

Abstract
The quantification of antituberculosis drug concentrations in multinational trials currently requires the collection of modest blood volumes, centrifugation, aliquoting of plasma, freezing, and keeping samples frozen during shipping. We prospectively enrolled healthy individuals into the Tuberculosis Trials Consortium Study 29B, a phase I dose escalation study of rifapentine, a rifamycin under evaluation in tuberculosis treatment trials. We developed a liquid chromatography-tandem mass spectrometry (LC-MS/MS) method for quantifying rifapentine in whole blood on dried blood spots (DBS) to facilitate pharmacokinetic/pharmacodynamic analyses in clinical trials. Paired plasma and whole-blood samples were collected by venipuncture, and whole blood was spotted on Whatman protein saver 903 cards. The methods were optimized for plasma and then validated for DBS. The analytical measuring range for quantification of rifapentine and its metabolite was 50 to 80,000 ng/ml in whole-blood DBS. The analyte was stable on the cards for 11 weeks with a desiccant at room temperature and protected from light. The method concordance for paired plasma and whole-blood DBS samples was determined after correcting for participant hematocrit or population-based estimates of bias from Bland-Altman plots. The application of either correction factor resulted in acceptable correlation between plasma and whole-blood DBS (Passing-Bablok regression corrected for hematocrit; y = 0.98x + 356). Concentrations of rifapentine may be determined from whole-blood DBS collected via venipuncture after normalization in order to account for the dilutional effects of red blood cells. Additional studies are focused on the application of this methodology to capillary blood collected by finger stick. The simplicity of processing, storage, shipping, and low blood volume makes whole-blood DBS attractive for rifapentine pharmacokinetic evaluations, especially in international and pediatric trials.
INTRODUCTION
Tuberculosis (TB) remains a global health threat, with an estimated 8.6 million incident cases and 1.3 million TB-related deaths occurring in 2012 (1). The current treatment of TB requires a minimum of 6 months of treatment to achieve high rates of cure. There is an urgent need for highly potent treatments that can cure TB disease in less time.
The development of novel agents and regimens for clinical use for TB and other disease areas requires a thorough understanding of concentration-response relationships. However, standard plasma pharmacokinetic (PK) sampling in clinical trials is limited by the need for adequate sample volume, transport on ice, rapid sample processing by trained laboratory technicians, freezer storage at −80°C, and shipping of potentially hazardous materials to centralized laboratories in a frozen state. These steps represent challenges to robust pharmacokinetic/pharmacodynamic (PK/PD) analyses in clinical trials, particularly in special populations, like children, and in resource-poor settings (2).
Dried blood spot (DBS) sampling is a popular alternative for the quantitative determination of drug concentrations (3–5). With DBS methodology, small blood volumes are used, processing and storage are simplified, and there is no biohazard risk or substantial expense associated with sending large numbers of samples via regular mail (6). DBS is particularly advantageous for the development of drugs for tropical or neglected diseases, like malaria, for which the affected population largely lives in areas that are not in close proximity to research laboratories or large hospitals (5, 7, 8). However, assay sensitivity, stability, and hematocrit effects using DBS as a collection device vary by drug, so assay validation using DBS must be performed prior to implementation and interpretation of the tool (9).
Rifapentine (RPT) is a rifamycin antibiotic with a longer half-life and lower mean inhibitory concentration (MIC) against Mycobacterium tuberculosis than those of rifampin (RIF), the rifamycin used in standard treatment for drug-sensitive TB disease. In humans, RPT is deacetylated by arylacetamide deacetylase to the metabolite desacetyl-rifapentine (desRPT), which is less active than the parent drug (10). RPT is also under investigation as a potent anti-TB drug that may help shorten treatment duration for TB disease. In animal studies, the treatment-shortening potential of RPT was found to increase with higher exposures, seemingly without plateau (11, 12). Initial PK/PD analyses of clinical trials data suggest both substantial variability in RPT concentrations between persons and a strong correlation between RPT exposures and time to sputum culture conversion (13). Clinical trials aimed at finding the optimal dose of RPT and evaluating the efficacy of a shorter-duration RPT-based regimen in patients with drug-sensitive TB disease are ongoing (14).
We incorporated DBS collection into a phase I dose escalation trial of RPT (15), with the goal of developing and validating an accurate and reproducible DBS assay to quantify RPT and desRPT, with the desRPT data supporting mathematical model building.
MATERIALS AND METHODS
Chemicals and materials.
RPT and desRPT were donated by Sanofi-Aventis (Gentilly, France). Isotopically labeled RIF ([2H3]rifampin) was purchased from Toronto Research Chemicals, Inc. (North York, Ontario, Canada). The structures of RPT, desRPT, and isotopically labeled rifampin are illustrated in Fig. 1. High-performance liquid chromatography (HPLC)-grade water, acetonitrile, and methanol were purchased from Fisher Scientific (Pittsburgh, PA). Ascorbic acid, dimethyl sulfoxide (DMSO), and ammonium formate were purchased from Sigma-Aldrich (St. Louis, MO). Whatman protein saver 903 cards and a Harris Uni-Core punch were obtained from GE Healthcare Life Sciences (Piscataway, NJ). Drug-free human plasma was acquired from Biological Specialty Corporation (Colmar, PA).
FIG 1.
Structures of rifapentine (A), desacetyl-rifapentine (B), and isotopically labeled rifampin (C), along with the chemical formulas and the mass/charge ratios of the compounds.
Preparation of calibrators and quality control (QC) materials.
Whole blood was collected in Vacutainer K2EDTA tubes from individuals not taking RPT under an independent institutional review board (IRB)-approved protocol for the collection of biological specimens from healthy volunteers through the Johns Hopkins University School of Medicine. On the day of collection, whole blood was pooled in 50-ml conicals and centrifuged at 2,000 × g for 10 min at 4°C. Initial hematocrit levels were determined by measuring the volume of red blood cells and the volume of plasma; hematocrit was adjusted to 40% through the addition of red blood cells or drug-free plasma. Resuspended whole blood with an empirically determined hematocrit of 40% was used to generate the calibrators and quality control solutions.
