MICs of many pump substrates dramatically (14). Molecular dynamics simulations suggested that F610A catalyzes a stronger substrate binding within the deep binding pocket, resulting in putative hindrance of the extrusion process (32). With our binding model showing NMP trapped close to F610 (Fig. 1 and 4), we postulate that the molecule modulates in a noncompetitive manner the substrate specificity of the large distal pocket by varying its binding capacity by affecting F610. Furthermore, the flexibility and ability to allow conformational changes of the pocket might be compromised by linking subdomains PN2 and PC1. A molecular dynamics simulation study outlined the peristaltic motion of subdomains PN2 and PC1 as being important for the substrate extrusion process (44). It remains unclear, however, why linezolid in particular is most affected by these changes.
We were unable to generate mutants with significant resistance to the PAβN EPI effects. This may indicate its different mode of action versus that of NMP. A strongly PAβN-resistant strain derived from our initial serial selection experiments had no AcrAB-TolC mutations but showed increased susceptibility to rifampin, perhaps in association with changed outer membrane properties. It is known that—besides efflux inhibition—PAβN enhances the permeability of the outer membrane (27, 45). Another antagonizing effect might be that, unlike NMP, PAβN itself is a substrate of AcrB, possibly enabling cells to adapt by increasing the efflux of the compound.
Some limitations of our approach should be mentioned. Variants derived from the in vitro random mutagenesis often exhibited enhanced NMP resistance with several drugs that was not completely reproducible in the corresponding site-directed-mutagenesis-generated strains. Whether additional mutations outside the addressed gene region are responsible for this observation is uncertain, and we also cannot exclude an additional mode of action of NMP. The mutagenesis rate of bacteria is known to be significantly elevated under stress conditions (46) that might originate from the transformation procedure. Synonymous nucleotide polymorphism (SNP) effects should be taken into account even with silent mutations (47), although such an SNP effect was not the reason for the striking frequency of the G288S mutation found here.
Another limitation of the present work and previous studies was the difficulty of directly measuring efflux of several pump substrates and control compounds with a reliable method suited to diverse compounds. MIC and synergy testing and cellular accumulation assays cannot differentiate between influx and efflux alterations and are insensitive for identifying and quantifying EPI resistance. Unfortunately, the 1,2′-DINA efflux assay we described earlier (42) was not suited to assays with NMP because of a relatively low efficacy of this EPI in combination with the dye. However, we believe that using a combination of methods was convincing enough to demonstrate the relevance of the double mutation for the relatively substrate-specific resistance to the pump inhibitory effects of NMP but also the substrate-dependent variably altered pump function associated with binding pocket mutations. This appears to be consistent with binding pocket flexibility, whereby it would be expected that a particular mutation may affect one substrate in a different and sometimes opposite way than another one.
In conclusion, with one passage of in vitro random mutagenesis we have detected mutations linked with relatively substrate-specific resistance to the EPI NMP located near the distal binding pocket. While this can be considered a strong indication that NMP targets the outer face of the binding pocket, further studies are needed to explore the mechanism of this NMP resistance, including a possible hindrance of the peristaltic movements of PN2 and PC1. The method can and should also be applied to assess targets of other EPIs and to use of these data for modeling and more rational EPI design.
Supplementary Material
ACKNOWLEDGMENT
This work was in part supported by a grant (to W.V.K.) from the German Federal Ministry of Education and Research (BMBF 01KI9951).
Footnotes
Published ahead of print 2 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03775-14.
