

Abstract
Pseudomonas aeruginosa is a major pathogen in cystic fibrosis (CF) lung disease. Children with CF are routinely exposed to P. aeruginosa from the natural environment, and by adulthood, 80% of patients are chronically infected. P. aeruginosa in the CF airway exhibits a unique biofilm-like structure, where it grows in small clusters or aggregates of bacteria in association with abundant polymers of neutrophil-derived components F-actin and DNA, among other components. These aggregates differ substantially in size and appearance compared to surface-attached in vitro biofilm models classically utilized for studies but are believed to share properties of surface-attached biofilms, including antibiotic resistance. However, little is known about the formation and function of surface-independent modes of biofilm growth, how they might be eradicated, and quorum sensing communication. To address these issues, we developed a novel in vitro model of P. aeruginosa aggregates incorporating human neutrophil-derived products. Aggregates grown in vitro and those found in CF patients' sputum samples were morphologically similar; viable bacteria were distributed in small pockets throughout the aggregate. The lasA quorum sensing gene was differentially expressed in the presence of neutrophil products. Importantly, aggregates formed in the presence of neutrophils acquired resistance to tobramycin, which was lost when the aggregates were dispersed with DNase, and antagonism of tobramycin and azithromycin was observed. This novel yet simple in vitro system advances our ability to model infection of the CF airway and will be an important tool to study virulence and test alternative eradication strategies against P. aeruginosa.
INTRODUCTION
From the time of early childhood, cystic fibrosis (CF) lung disease features increased inflammation and persistent neutrophil accumulation in the airways, independent of detectable infection (1). Pseudomonas aeruginosa from the environment routinely enters this neutrophil-rich milieu, intensifying the inflammatory response. During childhood, P. aeruginosa infections are often transient, with aggressive antibiotic treatment leading to eradication (2–4), but by adulthood nearly 80% of CF patients are chronically infected with P. aeruginosa (5). Central to the development of persistent infection is the formation of a biofilm, a well-recognized virulence mechanism that renders bacteria essentially impervious to host immune response or antibiotic treatment. Bacterial biofilms are traditionally defined as surface-attached communities of cells encased within a self-produced extracellular polysaccharide matrix. P. aeruginosa in the CF airway appears to exist primarily in a biofilm-like mode of growth (6), a conclusion supported by the inability of antibiotics and host defense mechanisms to eradicate the infection and the detection of quorum sensing signals (6–8).
It is now understood that P. aeruginosa biofilms in the CF airway possess a unique structure (6). Instead of surface-attached communities characteristic of most biofilm infection, the CF airway biofilm is comprised of small clusters or aggregates of P. aeruginosa and contains dead and dying neutrophils (6, 26). These aggregates appear to share many properties with conventional surface-attached biofilms, including resistance to antibiotics and the host immune system and dependence on quorum sensing (6). However, surface attachment of bacterial microcolonies to the bronchial wall is not observed in vivo (11, 12). Instead, P. aeruginosa growth in the CF airway occurs in the context of stagnant mucous plugs, which are lodged in the airway lumen (6, 11). The exuberance of the immune response results in excessive neutrophil accumulation that can overwhelm the capacity of the host to clear neutrophils from the tissue, resulting in sputum that is highly enriched in neutrophil-derived products implicated in CF lung pathogenesis (1, 14–19). Neutrophil survival in this setting is limited to just a few hours as a consequence of apoptosis and necrosis (20, 21). Previously, we have reported that necrotic neutrophils greatly accelerate P. aeruginosa biofilm formation (22–24); such neutrophils release DNA and F-actin, which polymerize via histones and cations though electrostatic attraction (25). We have found that P. aeruginosa uses these biopolymers as a scaffold for the formation of biofilms, taking the place of the “surface” that is traditionally thought to be required for biofilm development.
Nearly all studies of Pseudomonas biofilm formation and function have utilized in vitro biofilms that bear little resemblance to those observed in vivo (26). In such models, biofilms are grown on abiotic surfaces, typically producing biomass orders of magnitude larger than biofilms recovered from the CF lung. Moreover, such in vitro biofilms are generally devoid of airway components. While likely relevant to environmental or industrial applications, the remarkable surface-attached structures developed during the life cycle of in vitro biofilms in CF have never been observed in vivo; thus, the potential exists that, in some cases, conclusions drawn from these model systems may not be applicable to human infection (26).
We hypothesized that the presence of intracellular products from necrotic neutrophils enhances surface-independent aggregation of P. aeruginosa and that these aggregates display properties required for virulence in the CF airway. Herein, we describe an in vitro model system for the rapid formation of CF airway-like biofilms, using a laboratory strain of P. aeruginosa (PAO1) and also an isogenic pair of clinical strains isolated at different times in the course of CF airway infection. The model that we developed incorporates neutrophil-derived products in a surface-independent manner, similar to biofilms observed in the CF airway, allowing for more biologically relevant analysis of P. aeruginosa virulence and treatment response. Using this model, we sought to understand whether the presence of neutrophils would selectively promote the formation of aggregates morphologically similar to those recovered from the CF airway. We also sought to examine the properties of such aggregates with respect to antibiotic resistance and quorum sensing signaling and their dependence on neutrophil lysates.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
P. aeruginosa strains used were PAO1 (laboratory-adapted strain kindly provided by M. Vasil, University of Colorado, Denver, CO, and E. P. Greenberg, University of Washington, Seattle, WA) and PAO1 containing pMF230 (kindly provided by M. Franklin, Montana State University, Bozeman, MT), which expresses green fluorescent protein (PAO1-GFP) (22). Paired, isogenic isolates recovered from a CF patient at initial infection (AMT0027-1) and 12 years later (AMT0027-11) (27) were from Seattle Children's Research Institute. The early isolate has a nonmucoid phenotype, while the late isolate is mucoid. Planktonic cultures of the late strain are generally less sensitive to antibiotics, but both strains are equally sensitive to killing by the antipseudomonal antibiotic tobramycin and have equivalent lag phases and rates of growth to midpoint log phase (27). All bacterial stocks were stored in aliquots at −80°C. To obtain planktonic cultures, bacterial stocks were thawed at room temperature and cultured overnight in complete RPMI (RPMI 1640 medium [Cellgro] supplemented with l-glutamine and 2% pooled heat-inactivated platelet-poor human plasma [27]) at 37°C with moderate shaking. Complete RPMI was chosen as a medium more compatible with the host cell environment. Cultures were then adjusted to an optical density at 650 nm of 0.3 in a Beckman DU 640 spectrophotometer. CFU were quantified by plating serial dilutions on LB agar medium plates.
Neutrophil isolation.
Neutrophils were isolated from healthy human volunteers by the plasma Percoll method as previously described (28). This method provides a >95% pure, minimally activated population; furthermore, expression of CSF1R is negligible, indicating minimal contamination with monocytes. Neutrophils were washed and resuspended in Krebs-Ringer phosphate-buffered saline (PBS) with dextrose (154 mM NaCl, 5.6 mM KCl, 1.1 mM MgSO4, 2.2 mM CaCl, 0.85 mM NaH2PO4, 2.15 mM Na2HPO4, and 0.2% dextrose).