Master stock solutions of RPT and desRPT were prepared in DMSO at final concentrations of 10,000 μg/ml. A working stock solution was prepared in DMSO at a final concentration of 100 μg/ml. Whole blood (40% hematocrit) or drug-free plasma was spiked with master or working stock solutions to generate standards containing 50, 100, 250, 500, 1,000, 5,000, 10,000, 25,000, 50,000, and 80,000 ng/ml RPT and its metabolite. Quality control levels were prepared from independently weighed stock solutions at the lower limit of quantification (LLOQ), as well as at low, medium, and high levels. The final concentrations of both RPT and desRPT at the LLOQ, low, medium, and high QC levels were 50, 150, 2,500, and 70,000 ng/ml, respectively.
For the placement of whole blood on dried blood spot (DBS) cards, 25 μl of whole-blood calibrators or QC solution was spotted onto a Whatman protein saver 903 card. Additionally, RPT and desRPT were prepared in hematocrit-adjusted whole-blood DBS across a range of 20% to 70%. A hematocrit level of 45% was deemed standard, and the observed drug values at this hematocrit were normalized to 1.0. The samples prepared with various hematocrit levels were compared to the observed concentrations at a 45% hematocrit level. The cards were allowed to dry for ≥3 h, protected from light. The dried cards were stored at ambient temperature with a desiccant (silica) and away from light.
Analyte extraction.
For the plasma quantification of RPT and its metabolite, the drugs were extracted from 20 μl of heparinized plasma via protein precipitation with 0.5 ml acetonitrile fortified with 0.5 mg/ml ascorbic acid and 100 μl of 500 ng/ml isotopically labeled rifampin on a 96-well Captiva 0.45-μm protein precipitation plate (Agilent Technologies, CA). RPT and desRPT were eluted into the 96-well collection plate via vacuum filtration; the eluents were further diluted 2-fold with 5 mM ammonium formate in water fortified with 0.5 mg/ml ascorbic acid. One microliter was used for downstream liquid chromatography-tandem mass spectrometric (LC-MS/MS) analysis. For DBS whole-blood quantification of RPT and desRPT, the optimal punch size (between 3 and 6 mm) using a Harris Uni-Core punch was evaluated first by comparing samples spiked with 50 ng/ml RPT and desRPT. For drug extraction, a 6.0-mm spot was punched out of the Whatman protein saver 903 card and immediately transferred to a 96-deep-well plate. To each well, 20 μl of a 500 ng/ml solution of [2H3]rifampin was added, followed by 450 μl of an extraction solvent containing a 90:10 methanol-to-50 mM ammonium formate buffer ratio with 0.5 mg/ml ascorbic acid. The plates were then capped and mixed via a titer plate shaker for 1 h. Following extraction, a 400-μl aliquot was transferred to a new 96-well plate, and 10 μl was analyzed by LC-MS/MS.
LC-MS/MS conditions and instrumentation.
The LC-MS/MS method used for drug quantification was based on previously described methods (15). The analytes of interest were separated using a Waters BEH C8, 50- by 2.1-mm, and 1.7-μm particle size column at ambient temperature. The mobile phase system for both the loading and eluting pumps consisted of an aqueous mobile phase of 5 mM ammonium formate in water (mobile phase A) and an organic mobile phase containing 3% DMSO in acetonitrile (mobile phase B). The chromatographic method is illustrated in Table S1 in the supplemental material. Due to the unavailability of isotopic analogs for RPT and desRPT, isotopically labeled rifampin, a structural analog of RPT and desRPT, was used as an internal standard for both analytes to determine the peak area ratios (Fig. 1). The analytes were detected over a 4.0-min run using an AB Sciex QTRAP 5500 mass analyzer (Foster City, CA) interfaced with a Waters Acquity ultraperformance liquid chromatography (UPLC) system (Milford, MA). The instrument parameters were optimized for maximal ionization of the drug parent and product ions, and the analytes were monitored in selected reaction monitoring (SRM) mode. The ion transitions monitored for RPT, desRPT, and isotopically labeled rifampin were m/z 877.6→m/z 845.5, m/z 835.5→m/z 803.5, and m/z 826.6→m/z 749.5, respectively.
Assay validation.
A full description of the analytical methods employed during assay validation is included in the supplemental material. The LC-MS/MS methods were validated in accordance with recommendations by the Food and Drug Administration (FDA) “Guidance for industry: bioanalytical method validation” (16).
Accuracy and precision.
The validation metrics tested included interassay and intra-assay accuracy (% deviation) and precision (% coefficient of variation [%CV]), calibration curve analysis, recovery, sensitivity, matrix effects, extraction efficiency, selectivity, and stability. The other factors evaluated included the impact of paper type on recovery, punch location, punch size, hematocrit value, and temperature stability.
Calibration curve analysis.
A 20-point calibration curve was used for the validation of both the plasma and whole-blood assays. The calibration correlation of the analytes was assessed by dividing the peak area of each calibration point by the area of the internal standard, thereby obtaining a ratio. The calibration curves for both RPT and its metabolite were generated by plotting the peak area ratio versus the concentration. The calibration correlation of the curves was assessed from the r2 of a 1/x2 weighted quadratic regression analysis. Quadratic regression was required to cover the desired analytical measuring range.
Stability studies.
Stability studies were performed under a variety of conditions; the challenges were specimen source specific. The whole-blood samples as DBS were subjected to sample matrix stability at room temperature unprotected from light, injection matrix stability following analyte extraction from DBS, and subjected to high heat (45°C) and humidity (100%) while stored in plastic bags with desiccant and protected from light. The plasma samples were subjected to sample matrix stability at room temperature with and without protecting from light, injection matrix stability, and three freeze-thaw cycles. Sample matrix stability was defined as the stability of RPT and desRPT in plasma or whole blood as DBS at room temperature with or without protection from light. Injection matrix stability assessed analyte stability following extraction from the specimen source and in the sample injection solution. The stability challenges tested were performed based on both FDA recommendations and the types of conditions that samples may encounter during large clinical trials. Both whole-blood and plasma concentrations were deemed stable if there was ≤15% difference between the stability-challenged and freshly extracted specimens.