REFERENCES
- 1.Li XZ, Barre N, Poole K. 2000. Influence of the MexA-MexB-oprM multidrug efflux system on expression of the MexC-MexD-oprJ and MexE-MexF-oprN multidrug efflux systems in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 46:885–893. 10.1093/jac/46.6.885. [DOI] [PubMed] [Google Scholar]
- 2.Poole K. 2005. Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 56:20–51. 10.1093/jac/dki171. [DOI] [PubMed] [Google Scholar]
- 3.Fujihira E, Tamura N, Yamaguchi A. 2002. Membrane topology of a multidrug efflux transporter, AcrB, in Escherichia coli. J. Biochem. 131:145–151. 10.1093/oxfordjournals.jbchem.a003069. [DOI] [PubMed] [Google Scholar]
- 4.Murakami S, Nakashima R, Yamashita E, Yamaguchi A. 2002. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 419:587–593. 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- 5.Su CC, Li M, Gu R, Takatsuka Y, McDermott G, Nikaido H, Yu EW. 2006. Conformation of the AcrB multidrug efflux pump in mutants of the putative proton relay pathway. J. Bacteriol. 188:7290–7296. 10.1128/JB.00684-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seeger MA, von BC, Verrey F, Pos KM. 2009. Crucial role of Asp408 in the proton translocation pathway of multidrug transporter AcrB: evidence from site-directed mutagenesis and carbodiimide labeling. Biochemistry 48:5801–5812. 10.1021/bi900446j. [DOI] [PubMed] [Google Scholar]
- 7.Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. 2006. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443:173–179. 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- 8.Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM. 2006. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science 313:1295–1298. 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- 9.Sennhauser G, Amstutz P, Briand C, Storchenegger O, Grütter MG. 2007. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 5:e7. 10.1371/journal.pbio.0050007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nikaido H, Pages JM. 2012. Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria. FEMS Microbiol. Rev. 36:340–363. 10.1111/j.1574-6976.2011.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pos KM. 2009. Drug transport mechanism of the AcrB efflux pump. Biochim. Biophys. Acta 1794:782–793. 10.1016/j.bbapap.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 12.Elkins CA, Nikaido H. 2002. Substrate specificity of the RND-type multidrug efflux pumps AcrB and AcrD of Escherichia coli is determined predominantly by two large periplasmic loops. J. Bacteriol. 184:6490–6498. 10.1128/JB.184.23.6490-6499.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tikhonova EB, Wang Q, Zgurskaya HI. 2002. Chimeric analysis of the multicomponent multidrug efflux transporters from gram-negative bacteria. J. Bacteriol. 184:6499–6507. 10.1128/JB.184.23.6499-6507.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bohnert JA, Schuster S, Seeger MA, Fähnrich E, Pos KM, Kern WV. 2008. Site-directed mutagenesis reveals putative substrate binding residues in the Escherichia coli RND efflux pump AcrB. J. Bacteriol. 190:8225–8229. 10.1128/JB.00912-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wehmeier C, Schuster S, Fähnrich E, Kern WV, Bohnert JA. 2009. Site-directed mutagenesis reveals amino acid residues in the Escherichia coli RND efflux pump AcrB that confer macrolide resistance. Antimicrob. Agents Chemother. 53:329–330. 10.1128/AAC.00921-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takatsuka Y, Chen C, Nikaido H. 2010. Mechanism of recognition of compounds of diverse structures by the multidrug efflux pump AcrB of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 107:6559–6565. 10.1073/pnas.1001460107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vargiu AV, Nikaido H. 2012. Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc. Natl. Acad. Sci. U. S. A. 109:20637–20642. 10.1073/pnas.1218348109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eicher T, Cha HJ, Seeger MA, Brandstätter L, El-Delik J, Bohnert JA, Kern WV, Verrey F, Grütter MG, Diederichs K, Pos KM. 2012. Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc. Natl. Acad. Sci. U. S. A. 109:5687–5692. 10.1073/pnas.1114944109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao XQ, Kimura N, Murakami S, Takada S. 2013. Drug uptake pathways of multidrug transporter AcrB studied by molecular simulations and site-directed mutagenesis experiments. J. Am. Chem. Soc. 135:7474–7485. 10.1021/ja310548h. [DOI] [PubMed] [Google Scholar]