Human subjects.
The National Jewish Health Institutional Review Board approved these studies, and written informed consent approved by the National Jewish Health Institutional Review Board was obtained from all healthy neutrophil and CF sputum sample donors. The study was conducted in accordance with the Declaration of Helsinki.
Formation of bacterial aggregates in neutrophil lysates.
All experiments were performed in complete RPMI. Neutrophils were dispensed into 5-ml round-bottom polypropylene tubes (BD Falcon) at a concentration of 16.6 × 106/ml and were stored in individual aliquots at −80°C to promote lysis for up to 2 months without apparent change in aggregate-forming ability. Neutrophil lysates were thawed at 37°C immediately prior to the experiment. Bacteria were added to a final ratio of 5 to every 1 neutrophil equivalent. Aggregates were formed for 21 to 48 h at 37°C with oscillation set at 50 per min. Planktonic cultures were prepared in complete RPMI, and initial inoculum at time of treatment was 1 × 106/ml. Planktonic cultures and biofilms were treated with antibiotics and/or DNase (Pulmozyme; Genentech) for 90 min at 37°C, as indicated. In all cases, these agents were the same pharmaceuticals used in the clinical care of CF patients. Antibiotic concentrations tested in the biofilm and planktonic samples were 100 μg/ml tobramycin (TOBI; Novartis) and 20 μg/ml azithromycin (APP Pharmaceuticals), which are concentration ranges observed in CF sputum (29, 30). DNase concentration tested was 30 μg/ml. Antibiotics and/or DNase were removed by the washing of samples three times with 0.9% saline and centrifugation at 3,600 × g for 8 min. To determine viable bacteria, aggregates were disrupted by the addition of 0.01% Triton X-100 and three passes through a 25-gauge needle. Disruption of aggregates was confirmed by microscopy. Following serial dilution, bacteria were plated on LB plates for enumeration of colonies. Planktonic bacteria controls without neutrophils were treated in an identical manner, with the same antibiotics and DNase concentrations.
Sputum processing.
After collection, sputum samples were weighed in a tared collection cup, and each sputum sample was separated into two different tubes for use in RNA isolation and microscopy. Sputum processed for RNA isolation was weighed in a microcentrifuge tube, diluted with 2 volumes of PBS, and disrupted using 0.5-mm zirconium oxide beads in a Bullet Blender homogenizer (NextAdvance) at speed 1 for at least 1 min until homogenized. After centrifugation at 20,000 × g for 5 min at 4°C, the pellet was lysed in TriPure reagent (Roche) and processed for RNA isolation as described below.
Tubes containing sputum for microscopy were homogenized in PBS and 10% Sputolysin solution for 15 min at 37°C, following the CF Foundation Therapeutic Development Network standard operational procedure (31). Samples were then centrifuged in the StatSpin CytoFuge 2 at 850 rpm for 6 min and air dried.
Gram stain and bright-field microscopy.
To compare the morphology of aggregates recovered in sputum samples with bacterial aggregates formed in vitro, slides prepared by cytocentrifugation were analyzed by bright-field microscopy. Cytospin slides were made of planktonic bacteria and aggregates grown in the presence or absence of dead neutrophils. Each experiment was performed both with PAO1 and the isogenic early and late clinical isolates. Slides were prepared as described above by diluting each sample 2-fold and adding 200 μl of the dilution to each cuvette.
Gram stain was performed using the Gram safranin kit (BD) by following the manufacturer's protocol. Stained cytospins were examined using an Olympus BX51 microscope with a 60× oil immersion objective and photographed using Olympus DP controller software. A total of three images were taken from each slide; the locations on the slides were first selected randomly (left and right corners and center), and each location was kept consistent for each slide. Aggregate number and mean cross-sectional area were quantified using ImageJ software (32). Clusters were defined as larger than 2 μm2, the approximate size of planktonic bacteria.
RNA isolation and quantitative PCR of quorum sensing genes.
Planktonic P. aeruginosa and aggregates of P. aeruginosa grown in the presence or absence of lysed PMNs for 48 h were pelleted and solubilized in 1 ml TriPure reagent at 95°C for 5 min. Total RNA was isolated according to the manufacturer. cDNA was prepared from 1 μg RNA using Moloney murine leukemia virus reverse transcriptase (MMLV-RT; Invitrogen) and random hexamers. Quantitative PCR was performed using Sybr green PCR master mix (Applied Biosystems) and the following primers (IDT): lasA forward (5′-GGTCTGTTGCGAGAGGGGGC-3′) and reverse (5′-GGTTGCTCGGCTGCAGGAGT-3′), rhlA forward (5′-GTAGTCGAGCATCGCCTGGTTC-3′) and reverse (5′-CGAGGTCAATCACCTGGTCTCC-3′), ureB forward (5′-CCCGGCGATATCGAACTCAA-3′) and reverse (5′-GTGGTAGTGCGAGCCGACCT-3′), rpoD forward (5′-ATCCTGCGCAACCAGCAGAA-3′) and reverse (5′-GGGAGAAGGCTGCTCGTCGG-3′). Relative gene expression was determined by the threshold cycle (ΔCT) method using rpoD as the normalization gene (33). lasA and rhlA genes are quorum sensing genes directly regulated by lasR and rhlR, respectively, and sequentially expressed in early biofilm formation (34); ureB encodes a urease that is robustly upregulated in surface-attached biofilms (35) and appears critical for maintenance of pH homeostasis in biofilm cultures (36). Sputum Pseudomonas infection was confirmed using gyrB expression (37).
Confocal microscopy.
Aggregates of PAO1-GFP were assessed using confocal microscopy to visually confirm viability. Briefly, planktonic bacteria and aggregates grown for 48 h were incubated with propidium iodide for 15 min at room temperature. Samples were washed 3 times with 0.9% saline and fixed with 1.3% glutaraldehyde for 10 min at room temperature. Samples were then washed once, resuspended in saline, and transferred to a glass-bottom culture dish (MatTek Corp., United States) for visualization using an Axiovert 200M microscope (Carl Zeiss) for Cy3 (red filter for propidium iodide) and fluorescein isothiocyanate (FITC) (green filter for GFP) fluorescence. Images were obtained using 10× and 40× objectives.
Statistical analysis.
Data were analyzed using GraphPad Prism (version 6.02). Data of aggregate size obtained from microscopy, CFU, and gene expression were tested for normality using the D'Agostino-Pearson omnibus normality test and the Shapiro-Wilk normality test. One-way analysis of variance (ANOVA) was performed on normal data when making multiple comparisons for normally distributed data. The Kruskal-Wallis test was used to determine significance of more than two conditions for nonparametric data; Dunn's multiple-comparison test was utilized as noted. The Mann-Whitney test was used to determine significance of two conditions for unpaired, nonparametric data, and the Wilcoxon signed-rank test was used for testing paired, nonparametric data. Significance was set at a P value of <0.05.
RESULTS
Morphology of neutrophil-induced P. aeruginosa aggregates.