Matrix effect characterization.
Matrix effects were assessed quantitatively according to Matuszewski and colleagues (17). The quantitative determination of matrix effects, in addition to recovery and processing efficiency, was performed through the generation of solutions at QC concentrations across 6 different lots of whole blood or plasma. Three sets of conditions were tested. An unextracted (neat or never extracted) set was prepared by spiking RPT, desRPT, and isotopically labeled internal standard into 500 μl of acetonitrile with 0.5 mg/ml ascorbic acid and 500 μl of 5 mM ammonium formate with 0.5 mg/ml ascorbic acid (to mimic plasma conditions) or 450 μl of 90:10 methanol to 50 mM ammonium formate buffer with 0.5 mg/ml ascorbic acid (to mimic whole blood under dried blood conditions), to achieve final concentrations of 150, 2,500, and 70,000 ng/ml. The unextracted set was generated in the absence of plasma or whole blood as dried blood spot matrices but was subjected to the same dilution process implemented for injection into the LC-MS/MS system. For a postextracted set, multiple lots of plasma or whole blood as DBS were extracted as previously described but were spiked with stock solutions of RPT and its metabolite, as well as the internal standard postprocessing. A preextracted set was analyzed by processing the QC levels prepared in plasma or whole blood as dried blood spots, as previously described. A comparison of the peak responses of the unextracted, preextracted, and postextracted sample sets was performed to determine matrix effects, processing, and recovery efficiency, respectively.
Clinical experimental protocol.
The phase I open-label dose escalation study was aimed at defining the maximal tolerated dose of RPT in healthy volunteers when given orally once daily; the experimental design and findings have been reported elsewhere (15). In brief, 26 participants received oral RPT at doses ranging from 5 to 20 mg/kg of body weight daily for 14 days. PK sampling was performed after the first and 14th doses, and hematocrit levels were checked at each study visit. The exclusion criteria for the study participants included serum creatinine of >1.5 mg/dl, albumin of <3.5 g/dl, hemoglobin of <12.0 g/dl (men) or <11.0 g/dl (women), neutrophil count of <1,250/mm3, platelet count of <125,000/mm3, and/or a positive human chorionic gonadotropin (hCG) test. The study was approved by the IRBs of the Johns Hopkins University School of Medicine and the U.S. Centers for Disease Control and Prevention.
Clinical sample collection, processing, and storage.
Sampling for plasma PK analyses was performed predose and at 0.5, 1, 2, 4, 5, 8, 12, 24, 34, 48, and 72 h postdose. Whole blood was collected on DBS cards at a single randomly chosen time point during each PK visit, in parallel with the plasma collection. Venous blood was collected into a K2EDTA tube, and a 25-μl drop of whole blood (the optimal volume recommended by the card manufacturer) was applied to a Whatman protein saver 903 card using a positive displacement pipette. The experiments were performed in duplicate. The DBS cards were dried thoroughly on a drying rack for 3 h in the dark. The cards were individually wrapped in aluminum foil to protect them from the light, and each card was placed in a plastic zip-locked bag with a sachet of desiccant and stored in the freezer at −70°C.
Comparison of plasma and DBS assay results.
Forty-four matched plasma and whole-blood samples as DBS from participants receiving RPT were analyzed to assess the ability to use whole blood as a DBS for RPT and desRPT quantification, in accordance with the recommendations by the Clinical and Laboratory Standards Institute guidelines (18). The correction for individual hematocrit value was calculated as follows: [DBS]hematocrit = [DBS]/[1 − (hematocrit/100)]. The correction for population bias was defined as the geometric mean of the drug concentration in plasma and whole blood.
A comparison of the methods was carried out using Passing-Bablok analysis, with plasma drug concentrations plotted on the x axis (19). Bland-Altman plots were used to assess the agreement between the plasma and whole-blood values, with and without correction for individual hematocrit values or a population-based correction factor (20).
To assess bias and imprecision, the median percentage prediction error (MPPE) and median absolute percentage prediction error (MAPE) were calculated, respectively, comparing predicted RPT and desRPT concentrations from DBS, both raw and hematocrit corrected (Craw and Chct, respectively), to the drug levels from plasma (Cplasma). When the drug concentration levels were below the limit of quantification (BLQ), the drug level was set to 25 ng/ml (BLQ/2, as standard practice). The measures used for assessment were calculated as follows: MPPE = median [100% × (Craw or Chct − Cplasma)/Cplasma], and MAPE = median [100% × |(Craw or Chct − Cplasma)|/Cplasma]. MPPE and MAPE values of <15% were considered acceptable (21).
RESULTS
Chromatographic and mass spectrometric optimization.
The LC-MS/MS methodology used for separating RPT and desRPT from plasma and whole-blood samples was optimized in several ways. Adding ascorbic acid to the extraction solvents prevented the drugs from becoming oxidized to form quinones and thereby maintained the molecular character and m/z for the analytes. The increasing flow rate following compound elution minimized carryover over the dynamic range of the assays. The chromatographic gradient is illustrated in Table S1 in the supplemental material. For drugs extracted from both plasma and whole-blood DBS, RPT, desRPT, and deuterated RIF elute at 1.40 min, 1.21 min, and 1.25 min, respectively (Fig. 2). The mass spectrometric conditions were optimized via the direct infusion of the analytes into the mass analyzer. The product ions were identified and selected for monitoring based on intensity and abundance. The parent-to-product ion transitions are described in Materials and Methods.
FIG 2.

Chromatograms of drug-free and drug-spiked whole-blood DBS for the monitoring of ion transitions for rifapentine (A), desacetyl-rifapentine (B), and [2H3]rifampin (C). The drug-spiked whole blood concentration was 50 ng/ml (LLOQ) for both drugs, and the internal standard concentration spiked was 500 ng/ml.