- 20.Jellen-Ritter AS, Kern WV. 2001. Enhanced expression of the multidrug efflux pumps AcrAB and AcrEF associated with insertion element transposition in Escherichia coli mutants selected with a fluoroquinolone. Antimicrob. Agents Chemother. 45:1467–1472. 10.1128/AAC.45.5.1467-1472.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kern WV, Oethinger M, Jellen-Ritter AS, Levy SB. 2000. Non-target gene mutations in the development of fluoroquinolone resistance in Escherichia coli. Antimicrob. Agents Chemother. 44:814–820. 10.1128/AAC.44.4.814-820.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bohnert JA, Kern WV. 2005. Selected arylpiperazines are capable of reversing multidrug resistance in Escherichia coli overexpressing RND efflux pumps. Antimicrob. Agents Chemother. 49:849–852. 10.1128/AAC.49.2.849-852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bohnert JA, Schuster S, Fähnrich E, Trittler R, Kern WV. 2007. Altered spectrum of multidrug resistance associated with a single point mutation in the Escherichia coli RND-type MDR efflux pump YhiV (MdtF). J. Antimicrob. Chemother. 59:1216–1222. 10.1093/jac/dkl426. [DOI] [PubMed] [Google Scholar]
- 24.Choi CH. 2005. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 5:30. 10.1186/1475-2867-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel A, Tiwari AK, Chufan EE, Sodani K, Anreddy N, Singh S, Ambudkar SV, Stephani R, Chen ZS. 2013. PD173074, a selective FGFR inhibitor, reverses ABCB1-mediated drug resistance in cancer cells. Cancer Chemother. Pharmacol. 72:189–199. 10.1007/s00280-013-2184-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kern WV, Steinke P, Schumacher A, Schuster S, von BH, Bohnert JA. 2006. Effect of 1-(1-naphthylmethyl)-piperazine, a novel putative efflux pump inhibitor, on antimicrobial drug susceptibility in clinical isolates of Escherichia coli. J. Antimicrob. Chemother. 57:339–343. 10.1093/jac/dki445. [DOI] [PubMed] [Google Scholar]
- 27.Lomovskaya O, Warren MS, Lee A, Galazzo J, Fronko R, Lee M, Blais J, Cho D, Chamberland S, Renau T, Leger R, Hecker S, Watkins W, Hoshino K, Ishida H, Lee VJ. 2001. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob. Agents Chemother. 45:105–116. 10.1128/AAC.45.1.105-116.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martins M, Dastidar SG, Fanning S, Kristiansen JE, Molnar J, Pages JM, Schelz Z, Spengler G, Viveiros M, Amaral L. 2008. Potential role of non-antibiotics (helper compounds) in the treatment of multidrug-resistant Gram-negative infections: mechanisms for their direct and indirect activities. Int. J. Antimicrob. Agents 31:198–208. 10.1016/j.ijantimicag.2007.10.025. [DOI] [PubMed] [Google Scholar]
- 29.Hung LW, Kim HB, Murakami S, Gupta G, Kim CY, Terwilliger TC. 2013. Crystal structure of AcrB complexed with linezolid at 3.5 A resolution. J. Struct. Funct. Genomics 14:71–75. 10.1007/s10969-013-9154-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakashima R, Sakurai K, Yamasaki S, Nishino K, Yamaguchi A. 2011. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 480:565–569. 10.1038/nature10641. [DOI] [PubMed] [Google Scholar]
- 31.Husain F, Nikaido H. 2010. Substrate path in the AcrB multidrug efflux pump of Escherichia coli. Mol. Microbiol. 78:320–330. 10.1111/j.1365-2958.2010.07330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vargiu AV, Collu F, Schulz R, Pos KM, Zacharias M, Kleinekathöfer U, Ruggerone P. 2011. Effect of the F610A mutation on substrate extrusion in the AcrB transporter: explanation and rationale by molecular dynamics simulations. J. Am. Chem. Soc. 133:10704–10707. 10.1021/ja202666x. [DOI] [PubMed] [Google Scholar]