Previous reports have documented that P. aeruginosa biofilms recovered from the CF airway occur as aggregates of bacteria (9, 11, 12, 26, 38). In an attempt to form aggregates of P. aeruginosa in a CF-like environment, we grew bacteria in the presence of lysed neutrophils. Aggregates ranged in size up to 600 μm2 and were accompanied by planktonic bacteria. The aggregates appeared to include bacteria and other material likely originating from dead neutrophils (Fig. 1A), which contribute F-actin and DNA to the extracellular polymeric matrix (22, 24). We compared P. aeruginosa aggregates formed in vitro in the presence of neutrophil lysates to aggregates observed routinely from sputum samples recovered from CF patients infected with P. aeruginosa (Fig. 1B). Aggregates of P. aeruginosa strain PAO1 formed in the presence of neutrophil products were visually similar to aggregates found in sputum samples and to those previously reported (11). The CF sputum samples also contained similar aggregates of bacteria surrounded by extracellular material, cellular fragments, and intact neutrophils (Fig. 1B). In comparison, the initial inoculum of planktonic PAO1 showed little aggregation (Fig. 1C) and differed from lysed neutrophils (Fig. 1D).
FIG 1.

Morphology of P. aeruginosa aggregates. (A) Aggregation of strain PAO1 in vitro when combined with neutrophil debris for 48 h, demonstrating incorporation of extracellular material into the aggregate structure. (B) Representative sputum sample from 5 independent samples from a CF patient confirmed to be infected with P. aeruginosa. Intact neutrophils are apparent within the P. aeruginosa aggregate. (C) Planktonic culture of P. aeruginosa strain PAO1. (D) Neutrophil debris prepared by freeze-thawing, in the absence of bacteria. Samples were Gram stained and viewed with a 60× oil objective; length of size bar, 30 μm.
To investigate the distribution of living bacteria within in vitro aggregates, PAO1-GFP was combined with neutrophil lysates for 48 h. Aggregates consisted mostly of dead neutrophils (data not shown), extracellular DNA, and dead bacteria (stained red), with the majority of the live bacteria (green) being in the aggregate interior (Fig. 2A to C). The pattern of small pockets of viable cells enmeshed within fields of dead bacteria is consistent with the described organization of some CF strains of P. aeruginosa grown as microcolonies on a glass surface, although considerable variation in the pattern of distribution of live and dead cells is reported between various clinical isolates (39). The addition of DNase resulted in dispersion of aggregates into planktonic cells (Fig. 2D to F), indicating the contribution of extracellular DNA to this aggregation.
FIG 2.

Viable bacteria are present within P. aeruginosa aggregates formed in the presence of lysed neutrophils. Strain PAO1-GFP was combined with neutrophil lysates for 48 h. Samples were stained with propidium iodide to distinguish nonviable bacteria. (A and D) Viable bacteria (green) occur in clusters within a larger aggregate (A) but are dispersed by DNase treatment (D). (B and E) Nonviable bacteria and extracellular DNA identified by propidium iodide (red) in aggregates (B); treatment with DNase reduces propidium iodide staining (E). (C and F) Overlay of red fluorescent and green fluorescence bacteria. (D to F) Samples treated with DNase, resulting in near complete disruption of the aggregates. Viewed with a 40× objective; length of size bar, 10 μm. Representative images from 9 independent experiments.
Contribution of neutrophil products on the size of P. aeruginosa aggregates formed in vitro.
Sputum from CF airways has P. aeruginosa aggregates that range in diameter from 5 to 100 μm (9, 11, 12, 26, 38). P. aeruginosa in early stationary phase is comprised primarily of planktonic single-cell bacteria or small clusters of 2 or 3 cells. PAO1 and an isogenic pair of clinical isolates recovered from a CF patient soon after initial infection and 12 years later were compared. Mean cross-sectional areas of bacteria in these initial planktonic cultures range from 1.4 to 2.1 μm2 (Fig. 3A to C). To quantify the effect of neutrophil lysates on P. aeruginosa aggregation, we compared the size distributions of aggregates (≥2.0 μm2) formed in the presence and absence of neutrophils for 48 h. Following 21 h, single-cell bacteria remained predominant (not shown). All strains tested were capable of forming aggregates in either the presence or absence of dead neutrophils after 48 h; however, in the presence of neutrophil products, PAO1 and early CF isolates formed significantly larger aggregates than in the absence of neutrophil lysates (Fig. 3A and B). The presence of neutrophils did not induce larger aggregates in the late CF isolate (Fig. 3C). Compared to small aggregates, large aggregates may have a greater potential to occlude small airways, may represent more developed colonies, and may present a greater barrier to antibiotics; therefore, the largest-sized aggregates were considered by comparing the upper quartile under each condition tested. The effect of neutrophil products on inducing large P. aeruginosa aggregates was apparent for all strains tested, including the late CF isolate (Fig. 3D to F). These data suggest that neutrophil products promote the formation of larger bacterial aggregates.
FIG 3.

Distribution of aggregate size in the presence and absence of neutrophil lysates. Size of aggregates (μm2) plotted on a log2 scale formed within the initial inoculum (○) and aggregates formed after 48 h in the absence (△) or presence (▽) of neutrophil products. (A and D) Strain PAO1; (B and E) early CF strain; (C and F) isogenic late CF strain; (D to F) larger aggregates were measured in the top quartile of aggregate size. Red bars represent the median for each condition. Analysis of variance performed for each strain. ****, P < 00001; *, P < 0.05, by Kruskal-Wallis test.
Neutrophil-dependent aggregates do not confer a growth advantage.
To quantify the effect of neutrophil lysates on bacterial growth, aggregates formed after 48 h in the presence or absence of neutrophil lysates were disrupted, and the quantities of viable bacteria were compared (Fig. 4). The presence of neutrophil products did not enhance bacterial number at 48 h for any strain tested, demonstrating that the larger aggregate size achieved in the presence of neutrophils was not due to increased bacterial numbers.
FIG 4.
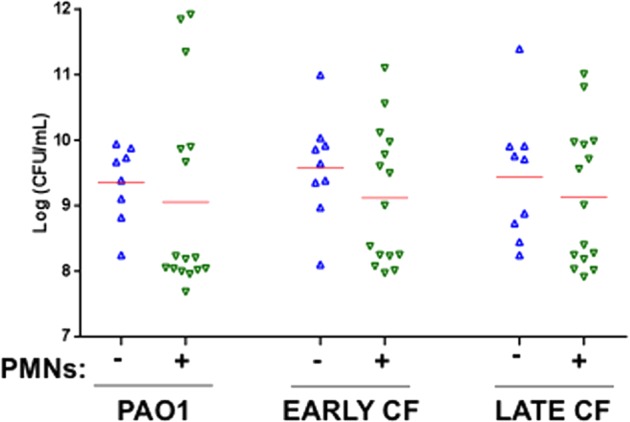
P. aeruginosa growth is not altered by the presence of neutrophil lysates. Viable bacteria after 48 h in the absence (△) or presence (▽) of neutrophil (PMN) products for PAO1 and early and late CF isolates. Aggregates were disrupted, and bacteria were quantified by colony counts following serial dilution and plating. Initial inoculum was approximately 5 × 105 CFU. Red bars represent the median for each condition, which were not different by Kruskal-Wallis test.