The initial development of the methods for extraction from whole-blood DBS involved spiking whole blood with various RPT concentrations and applying it to DMPK-A, DMPK-B, DMPK-C, and Whatman 903 filter papers. The Whatman 903 and DMPK-C DBS papers had the highest recovery of observed RPT and desRPT to the analytes prepared in solvent and were deemed optimal (see Table S2 in the supplemental material). Full bioanalytical validation was performed using the less expensive Whatman 903 DBS paper. Punch size comparisons demonstrated that the 6-mm punch was the optimal size for drug quantification, providing increased recovery and improved signal intensity on the mass analyzer (see Table S3 in the supplemental material).
Accuracy and precision.
The precision and accuracy for RPT and desRPT quantified in plasma and whole blood were acceptable according to FDA recommendations (Table 1). Within-run (intra-assay) and between-run (interassay) precision and accuracy were comparable for RPT and desRPT isolated from plasma and whole blood at all QC levels tested (Table 1).
TABLE 1.
Intra-assay and interassay precision (%CV) and accuracy (%DEV) results for rifapentine and desacetyl-rifapentine quality control samples prepared in plasma and in whole-blood dried blood spots
| QC levela | Intra-assay precision and accuracy |
Interassay precision and accuracy |
||||||
|---|---|---|---|---|---|---|---|---|
| Mean (ng/ml) | SD (ng/ml) | %CV | %DEV | Mean (ng/ml) | SD (ng/ml) | %CV | %DEV | |
| RPT | ||||||||
| DBS | ||||||||
| LLOQ | 53.5 | 3.46 | 6.47 | 6.96 | 51.3 | 3.48 | 6.78 | 2.63 |
| Low | 158 | 5.15 | 3.26 | 5.33 | 155 | 9.65 | 6.23 | 3.33 |
| Medium | 2,644 | 94.5 | 3.57 | 5.76 | 2,625 | 95.7 | 3.64 | 5.01 |
| High | 70,100 | 3,979 | 5.68 | 0.14 | 70,367 | 6,706 | 9.53 | 0.52 |
| Plasma | ||||||||
| LLOQ | 56.6 | 4.47 | 7.88 | 13.3 | 53.6 | 4.64 | 8.66 | 7.19 |
| Low | 136 | 3.73 | 2.75 | −9.67 | 133 | 5.92 | 4.45 | −11.3 |
| Medium | 2,475 | 187 | 7.57 | −1.00 | 2,516 | 136 | 5.41 | 0.64 |
| High | 73,533 | 2,183 | 2.97 | 5.05 | 74,306 | 3,141 | 4.23 | 6.15 |
| desRPT | ||||||||
| DBS | ||||||||
| LLOQ | 49.8 | 2.77 | 5.56 | −0.32 | 47.1 | 4.32 | 9.19 | −5.89 |
| Low | 149 | 6.78 | 4.55 | −0.67 | 146 | 8.66 | 5.94 | −2.80 |
| Medium | 2,424 | 65.8 | 2.71 | −3.04 | 2,424 | 97.8 | 4.04 | −3.04 |
| High | 70,220 | 6,491 | 9.24 | 0.31 | 69,420 | 7,175 | 10.3 | −0.83 |
| Plasma | ||||||||
| LLOQ | 51.4 | 3.55 | 6.91 | 2.70 | 52.9 | 4.47 | 8.46 | 5.70 |
| Low | 136 | 4.62 | 3.39 | −9.22 | 137 | 14.8 | 10.8 | −8.41 |
| Medium | 2,685 | 99.1 | 3.69 | 7.40 | 2,601 | 131 | 5.04 | 4.02 |
| High | 72,717 | 2,651 | 3.65 | 3.88 | 72,911 | 3,258 | 4.47 | 4.16 |
The quality control (QC) levels are defined as follows: LLOQ, 50 ng/ml; low, 150 ng/ml; medium, 2,500 ng/ml; and high, 70,000 ng/ml. RPT, rifapentine; DBS, dried blood spots; desRPT, desacetyl-rifapentine.
Calibration curve analysis.
Calibration curves for both RPT and its metabolite were generated by plotting the peak area ratio versus drug concentration. The calibration correlation of the curves was assessed from the r2 of a 1/x2 weighted quadratic regression analysis. Quadratic regression was required to cover the desired analytical measuring range. The average r2 for both analytes was ≥0.996 for plasma and ≥0.997 for whole-blood DBS. Representative calibration curves in whole blood are shown in Fig. 3.
FIG 3.
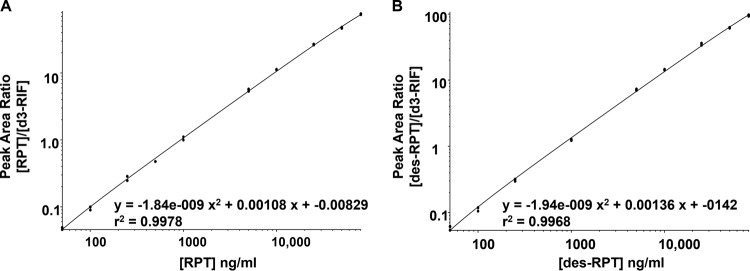
Representative calibration curve for rifapentine and desacetyl-rifapentine from whole-blood DBS. The analytical measuring range for both drugs is 50 to 80,000 ng/ml, and the curves were fit to the calibrators using a quadratic regression with 1/x2 weighting. The calibration curves are displayed on a log-log scale so that all calibration points would be visible. The y axis is the peak area ratio of the analyte to the internal standard, and the x axis demarcates analyte concentration.
RPT and desRPT stability studies.
The results from the sample stability challenges are summarized in Table 2. RPT and desRPT in whole blood applied to DBS, incubated at room temperature, and protected from light for 48 h showed a percent difference in the mean concentration ranging from −4.97% to −3.89% and −1.21% to 4.72%, respectively. The heat- and humidity-challenged samples exhibited <4.13% (positive or negative) difference for both RPT and its metabolite between whole-blood samples as DBS incubated at 45°C and 100% humidity for 24 h and nonchallenged samples (Table 2). Also, the stability of the DBS samples under heat and humidity challenges was only acceptable if the cards were stored in plastic bags with a desiccant. Long-term stability studies indicated that RPT and desRPT were stable for at least 11 weeks protected from light stored in a plastic bag with a desiccant but were unstable after 9 months under these storage conditions (data not shown).