- 33.Nakashima R, Sakurai K, Yamasaki S, Hayashi K, Nagata C, Hoshino K, Onodera Y, Nishino K, Yamaguchi A. 2013. Structural basis for the inhibition of bacterial multidrug exporters. Nature 500:102–106. 10.1038/nature12300. [DOI] [PubMed] [Google Scholar]
- 34.Turner NJ. 2009. Directed evolution drives the next generation of biocatalysts. Nat. Chem. Biol. 5:567–573. 10.1038/nchembio.203. [DOI] [PubMed] [Google Scholar]
- 35.Augustus AM, Celaya T, Husain F, Humbard M, Misra R. 2004. Antibiotic-sensitive TolC mutants and their suppressors. J. Bacteriol. 186:1851–1860. 10.1128/JB.186.6.1851-1860.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malle E, Zhou H, Neuhold J, Spitzenberger B, Klepsch F, Pollak T, Bergner O, Ecker GF, Stolt-Bergner PC. 2011. Random mutagenesis of the prokaryotic peptide transporter YdgR identifies potential periplasmic gating residues. J. Biol. Chem. 286:23121–23131. 10.1074/jbc.M111.239657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lluis MW, Godfroy JI, III, Yin H. 2013. Protein engineering methods applied to membrane protein targets. Protein Eng. Des. Sel. 26:91–100. 10.1093/protein/gzs079. [DOI] [PubMed] [Google Scholar]
- 38.Gros Y, Schuldiner S. 2010. Directed evolution reveals hidden properties of VMAT, a neurotransmitter transporter. J. Biol. Chem. 285:5076–5084. 10.1074/jbc.M109.081216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamicker BJ, Sweeney MT, Kaczmarek F, Dib-Hajj F, Shang W, Crimin K, Duignan J, Gootz TD. 2008. Bacterial efflux pump inhibitors. Methods Mol. Med. 142:187–204. 10.1007/978-1-59745-246-5_15. [DOI] [PubMed] [Google Scholar]
- 40.Ehrlich M, Trittler R, Daschner FD, Kümmerer K. 2001. A new and rapid method for monitoring the new oxazolidinone antibiotic linezolid in serum and urine by high performance liquid chromatography-integrated sample preparation. J. Chromatogr. B Biomed. Sci. Appl. 755:373–377. 10.1016/S0378-4347(01)00115-3. [DOI] [PubMed] [Google Scholar]
- 41.Schumacher A, Trittler R, Bohnert JA, Kümmerer K, Pages JM, Kern WV. 2007. Intracellular accumulation of linezolid in Escherichia coli, Citrobacter freundii and Enterobacter aerogenes: role of enhanced efflux pump activity and inactivation. J. Antimicrob. Chemother. 59:1261–1264. 10.1093/jac/dkl380. [DOI] [PubMed] [Google Scholar]
- 42.Bohnert JA, Schuster S, Szymaniak-Vits M, Kern WV. 2011. Determination of real-time efflux phenotypes in Escherichia coli AcrB binding pocket phenylalanine mutants using a 1,2′-dinaphthylamine efflux assay. PLoS One 6:e21196. 10.1371/journal.pone.0021196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH. 2013. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 41:D348–D352. 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schulz R, Vargiu AV, Collu F, Kleinekathöfer U, Ruggerone P. 2010. Functional rotation of the transporter AcrB: insights into drug extrusion from simulations. PLoS Comput. Biol. 6:e1000806. 10.1371/journal.pcbi.1000806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lamers RP, Cavallari JF, Burrows LL. 2013. The efflux inhibitor phenylalanine-arginine beta-naphthylamide (PAβN) permeabilizes the outer membrane of gram-negative bacteria. PLoS One 8:e60666. 10.1371/journal.pone.0060666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ryall B, Eydallin G, Ferenci T. 2012. Culture history and population heterogeneity as determinants of bacterial adaptation: the adaptomics of a single environmental transition. Microbiol. Mol. Biol. Rev. 76:597–625. 10.1128/MMBR.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, Gottesman MM. 2007. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315:525–528. 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.