Quorum sensing signaling by P. aeruginosa aggregates.
Quorum sensing is a mechanism that coordinates gene expression according to the density of bacteria in a local population; its activation is indicative for the biofilm phenotype. Expression was tested for three quorum sensing-regulated genes involved in various stages of the development of surface-attached biofilms. Activation of the Las quorum sensing system, which is associated with production of N-(3-oxododecanoyl)-l-homoserine lactone, has been linked to the stage of biofilm development when motile bacteria become irreversibly attached to a surface and small clusters of bacteria are formed (34). In the presence of neutrophil products, lasA was significantly upregulated in the clinical strains but not in environmental laboratory strain PAO1 (Fig. 5A). lasA was also upregulated in sputum samples from P. aeruginosa-infected CF patients compared to that in PAO1; interestingly, a small population of individuals harbor strains with high levels of lasA expression.
FIG 5.
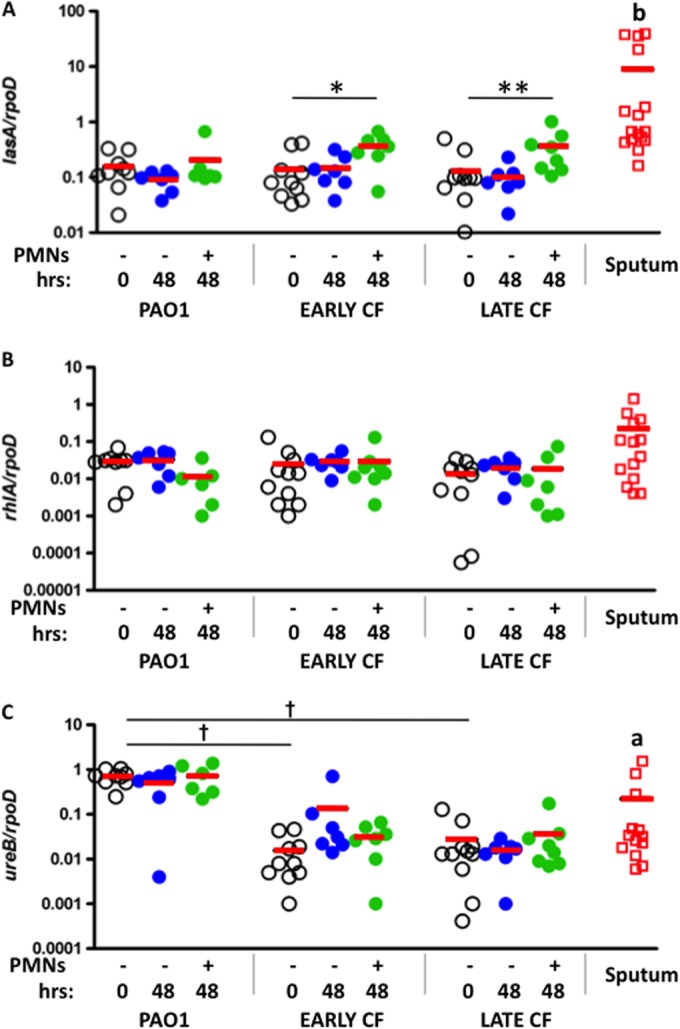
Quorum sensing gene expression in aggregates and sputum. Expression of genes involved in quorum sensing in PAO1. Early and late CF isolates of planktonic cultures (open circles) were compared to P. aeruginosa grown for 48 h in the absence (blue circles) or presence (green circles) of neutrophil products and sputum samples from CF patients (red squares). Solid lines represent the mean. (A) Expression of lasA normalized to expression of rpoD; (B) expression of rhlA normalized to expression of rpoD; (C) expression of ureB normalized to expression of rpoD. *, P < 0.05; **, P < 0.01, by Wilcoxon signed-rank test. †, P < 0.0001, by unpaired t test comparing planktonic bacteria. a, P < 0.05; b, P < 0.01, by Mann-Whitney test for sputum samples compared to planktonic PAO1.
Activation of the Rhl quorum sensing system is associated with production of N-butanoyl-l-homoserine lactone. It has been linked to surface-attached biofilm development and integrity, when cell clusters become progressively layered, and also to rhamnolipid synthesis (34, 40). In all 3 strains tested, rhlA was not changed in the absence or presence of neutrophil products (Fig. 5B). The expression of rhlA was not significantly upregulated in the sputum samples.
Expression of ureB, a gene encoding a urease that is induced in surface-attached biofilms and appears critical for pH homeostasis in biofilm cultures, was also analyzed (35, 36). Expression of ureB was not altered after 48 h for any strain, in the presence or absence of neutrophil products. However, planktonic expression of ureB was noted to be approximately 100-fold higher in PAO1 than in the isogenic CF strains and selective sputum samples (Fig. 5C). Similar to the CF isolates, sputum samples expressed lower levels of ureB than PAO1; although a subpopulation of patients' samples expressed levels similar to those of the environmental strain, four samples had no detectable ureB.
Effects of in vivo aggregation on P. aeruginosa resistance to tobramycin.
Antibiotic resistance is a clinically important feature evoked by biofilm formation (8, 41, 42). To determine if P. aeruginosa aggregates acquire relative antibiotic resistance, planktonic cultures and aggregates formed in the presence of neutrophil products were exposed to tobramycin for 90 min. As expected, exposure of planktonic bacteria to tobramycin resulted in >98% killing of all strains tested (Fig. 6). P. aeruginosa aggregates formed from the clinical isolates demonstrated greater survival than PAO1 following exposure to the antibiotic, with the largest resistance achieved by the early CF strain (Fig. 6). No enhanced killing by tobramycin was achieved by the addition of DNase to planktonic bacteria and the PAO1 aggregates (Fig. 6). In contrast, the antibiotic property of tobramycin was improved following DNase treatment of aggregates of CF clinical strains formed in the presence of neutrophil products (Fig. 6). Exposure of the aggregates to DNase resulted in near complete disruption of these structures (Fig. 2), corroborating the ability of the aggregate structure to confer antibiotic resistance.
FIG 6.

Aggregate structure allows for antibiotic resistance to tobramycin. (A) Planktonic bacterial survival when exposed to tobramycin (TOB; open bars) and tobramycin and DNase (hatched bars). (B) Survival of P. aeruginosa aggregates formed in the presence of neutrophil products and exposed to tobramycin (open bars) and tobramycin and DNase (hatched bars). Survival (%) was calculated by dividing the CFU of each treatment by the CFU of an untreated sample. The means ± standard errors of the means (SEM) are depicted from 8 independent experiments. Mann-Whitney t test between aggregates treated with tobramycin and those treated with tobramycin plus DNase. *, P < 0.05.
Azithromycin induces antagonism against antibiotic effects of tobramycin in P. aeruginosa aggregates.