TABLE 2.
RPT and desRPT whole-blood DBS and plasma stability challenges
| QC levela | Sample matrix stabilityb |
Injection matrix stabilityc |
Heat stability (whole-blood DBS) or freeze-thaw cycles (plasma)d |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Control mean | Treated mean | % Difference | Control mean | Treated mean | % Difference | Control mean | Treated mean | % Difference | |
| Whole-blood DBS | |||||||||
| RPT | |||||||||
| Low | 161 | 153 | −4.97 | 156 | 156 | −0.51 | 144 | 142 | −1.32 |
| Medium | 2,670 | 2,554 | −4.34 | 2,448 | 2,620 | 7.03 | 2,474 | 2,468 | −0.24 |
| High | 65,320 | 62,780 | −3.89 | 66,720 | 68,300 | 2.37 | 67,160 | 66,380 | −1.16 |
| desRPT | |||||||||
| Low | 149 | 147 | −1.21 | 142 | 147 | 3.38 | 158 | 164 | 4.13 |
| Medium | 2,374 | 2,486 | 4.72 | 2,278 | 2,308 | 1.32 | 2,656 | 2,734 | 2.94 |
| High | 61,060 | 63,640 | 4.23 | 64,220 | 67,660 | 5.36 | 73,920 | 73,880 | −0.05 |
| Plasma | |||||||||
| RPT | |||||||||
| Low | 131 | 141 | 7.65 | 130 | 138 | 6.63 | 148 | 148 | 0.11 |
| Medium | 2,510 | 2,265 | −9.76 | 2,548 | 2,535 | −0.52 | 2,635 | 2,435 | −7.59 |
| High | 70,150 | 66,625 | −5.02 | 72,550 | 67,650 | −6.75 | 70,150 | 68,200 | −2.78 |
| desRPT | |||||||||
| Low | 137 | 133 | −2.74 | 145 | 136 | −6.59 | 130 | 135 | 3.85 |
| Medium | 2,743 | 2,605 | −5.01 | 2,652 | 2,682 | 1.13 | 2,775 | 2,588 | −6.76 |
| High | 69,400 | 67,800 | −2.31 | 70,933 | 66,200 | −6.67 | 69,400 | 69,300 | −0.14 |
QC levels are defined as follows: low, 150 ng/ml; medium, 2,500 ng/ml; high, 70,000 ng/ml.
n = 5 for whole-blood DBS (2 days in the light at room temperature) and n = 4 for plasma (1 day in the light at room temperature).
n = 5 for whole-blood DBS (3 days in injection matrix on a reanalyzed curve) and n = 6 for plasma (5 days in injection matrix on a reanalyzed curve).
n = 5 for whole-blood DBS (1 day at 45°C with 100% humidity with samples stored in plastic bag with desiccant) and n = 4 for plasma (3 freeze-thaw cycles).
The QC levels prepared in plasma were stable in sample matrix for 1 day unprotected from light and 2 days protected from light, but the samples were not stable for longer periods of time (Table 2 and data not shown). Across the tested QC levels, the sample matrix stability for RPT and desRPT ranged in the percent differences of tested concentrations in freshly prepared samples from −9.76% to 7.65% and −5.01% to −2.31%, respectively. The plasma samples were stable in injection matrix for 5 days and for 3 freeze-thaw cycles (Table 2).
Matrix effects, recovery, and processing efficiency.
The average peak areas for the analytes and internal standard, along with matrix effects, recovery, and processing efficiency, are summarized in Table 3. RPT and desRPT exhibited no ion suppression or enhancement in plasma, but ion suppression was observed in whole blood. While plasma had nearly 100% recovery for both the analytes and internal standard, whole blood demonstrated a lower recovery for both RPT and its metabolite. However, the internal standard recovery remained at nearly 100%. This was expected, since it is not added at the same time that the sample matrix is added to the collection device. The average matrix effects, recovery efficiency, and processing efficiency are described in Table 3.
TABLE 3.
Matrix effects, recovery efficiency, and processing efficiency of RPT and desRPT and deuterated rifampin internal standard ([2H3]RIF) in whole-blood DBS and plasma
| QC levela | Analyte peak area |
Internal standard peak area |
Matrix effects (%) |
Recovery efficiency (%) |
Processing efficiency (%) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unextracted | Postextracted | Preextracted | Unextracted | Postextracted | Preextracted | RPT/desRPT | [2H3]RIF | RPT/desRPT | [2H3]RIF | RPT/desRPT | [2H3]RIF | |
| RPT | ||||||||||||
| Whole-blood DBS | ||||||||||||
| Low | 159,083 | 123,997 | 52,949 | 524,902 | 439,316 | 441,389 | 77.9 | 83.7 | 42.7 | 100 | 33.3 | 84.1 |
| Medium | 2,431,360 | 1,964,432 | 871,343 | 526,875 | 442,606 | 425,975 | 80.8 | 84.0 | 44.4 | 86.2 | 35.8 | 80.8 |
| High | 33,619,579 | 30,464,600 | 19,662,724 | 476,178 | 413,739 | 429,832 | 90.6 | 86.9 | 64.5 | 104 | 58.5 | 90.3 |
| Plasma | ||||||||||||
| Low | 110,831 | 110,340 | 103,961 | 588,046 | 571,036 | 558,721 | 99.6 | 97.1 | 94.2 | 97.8 | 93.8 | 95.0 |
| Medium | 1,762,204 | 1,748,141 | 1,660,195 | 556,866 | 557,608 | 535,108 | 99.2 | 100 | 95.0 | 96.0 | 94.2 | 96.1 |
| High | 25,146,811 | 23,248,023 | 22,901,247 | 489,608 | 459,811 | 452,989 | 92.4 | 93.9 | 98.5 | 98.5 | 91.1 | 92.5 |
| desRPT | ||||||||||||
| Whole-blood DBS | ||||||||||||
| Low | 171,432 | 134,383 | 76,003 | 524,902 | 439,316 | 441,389 | 78.4 | 83.7 | 56.6 | 100 | 44.3 | 84.1 |
| Medium | 2,583,735 | 2,169,563 | 1,177,477 | 526,875 | 442,606 | 425,975 | 84.0 | 84.0 | 54.3 | 86.2 | 45.6 | 80.8 |
| High | 36,721,841 | 33,947,496 | 23,993,094 | 476,178 | 413,739 | 429,832 | 92.4 | 86.9 | 70.7 | 104 | 65.3 | 90.3 |
| Plasma | ||||||||||||
| Low | 125,750 | 126,934 | 122,424 | 588,046 | 571,036 | 558,721 | 101 | 97.1 | 96.4 | 97.8 | 97.4 | 95.0 |
| Medium | 1,960,483 | 1,900,277 | 1,830,786 | 556,866 | 557,608 | 535,108 | 96.9 | 100 | 96.3 | 96.0 | 93.4 | 96.1 |
| High | 29,771,360 | 27,575,837 | 27,611,673 | 489,608 | 459,811 | 452,989 | 92.6 | 93.9 | 100 | 98.5 | 92.7 | 92.5 |
QC levels are defined as follows: low, 150 ng/ml; medium, 2,500 ng/ml; high, 70,000 ng/ml.