Azithromycin has recently been found to antagonize the antipseudomonal activity of tobramycin in abiotic biofilm and mouse infection models (43–46). Therefore, we tested P. aeruginosa planktonic cultures and neutrophil-enhanced aggregates for this drug interaction. In planktonic cultures, tobramycin had profound antipseudomonal activity for all strains tested (Fig. 7A), whereas azithromycin was significantly effective only against the early CF strain. However, no antagonism was observed when planktonic cultures were treated with both antibiotics. When P. aeruginosa strains were grown in aggregates in the presence of neutrophils, azithromycin had a significant antibiotic effect against PAO1 aggregates but not against aggregates formed by the clinical isolates (Fig. 7B). For aggregates of the early CF strain, azithromycin significantly decreased the antipseudomonal activity of tobramycin (Fig. 7B). A similar trend was seen for the late CF strain. Therefore, growth of clinical strains of P. aeruginosa in biofilm-like conditions can promote antagonism between tobramycin and azithromycin.
FIG 7.

Antagonistic effect of azithromycin toward tobramycin-induced killing of P. aeruginosa aggregates. (A) Planktonic bacteria survival when exposed to tobramycin (TOB), azithromycin (AZM), or the combination. (B) Survival of P. aeruginosa aggregates formed for 48 h in the presence of neutrophil products from PAO1, and in the early and late CF strains, and exposed to tobramycin (TOB), azithromycin (AZM), or the combination. Survival (%) was calculated by dividing CFU from each treatment by CFU of cultures from the respective strain prior to antibiotic treatment. The means ± SEM are depicted from 4 to 8 independent experiments. Lines represent Dunn's multiple-comparison test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
Environmental strains of P. aeruginosa likely enter the airway routinely and are nonpathogenic for the normal host. In CF patients, the lung typically withstands chronic P. aeruginosa infection for years, and with modern eradication strategies, persistent P. aeruginosa infection can be delayed into late adolescence or adulthood (5). The mechanism(s) that allows a small inoculum of planktonic P. aeruginosa to survive in the CF airway and acquire the biofilm phenotype is not completely understood. Herein, we report that the presence of neutrophil products, a central feature of the CF airway, enhances P. aeruginosa aggregate formation. Neutrophil-induced P. aeruginosa aggregates formed in vitro are morphologically similar to those recovered from the CF airway. Moreover, aggregate formation confers antibiotic resistance and selective upregulation of quorum sensing signaling, two features essential to the enhanced virulence of the biofilm phenotype. These early biofilms are extremely sensitive to dispersion by DNase, which restored antibiotic sensitivity. Interestingly, these simple aggregates, but not planktonic cultures, demonstrate azithromycin-induced antagonism toward tobramycin, as previously reported in other P. aeruginosa biofilm systems and in vivo (43–46). Together, these findings support the usefulness of combining neutrophil products with P. aeruginosa to represent the biofilm phenotype within the CF airway more accurately.
A number of in vitro biofilm models have been described, providing the foundation of our current understanding of biofilm development and regulation. The static 96-well format can be used to test for biofilm growth by staining the biomass with crystal violet or for viability with LIVE/DEAD staining (47, 48). Other models use flow cells, growth on synthetic membranes, drip flow reactors, or rotating disk reactors (49–53). In many of these systems, biofilm structure can be visualized by microscopy, but throughput is limited and generally requires a surface (26). In addition, these models generally produce biofilms in the range of 1 cm2 in the microtiter plate assay to 10 cm2 in flow cells, and biofilm thickness can reach 300 μm (26). In contrast to the CF airway, where biofilms are lodged within stagnant mucous plugs of the small and medium-sized bronchi (11, 12), surface-attached biofilm models are commonly exposed to air or freshly supplied media. Moreover, adherence of bacterial microcolonies to the bronchial wall surface has never been observed in vivo (11, 12), making it unclear if surface-attached biofilm models accurately represent in vivo aggregates. Thus, although surface-attached in vitro systems have been instrumental in testing many biofilm characteristics, they differ considerably from the growth conditions pathogens actually confront in the CF airway.
Strains of P. aeruginosa can aggregate to various degrees in the absence of neutrophil products (54). However, products released from necrotic neutrophils enhance biofilm development, an advantage that arises in part through neutrophil-derived F-actin and DNA (22–24). In addition to F-actin and DNA, incorporation of additional neutrophil products likely alters survival properties of P. aeruginosa (55, 56). The presence of neutrophils dramatically enhances biofilm mass in the first 24 to 48 h (22–24, 57), the time frame when the planktonic bacteria are generally most vulnerable to eradication by host defenses and antibiotic treatment. The early survival advantage afforded by the presence of dying neutrophils may explain why biofilm-associated P. aeruginosa infection typically occurs in the setting of intense preexisting inflammation, such as burns, wounds, corneal damage, and the CF airway. A comprehensive understanding of the initial formation of P. aeruginosa aggregates in the CF airway remains elusive. While it is recognized that environmental factors and available nutrients regulate many facets of biofilm development and metabolism, models that incorporate aspects of the host inflammatory response promise to enhance our understanding of this mode of growth.
Differences between the isogenic early and late CF strains utilized in this study support the conclusion that neutrophil products are particularly important in early biofilm development. The clinical strain recovered early from a child with CF displayed greater enhancement of aggregate size in the presence than in the absence of neutrophil products, as did PAO1, which resembles environmental strains with regard to its lack of active virulence factor expression. In contrast, the isogenic strain recovered 12 years later from the same CF patient formed aggregates similarly in the absence or presence of neutrophil products. Importantly, the sizes of the aggregates are similar to those seen in CF sputum (12). Recently, Staudinger et al. (56) demonstrated P. aeruginosa aggregate formation on solid medium in the presence of CF sputum supernatants and attributed aggregate formation to loss of bacterial motility and cleavage of flagellin by human neutrophil elastase; aggregate formation of late CF isolates is also associated with greater selective pressure and intrinsic motility defects (56, 58). These investigators also delineate between biofilm formation, for which late, mucoid strains are deficient, and aggregate formation, for which late strains are proficient (56); therefore, our data similarly support a role of mucoid strains to allow aggregate formation in CF-like conditions that is distinct from surface-attached biofilm formation.
Upregulation of quorum sensing-related genes suggests that aggregate formation shares some features of surface-attached biofilms. In our system, lasA induction for the CF isolates was observed only in the presence of neutrophil products. It is unclear if lasA, which codes for an elastase, promotes aggregate formation in a manner similar to that observed for human neutrophil elastase (56). The finding that all strains tested formed aggregates to some extent in the absence of neutrophil products indicates that lasA induction is not required for aggregate formation but may enhance it.