Relationship of RPT and desRPT concentrations in DBS versus plasma.
A lower recovery rate was observed in the QC whole-blood samples with lower hematocrit levels; specifically, in whole-blood samples adjusted to 20% hematocrit, recovery was <80% of the expected concentrations of RPT and desRPT. While higher-level hematocrit samples showed increased drug concentrations, the degree of increase was not outside the variation of the assay (data not shown).
For the samples from healthy volunteers, a comparison of the matched plasma and whole-blood DBS was performed. The MPPE and MAPE values for comparisons of RPT (MPPE, −38.9%; MAPE, 39.2%) and desRPT (MPPE, −49.7%; MAPE, 49.8%) DBS to plasma suggest unacceptable bias and imprecision. When individually based hematocrit levels were used to correct the DBS drug concentrations, both bias and imprecision improved. Both were acceptable for RPT (MPPE, 0.7%; MAPE, 6.0%), and only bias was acceptable for desRPT (MPPE, −14.3%; MAPE, 17.5%).
Graphical comparisons between the methods are shown in Fig. 4. Bland-Altman plots illustrate a 0.63 (95% confidence interval [CI], 0.58 to 0.68) bias for RPT and a 0.53 (95% CI, 0.46 to 0.61) bias for desRPT. When individually based hematocrit levels were used to correct the drug concentrations, the bias for RPT was 1.07 (95% CI, 0.99 to 1.12), and the bias for desRPT was 0.91 (95% CI, 0.79 to 1.03).
FIG 4.

Bland-Altman plots of rifapentine (A and C) and desacetyl-rifapentine (B and D) concentration ratios of whole-blood DBS to plasma versus the average of the two concentrations (whole blood and plasma). The dark gray solid line is the mean ratio (bias) and the dashed lines are the limits of agreement (mean ratio ± 1.96 × standard deviation of the ratio). Shown are the whole-blood concentrations (A and B) and a comparison of participant-corrected hematocrit whole-blood concentrations with plasma concentrations (C and D). The one outlier seen in all four plots is from the same subject at the same time point. The most logical reason for the outlier status of this sample is that the plasma sample was drawn 23 min prior to the DBS sample, and therefore, the DBS concentrations were higher than those of plasma.
A scatter plot analysis of plasma concentrations compared to whole-blood drug concentrations as extracted from DBS was performed using a Passing-Bablok fit. Figure 5 illustrates the concordance of whole blood as DBS and plasma drug concentrations using Passing-Bablok fit for three scenarios: unadjusted DBS concentrations, adjustment of whole-blood values for population-based estimates of Bland-Altman bias, and adjustment of whole blood as DBS values for individual patient hematocrit values. The unadjusted whole blood as DBS has a proportional bias compared to plasma, as the slopes were 0.56 (95% CI, 0.50 to 0.64) for RPT and 0.50 (95% CI, 0.47 to 0.55) for desRPT (a slope of 1.00 would show no bias). When the Bland-Altman bias is used as a general population-based correction factor, the slope for RPT was 0.90 (95% CI, 0.80 to 1.01) and the desRPT slope was 0.95 (95% CI, 0.89 to 1.03). After adjusting for patient-specific hematocrit, a comparison of whole blood to plasma showed a slope of 0.98 (95% CI, 0.87 to 1.10) for RPT and 0.86 (95% CI, 0.81 to 0.93) for desRPT.
FIG 5.

Passing-Bablok regression between concentrations in whole-blood DBS and plasma are displayed. The dark solid line is the Passing-Bablok fit, and the dashed lines are the 95% CI bands. The scatter plots illustrate uncorrected whole-blood rifapentine (A) and desacetyl-rifapentine (B) versus plasma drug concentrations, with regression line slopes and intercepts of 0.56 (95% CI, 0.50 to 0.64) and 303 (95% CI, −188 to 1,142), and 0.50 (95% CI, 0.47 to 0.55) and −8.84 (95% CI, −239 to 133), respectively. (C and D) Bland-Altman-corrected whole-blood rifapentine (C) and desacetyl-rifapentine (D) drug concentrations compared to those in plasma, with regression line slopes and intercepts of 0.90 (95% CI, 0.80 to 1.01) and 443 (95% CI, −273 to 1,652), and 0.95 (95% CI, 0.89 to 1.03) and −31.7 (95% CI, −489 to 185), respectively. (E and F) Participant-specific hematocrit-corrected whole-blood rifapentine (E) and desacetyl-rifapentine (F) concentrations compared to those in plasma, with regression line slopes and intercepts of 0.98 (95% CI, 0.87 to 1.10) and 356 (95% CI, −452 to 1,827), and 0.86 (95% CI, 0.81 to 0.93) and −144 (95% CI, −500 to 73.1), respectively.