In contrast to lasA, rhlA was not induced under any conditions, suggesting that quorum sensing signaling in CF-like conditions is specific for the Las system. It is possible that sampling at different times would uncover changes in rhlA expression. Although a global analysis of the longitudinal effect of neutrophil products on biofilm regulation extends beyond the scope of the current report, significant differences in quorum sensing genes suggest a potentially broad range of functional consequences. While aggregate formation in other systems may not require biofilm-associated genes (56, 59–61), the same pattern of dependence on the Las system, but not the Rhl system, is observed in microcolony formation in artificial CF sputum that contains DNA (62). In addition, quorum sensing genes are expressed in CF sputum samples at levels similar to those seen in the presence of lysed neutrophils (Fig. 5). The functional consequences of deficiency of ureB expression apparent in the CF isolates and the majority of CF sputum samples are not known but worthy of future study; although mutations in the primer regions could result in an apparent decrease in ureB expression, a survey of P. aeruginosa genomes indicated no mismatches in these regions. Furthermore, these findings are consistent with the previous observation that quorum sensing signals are observed in CF sputum (6). Overall, these data support the conclusion that quorum sensing plays a role in aggregate formation in the CF environment.
Finally, the mechanism of DNase has been attributed to enhanced airway sputum clearance (63), without an effect on availability of sputum-bound antibiotics (64). However, our findings illustrate the potential of DNase to augment antibiotic activity by disrupting P. aeruginosa aggregates within the airways. These results indicate a more direct mechanism through which DNase contributes to antimicrobial activity and thus may be considered an additional component to enhance treatment regimens designed to eradicate or suppress P. aeruginosa infection in the CF airway.
While any one in vitro system is not likely to recreate the complexity of the lung environment, our data suggest that incorporation of host products into the biofilm is an important step toward obtaining a more sophisticated understanding of the early pathogenesis of infection. As with any model, limitations exist. Bacterial aggregate formation occurs in a largely anaerobic environment, which may only partially mimic the CF airway. Other host factors, such as mucin and surfactant, may modify aggregate formation. The use of frozen aliquots of neutrophils greatly simplifies the model but does not accurately depict the constant influx of live neutrophils into the infected airway. Furthermore, this study utilized a single isogenic pair of clinical strains, although this model is adaptable for comparative studies of bacterial virulence between isolates. We developed this model using peripheral neutrophils from healthy adults. Although our data are consistent with the importance of DNA in establishing aggregates, it is possible that unique properties of CF airway neutrophils from children and adults differently model in vivo aggregate formation; however, we have shown that neutrophils from CF patients and healthy subjects release neutrophil extracellular traps equally well (27). In addition, in vitro antibiotic susceptibility testing and morphological and molecular analyses of bacterial responses can be examined. Furthermore, other components of the immune response, such as viable host cells, antibodies, and other host factors, can be tested for modification of aggregate formation, structure, and bacterial responses.
With the current model, surface-free aggregates are easily formed that morphologically resemble the CF airway biofilms and share functional properties recognized as central to biofilm survival. Sputum samples showed morphology and quorum sensing signaling similar to those of aggregates formed in vitro in the presence of neutrophils, supporting the clinical relevance of this model of biofilm growth. Stored aliquots of neutrophils are sufficient for this model, precluding neutrophil isolation at the time of the experiment. The neutrophil-dependent aggregate model presented here and a model of artificial CF sputum share a dependency on DNA and a role for the Las quorum sensing system (62); therefore, models that incorporate in vivo growth components may provide a way to understand the biology of early P. aeruginosa growth and therapeutic strategies. In summary, these studies of surface-independent P. aeruginosa aggregates of the appropriate size, formed through incorporation of the principal components of the CF airway, represent a substantial advance toward defining clinically relevant mechanisms of early biofilm formation.
ACKNOWLEDGMENTS
Support for this work was provided by an Investigator Support Trial grant from Genentech and the Rebecca Runyon Bryan Chair for Cystic Fibrosis (J.A.N.) and NIH HL34303 (D.L.B.).
Footnotes
Published ahead of print 2 September 2014
REFERENCES
- 1.Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. 1995. Early pulmonary inflammation in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 151:1075–1082. 10.1164/ajrccm.151.4.7697234. [DOI] [PubMed] [Google Scholar]
- 2.Burns JL, Gibson RL, McNamara S, Yim D, Emerson J, Rosenfeld M, Hiatt P, McCoy K, Castile R, Smith AL, Ramsey BW. 2001. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J. Infect. Dis. 183:444–452. 10.1086/318075. [DOI] [PubMed] [Google Scholar]
- 3.Rosenfeld M, Gibson RL, McNamara S, Emerson J, Burns JL, Castile R, Hiatt P, McCoy K, Wilson CB, Inglis A, Smith A, Martin TR, Ramsey BW. 2001. Early pulmonary infection, inflammation, and clinical outcomes in infants with cystic fibrosis. Pediatr. Pulmonol. 32:356–366. 10.1002/ppul.1144. [DOI] [PubMed] [Google Scholar]
- 4.Frederiksen B, Koch SLC, Hoiby N. 1996. Improved survival in the Danish center-treated CF patients: results of aggressive treatment. Pediatr. Pulmonol. 21:153–158. . [DOI] [PubMed] [Google Scholar]
- 5.Cystic Fibrosis Foundation. 2013. Cystic Fibrosis Foundation patient registry. 2012 annual data report to the center directors Cystic Fibrosis Foundation, Bethesda, MD. [Google Scholar]
- 6.Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, Greenberg EP. 2000. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407:762–764. 10.1038/35037627. [DOI] [PubMed] [Google Scholar]
- 7.Aaron SD, Ferris W, Ramotar K, Vandemheen K, Chan F, Saginur R. 2002. Single and combination antibiotic susceptibilities of planktonic, adherent, and biofilm-grown Pseudomonas aeruginosa isolates cultured from sputa of adults with cystic fibrosis. J. Clin. Microbiol. 40:4172–4179. 10.1128/JCM.40.11.4172-4179.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drenkard E, Ausubel FM. 2002. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 416:740–743. 10.1038/416740a. [DOI] [PubMed] [Google Scholar]
- 9.Reference deleted.
- 10.Lam J, Chan R, Lam K, Costerton JW. 1980. Production of mucoid microcolonies by Pseudomonas aeruginosa within infected lungs in cystic fibrosis. Infect. Immun. 28:546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bjarnsholt T, Jensen PO, Fiandaca MJ, Pedersen J, Hansen CR, Andersen CB, Pressler T, Givskov M, Hoiby N. 2009. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr. Pulmonol. 44:547–558. 10.1002/ppul.21011. [DOI] [PubMed] [Google Scholar]
- 12.Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, Birrer P, Bellon G, Berger J, Weiss T, Botzenhart K, Yankaskas JR, Randell S, Boucher RC, Doring G. 2002. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J. Clin. Invest. 109:317–325. 10.1172/JCI13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reference deleted.
- 14.Perks B, Shute JK. 2000. DNA and actin bind and inhibit interleukin-8 function in cystic fibrosis sputa: in vitro effects of mucolytics. Am. J. Respir. Crit. Care Med. 162:1767–1772. 10.1164/ajrccm.162.5.9908107. [DOI] [PubMed] [Google Scholar]
- 15.Sheils CA, Kas J, Travassos W, Allem PG, Janmey PA, Wohl ME, Stossel TP. 1996. Actin filaments mediate DNA fiber formation in chronic inflammatory airway disease. Am. J. Pathol. 148:919–927. [PMC free article] [PubMed] [Google Scholar]
- 16.Lethem MI, James SL, Marriott C, Burke JF. 1990. The origin of DNA associated with mucus glycoproteins in cystic fibrosis sputum. Eur. Respir. J. 3:19–23. [PubMed] [Google Scholar]
- 17.Roum JH, Buhl R, McElvaney NG, Borok Z, Crystal RG. 1993. Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. 75:2419–2424. [DOI] [PubMed] [Google Scholar]
- 18.Balfour-Lynn IM. 1999. The protease-antiprotease battle in the cystic fibrosis lung. J. R Soc. Med. 92(Suppl 37):S23–S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sagel SD, Sontag MK, Accurso FJ. 2009. Relationship between antimicrobial proteins and airway inflammation and infection in cystic fibrosis. Pediatr. Pulmonol. 44:402–409. 10.1002/ppul.21028. [DOI] [PubMed] [Google Scholar]
- 20.Lee A, Whyte MKB, Haslett C. 1993. Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J. Leukocyte Biol. 54:283–288. [PubMed] [Google Scholar]
- 21.Edwards SW, Moulding DA, Derouet M, Moots RJ. 2003. Regulation of neutrophil apoptosis, p 204–224 In Cassatella MA. (ed), The neutrophil, vol 83 Karger, Berlin, Germany. [DOI] [PubMed] [Google Scholar]
- 22.Walker TS, Worthen GS, Poch KR, Lieber JG, Saavedra MT, Malcolm KC, Fessler MB, Vasil ML, Nick JA. 2005. Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infect. Immun. 73:3693–3701. 10.1128/IAI.73.6.3693-3701.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robertson DM, Parks QM, Young RL, Kret J, Poch KR, Malcolm KC, Nichols DP, Nichols M, Zhu M, Cavanagh HD, Nick JA. 2010. Disruption of contact lens-associated Pseudomonas aeruginosa biofilms formed in the presence of neutrophils. Invest. Ophthalmol. Vis. Sci. 52:2844–2850. 10.1167/iovs.10-6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parks QM, Young RL, Poch KR, Malcolm KC, Vasil ML, Nick JA. 2009. Neutrophil enhancement of Pseudomonas aeruginosa biofilm development: human F-actin and DNA as targets for therapy. J. Med. Microbiol. 58:492–502. 10.1099/jmm.0.005728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang JX, Wen Q, Bennett A, Kim B, Sheils CA, Bucki R, Janmey PA. 2005. Anionic poly(amino acid)s dissolve F-actin and DNA bundles, enhance DNase activity, and reduce the viscosity of cystic fibrosis sputum. Am. J. Physiol. Lung Cell. Mol. Physiol. 289:L599–L605. 10.1152/ajplung.00061.2005. [DOI] [PubMed] [Google Scholar]
- 26.Bjarnsholt T, Alhede M, Alhede M, Eickhardt-Sorensen SR, Moser C, Kuhl M, Jensen PO, Hoiby N. 2013. The in vivo biofilm. Trends Microbiol. 21:466–474. 10.1016/j.tim.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 27.Young RL, Malcolm KC, Kret JE, Caceres SM, Poch KR, Nichols DP, Taylor-Cousar JL, Saavedra MT, Randell SH, Vasil ML, Burns JL, Moskowitz SM, Nick JA. 2011. Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PLoS One 6:e23637. 10.1371/journal.pone.0023637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haslett C, Guthrie LA, Kopaniak M, Johnston RB, Jr, Henson PM. 1985. Modulation of multiple neutrophil functions by trace amounts of bacterial LPS and by preparative methods. Am. J. Pathol. 119:101–110. [PMC free article] [PubMed] [Google Scholar]
- 29.Ruddy J, Emerson J, Moss R, Genatossio A, McNamara S, Burns JL, Anderson G, Rosenfeld M. 2013. Sputum tobramycin concentrations in cystic fibrosis patients with repeated administration of inhaled tobramycin. J. Aerosol Med. Pulm. Drug Deliv. 26:69–75. 10.1089/jamp.2011.0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilms EB, Touw DJ, Heijerman HG. 2006. Pharmacokinetics of azithromycin in plasma, blood, polymorphonuclear neutrophils and sputum during long-term therapy in patients with cystic fibrosis. Ther. Drug Monit. 28:219–225. 10.1097/01.ftd.0000195617.69721.a5. [DOI] [PubMed] [Google Scholar]
- 31.Cystic Fibrosis Therapeutics Development Network Coordinating Center. 2013. Sputum processing for cytology and inflammatory markers: SOP 508. Cystic Fibrosis Therapeutics Development Network Coordinating Center, Bethesda, MD. [Google Scholar]
- 32.Rasband WS. 2012. ImageJ. U.S. National Institutes of Health, Bethesda, MD: http://imagej.nih.gov/ij/. [Google Scholar]
- 33.Savli H, Karadenizli A, Kolayli F, Gundes S, Ozbek U, Vahaboglu H. 2003. Expression stability of six housekeeping genes: a proposal for resistance gene quantification studies of Pseudomonas aeruginosa by real-time quantitative RT-PCR. J. Med. Microbiol. 52:403–408. 10.1099/jmm.0.05132-0. [DOI] [PubMed] [Google Scholar]
- 34.Sauer K, Camper AK, Ehrlich GD, Costerton JW, Davies DG. 2002. Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J. Bacteriol. 184:1140–1154. 10.1128/jb.184.4.1140-1154.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whiteley M, Bangera MG, Bumgarner RE, Parsek MR, Teitzel GM, Lory S, Greenberg EP. 2001. Gene expression in Pseudomonas aeruginosa biofilms. Nature 413:860–864. 10.1038/35101627. [DOI] [PubMed] [Google Scholar]
- 36.Musken M, Di Fiore S, Dotsch A, Fischer R, Haussler S. 2010. Genetic determinants of Pseudomonas aeruginosa biofilm establishment. Microbiology 156:431–441. 10.1099/mic.0.033290-0. [DOI] [PubMed] [Google Scholar]
- 37.Lee CS, Wetzel K, Buckley T, Wozniak D, Lee J. 2011. Rapid and sensitive detection of Pseudomonas aeruginosa in chlorinated water and aerosols targeting gyrB gene using real-time PCR. J. Appl. Microbiol. 111:893–903. 10.1111/j.1365-2672.2011.05107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baltimore RS, Christie CD, Smith GJ. 1989. Immunohistopathologic localization of Pseudomonas aeruginosa in lungs from patients with cystic fibrosis. Implications for the pathogenesis of progressive lung deterioration. Am. Rev. Resp. Dis. 140:1650–1661. [DOI] [PubMed] [Google Scholar]
- 39.Kirov SM, Webb JS, O'May CY, Reid DW, Woo JK, Rice SA, Kjelleberg S. 2007. Biofilm differentiation and dispersal in mucoid Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Microbiology 153:3264–3274. 10.1099/mic.0.2007/009092-0. [DOI] [PubMed] [Google Scholar]