DISCUSSION
This work describes the development and validation of a high-throughput assay for the quantification of the anti-TB drug RPT and its metabolite, desRPT, in plasma and whole blood as DBS. Extensive precision, accuracy, and stability studies were performed. The analytical measuring ranges of the assays for both RPT and desRPT, which were 50 to 80,000 ng/ml, were sufficient to encompass clinically relevant drug concentrations, as indicated by previously described single- and multiple-dose studies (14, 22). The plasma and individual hematocrit-corrected whole-blood DBS values for RPT and desRPT correlated well (slopes of 0.98 [95% CI, 0.87 to 1.10] and 0.86 [95% CI, 0.81 to 0.93], respectively), the limits of quantification were acceptable for both matrices, and both RPT and desRPT were stable in whole-blood DBS for ≥11 weeks at room temperature. Of note, full validation of whole blood as DBS was performed in the whole-blood set at a hematocrit of 40%. While the hematocrit used for validation is reflective of a healthy population as opposed to the volume of red blood cells typically observed in a TB-infected population (i.e., hematocrit, 35%), the ruggedness of the assay was subsequently demonstrated for both RPT and its metabolite over a wide hematocrit range. Therefore, the described method is appropriate for both healthy populations and TB-infected individuals.
Whole-blood DBSs are increasingly being utilized by pharmaceutical companies from the earliest stages of drug development, including animal toxicology studies, preclinical efficacy models, first-in-human, and treatment efficacy trials (23, 24), and the cost savings can be substantial (25, 26). Whole-blood DBS, however, cannot be used for all drugs. Notably, whole-blood DBS cannot be used for drug development if any of the following conditions are true: the lower limit of quantification is too high (poor sensitivity), blood/plasma partitioning is concentration or time dependent, the drug or its metabolites are not stable on the filter paper, there is puncher carryover, or the unbound drug fraction is not constant in plasma over a wide range of concentrations (27).
Most drugs are quantified in plasma or serum, which are aqueous specimen sources. The protein content of whole blood, though, results in a differential drug partitioning of water-soluble drugs, such as RPT. Thus, the physiological differences between whole blood and plasma must be taken into account in the quantification and interpretation of drug concentrations, and each drug must be tested individually to ensure that whole blood collected as a DBS can be used for PK evaluations. While the MPPE and Bland-Altman plots showed a negative bias in a comparison of whole blood as DBS to plasma for RPT and its metabolite, the correction for hematocrit significantly improves the bias, resulting in an overall concordance between the specimen types and an acceptable bias (Passing-Bablok slopes, 0.98 [95% CI, 0.87 to 1.10] and 0.86 [95% CI, 0.81 to 0.93] and intercepts of 356 [95% CI, −452 to 1,827] and −144 [95% CI, −500 to 73.1]; MPPE of 0.7% and −14.3% for RPT and its metabolite, respectively). Imprecision, as measured by MAPE, also markedly improves to an acceptable level upon correction for hematocrit for RPT. Correction for hematocrit appears to impact RPT DBS bias and imprecision more than that for desRPT, as desRPT MAPE was higher than acceptable (17.5%) even after hematocrit correction. As RPT exposures are used for PK/PD analyses assessing concentration-effect and dose-toxicity relationships, and desRPT concentrations are not predictive of treatment response or toxicity, an accurate estimation of RPT concentrations is more clinically relevant than that for desRPT.
The requirement of knowing hematocrit is a disadvantage of this assay method. However other investigators have shown that it may be possible to estimate hematocrit from whole-blood DBS samples via measurement of potassium using routine clinical analyzers, circumventing the need for a separate blood draw for hematocrit determination (28). Potassium is an intracellular electrolyte found predominantly in erythrocytes, with serum, plasma, and other cells found in whole blood contributing a minimal amount of the total blood potassium concentrations. Thus, the measurement of potassium in DBS can be used to predict hematocrit, allowing for patient-specific hematocrit corrections postcollection. Alternatively, to avoid the requirement for patient-specific hematocrit corrections, a correlation between plasma and whole blood collected as DBS samples can be assessed in other settings to evaluate whether or not the population-based Bland-Altman bias is consistent across populations. If that is the case, individual hematocrit values may not be needed, and a standard correction could be applied. Evaluating DBS in other settings would have the benefit of allowing an additional assessment of matrix effects in the whole blood of patients with TB receiving treatment in different geographic areas. In addition, validation of a method for the quantification of RPT from capillary blood collected by finger stick would be needed prior to the implementation of finger stick for specimen collection on DBS.
Of note, whole-blood DBS methods have been investigated for therapeutic drug monitoring (TDM) of TB drugs (29). The utility of patient- and clinic-friendly sample collection techniques for TDM is particularly evident for drugs with a narrow therapeutic margin, like linezolid (30, 31). The efficacy of TB treatment is highly dependent on achieving adequate drug concentrations of rifamycin antibiotics, as these drugs have unparalleled sterilizing activity against M. tuberculosis when adequate exposure is achieved. For reasons that are only partially understood, the concentrations of rifamycins in the blood of TB patients vary substantially (32, 33). The DBS method for RPT enhances the potential for identifying patients in clinical trials with microbiological, radiographic, and pharmacokinetic characteristics that may allow for TB treatment of <6 months. Thus, the development of whole-blood DBS methods for measuring rifampin and RPT may be of high value for both drug optimization efforts and eventual TDM once the most effective doses of these drugs are established (13, 14, 29, 34–36).