- 40.Lequette Y, Greenberg EP. 2005. Timing and localization of rhamnolipid synthesis gene expression in Pseudomonas aeruginosa biofilms. J. Bacteriol. 187:37–44. 10.1128/JB.187.1.37-44.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stewart PS, Costerton JW. 2001. Antibiotic resistance of bacteria in biofilms. Lancet 358:135–138. 10.1016/S0140-6736(01)05321-1. [DOI] [PubMed] [Google Scholar]
- 42.Mah TF, Pitts B, Pellock B, Walker GC, Stewart PS, O'Toole GA. 2003. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 426:306–310. 10.1038/nature02122. [DOI] [PubMed] [Google Scholar]
- 43.Nichols DP, Caceres S, Caverly L, Fratelli C, Kim SH, Malcolm K, Poch KR, Saavedra M, Solomon G, Taylor-Cousar J, Moskowitz S, Nick JA. 2013. Effects of azithromycin in Pseudomonas aeruginosa burn wound infection. J. Surg. Res. 183:767–776. 10.1016/j.jss.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tre-Hardy M, Nagant C, El Manssouri N, Vanderbist F, Traore H, Vaneechoutte M, Dehaye JP. 2010. Efficacy of the combination of tobramycin and a macrolide in an in vitro Pseudomonas aeruginosa mature biofilm model. Antimicrob. Agents Chemother. 54:4409–4415. 10.1128/AAC.00372-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dales L, Ferris W, Vandemheen K, Aaron SD. 2009. Combination antibiotic susceptibility of biofilm-grown Burkholderia cepacia and Pseudomonas aeruginosa isolated from patients with pulmonary exacerbations of cystic fibrosis. Eur. J. Clin. Microbiol. Infect. Dis. 28:1275–1279. 10.1007/s10096-009-0774-9. [DOI] [PubMed] [Google Scholar]
- 46.Nick JA, Moskowitz SM, Chmiel JF, Forssen AV, Kim SH, Saavedra MT, Saiman L, Taylor-Cousar JL, Nichols DP. 2014. Azithromycin may antagonize inhaled tobramycin when targeting P. aeruginosa in cystic fibrosis. Ann. Am. Thorac. Soc. 11:342–350. 10.1513/AnnalsATS.201310-352OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O'Toole GA, Kolter R. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 28:449–461. 10.1046/j.1365-2958.1998.00797.x. [DOI] [PubMed] [Google Scholar]
- 48.Ceri H, Olson ME, Stremick C, Read RR, Morck D, Buret A. 1999. The Calgary biofilm device: new technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J. Clin. Microbiol. 37:1771–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pamp SJ, Sternberg C, Tolker-Nielsen T. 2009. Insight into the microbial multicellular lifestyle via flow-cell technology and confocal microscopy. Cytometry A 75:90–103. 10.1002/cyto.a.20685. [DOI] [PubMed] [Google Scholar]
- 50.Christensen BB, Sternberg C, Andersen JB, Palmer RJ, Jr, Nielsen AT, Givskov M, Molin S. 1999. Molecular tools for study of biofilm physiology. Methods Enzymol. 310:20–42. 10.1016/S0076-6879(99)10004-1. [DOI] [PubMed] [Google Scholar]
- 51.Anderl JN, Franklin MJ, Stewart PS. 2000. Role of antibiotic penetration limitation in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob. Agents Chemother. 44:1818–1824. 10.1128/AAC.44.7.1818-1824.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zelver N, Hamilton M, Pitts B, Goeres D, Walker D, Sturman P, Heersink J. 1999. Measuring antimicrobial effects on biofilm bacteria: from laboratory to field. Methods Enzymol. 310:608–628. 10.1016/S0076-6879(99)10047-8. [DOI] [PubMed] [Google Scholar]
- 53.Goeres DM, Hamilton MA, Beck NA, Buckingham-Meyer K, Hilyard JD, Loetterle LR, Lorenz LA, Walker DK, Stewart PS. 2009. A method for growing a biofilm under low shear at the air-liquid interface using the drip flow biofilm reactor. Nat. Protoc. 4:783–788. 10.1038/nprot.2009.59. [DOI] [PubMed] [Google Scholar]
- 54.Schleheck D, Barraud N, Klebensberger J, Webb JS, McDougald D, Rice SA, Kjelleberg S. 2009. Pseudomonas aeruginosa PAO1 preferentially grows as aggregates in liquid batch cultures and disperses upon starvation. PLoS One 4:e5513. 10.1371/journal.pone.0005513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lopez-Boado YS, Espinola M, Bahr S, Belaaouaj A. 2004. Neutrophil serine proteinases cleave bacterial flagellin, abrogating its host response-inducing activity. J. Immunol. 172:509–515. 10.4049/jimmunol.172.1.509. [DOI] [PubMed] [Google Scholar]
- 56.Staudinger BJ, Muller JF, Halldorsson S, Boles B, Angermeyer A, Nguyen D, Rosen H, Baldursson O, Gottfreethsson M, Guethmundsson GH, Singh PK. 2014. Conditions associated with the cystic fibrosis defect promote chronic Pseudomonas aeruginosa infection. Am. J. Respir. Crit. Care Med. 189:812–824. 10.1164/rccm.201312-2142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Malcolm KC, Nichols EM, Caceres SM, Kret JE, Martiniano SL, Sagel SD, Chan ED, Caverly L, Solomon GM, Reynolds P, Bratton DL, Taylor-Cousar JL, Nichols DP, Saavedra MT, Nick JA. 2013. Mycobacterium abscessus induces a limited pattern of neutrophil activation that promotes pathogen survival. PLoS One 8:e57402. 10.1371/journal.pone.0057402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luzar MA, Thomassen MJ, Montie TC. 1985. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: relationship to patient clinical condition. Infect. Immun. 50:577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cabrol S, Olliver A, Pier GB, Andremont A, Ruimy R. 2003. Transcription of quorum-sensing system genes in clinical and environmental isolates of Pseudomonas aeruginosa. J. Bacteriol. 185:7222–7230. 10.1128/JB.185.24.7222-7230.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D'Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, Burns JL, Kaul R, Olson MV. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. U. S. A. 103:8487–8492. 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schaber JA, Triffo WJ, Suh SJ, Oliver JW, Hastert MC, Griswold JA, Auer M, Hamood AN, Rumbaugh KP. 2007. Pseudomonas aeruginosa forms biofilms in acute infection independent of cell-to-cell signaling. Infect. Immun. 75:3715–3721. 10.1128/IAI.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sriramulu DD, Lunsdorf H, Lam JS, Romling U. 2005. Microcolony formation: a novel biofilm model of Pseudomonas aeruginosa for the cystic fibrosis lung. J. Med. Microbiol. 54:667–676. 10.1099/jmm.0.45969-0. [DOI] [PubMed] [Google Scholar]
- 63.Shak S, Capon DJ, Hellmiss R, Marsters SA, Baker CL. 1990. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc. Natl. Acad. Sci. U. S. A. 87:9188–9192. 10.1073/pnas.87.23.9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hunt BE, Weber A, Berger A, Ramsey B, Smith AL. 1995. Macromolecular mechanisms of sputum inhibition of tobramycin activity. Antimicrob. Agents Chemother. 39:34–39. 10.1128/AAC.39.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]