Although this work demonstrates the potential utility of DBS analysis as a mechanism for drug quantification, particularly in resource-limited environments, there are limitations to the use of DBS in general that should be kept in mind. In addition to the standard requirements for validating bioanalytical assays, additional steps are required for DBS method development, such as an assessment and definition of sampling methods and evaluation of the effects of hematocrit and blood volume on assay results, as well as an assessment of matrix effects and recovery from DBS (9, 30). Further validation steps are required to ensure the agreement of whole-blood and serum or plasma drug concentrations and the proper interpretation of results generated from DBS analysis. In addition, extra training of both the individuals collecting the blood and laboratory technicians is mandatory.
In conclusion, we developed an accurate and reproducible method to quantify concentrations of RPT, a potent anti-TB drug, and its metabolite, collected as DBS on Whatman 903 filter paper from venipuncture, using LC-MS/MS analysis. While further studies are required to recapitulate the described findings in capillary blood collected by finger stick, this methodology may be helpful for drug quantification in upcoming international clinical trials and drug evaluations in small children. Additional considerations for DBS use in upcoming studies are focused on the mode of collection and participant-specific hematocrit collection. Yet, the lower cost, ease of processing, storage, and shipping, and stability may allow for enhanced PK/PD analyses as this promising anti-TB drug is evaluated further.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the contributions of James Johnson, Yanhui Lu, and Walter Hubbard from the Clinical Pharmacology Analytical Laboratory at Johns Hopkins. We thank Stephanie Everts and Elizabeth Purdy of the Drug Development Unit in the Division of Clinical Pharmacology at Johns Hopkins, who aided with the design and collection of the clinical components of this study.
This study was sponsored by the Centers for Disease Control and Prevention and the CDC Foundation. Other support was provided by grant K23AI080842 (to K.E.D.). The ABI-Sciex API5500 quadrupole linear ion-trap console used for the quantification of RPT was purchased with the proceeds of grant 1S10 RR 27733 awarded to Walter C. Hubbard. The Waters Acquity ultraperformance liquid chromatograph (UPLC) interfaced with the API-5500 was purchased with funds from Pendleton Enterprises awarded to Craig W. Hendrix. RPT and desRPT were provided by Sanofi.
We declare no conflicts of interest.
Footnotes
Published ahead of print 2 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03607-14.
REFERENCES
- 1.World Health Organization. 2013. Global tuberculosis report 2013. WHO/HTM/TB/2013.11. World Health Organization, Geneva, Switzerland: http://apps.who.int/iris/bitstream/10665/91355/1/9789241564656_eng.pdf?ua=1. [Google Scholar]
- 2.Hietala SF, Bhattarai A, Msellem M, Röshammar D, Ali AS, Strömberg J, Hombhanje FW, Kaneko A, Björkman A, Ashton M. 2007. Population pharmacokinetics of amodiaquine and desethylamodiaquine in pediatric patients with uncomplicated falciparum malaria. J. Pharmacokinet. Pharmacodyn. 34:669–686. 10.1007/s10928-007-9064-2. [DOI] [PubMed] [Google Scholar]
- 3.Hoogtanders K, van der Heijden J, Christiaans M, Edelbroek P, van Hooff JP, Stolk LM. 2007. Therapeutic drug monitoring of tacrolimus with the dried blood spot method. J. Pharm. Biomed. Anal. 44:658–664. 10.1016/j.jpba.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 4.Koal T, Burhenne H, Römling R, Svoboda M, Resch K, Kaever V. 2005. Quantification of antiretroviral drugs in dried blood spot samples by means of liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 19:2995–3001. 10.1002/rcm.2158. [DOI] [PubMed] [Google Scholar]
- 5.Taneja I, Erukala M, Raju KS, Singh SP, Wahajuddin 2013. Dried blood spots in bioanalysis of antimalarials: relevance and challenges in quantitative assessment of antimalarial drugs. Bioanalysis 5:2171–2186. 10.4155/bio.13.180. [DOI] [PubMed] [Google Scholar]
- 6.Knudsen RC, Slazyk WE, Richmond JY, Hannon WH. 2012. Guidelines for the shipment of dried blood spot specimens. Centers for Disease Control and Prevention Office of Health and Safety, Biosafety Branch, Atlanta, GA. [Google Scholar]
- 7.Blessborn D, Römsing S, Annerberg A, Sundquist D, Björkman A, Lindegardh N, Bergqvist Y. 2007. Development and validation of an automated solid-phase extraction and liquid chromatographic method for determination of lumefantrine in capillary blood on sampling paper. J. Pharm. Biomed. Anal. 45:282–287. 10.1016/j.jpba.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 8.Malm M, Lindegårdh N, Bergqvist Y. 2004. Automated solid-phase extraction method for the determination of piperaquine in capillary blood applied onto sampling paper by liquid chromatography. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 809:43–49. 10.1016/j.jchromb.2004.05.032. [DOI] [PubMed] [Google Scholar]
- 9.Vu DH, Alffenaar JW, Edelbroek PM, Brouwers JR, Uges DR. 2011. Dried blood spots: a new tool for tuberculosis treatment optimization. Curr. Pharm. Des. 17:2931–2939. 10.2174/138161211797470174. [DOI] [PubMed] [Google Scholar]
- 10.Nakajima A, Fukami T, Kobayashi Y, Watanabe A, Nakajima M, Yokoi T. 2011. Human arylacetamide deacetylase is responsible for deacetylation of rifamycins: rifampicin, rifabutin, and rifapentine. Biochem. Pharmacol. 82:1747–1756. 10.1016/j.bcp.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Rosenthal IM, Zhang M, Williams KN, Peloquin CA, Tyagi S, Vernon AA, Bishai WR, Chaisson RE, Grosset JH, Nuermberger EL. 2007. Daily dosing of rifapentine cures tuberculosis in three months or less in the murine model. PLoS Med. 4:e344. 10.1371/journal.pmed.0040344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenthal IM, Tasneen R, Peloquin CA, Zhang M, Almeida D, Mdluli KE, Karakousis PC, Grosset JH, Nuermberger EL. 2012. Dose-ranging comparison of rifampin and rifapentine in two pathologically distinct murine models of tuberculosis. Antimicrob. Agents Chemother. 56:4331–4340. 10.1128/AAC.00912-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Savic RM, Weiner M, Mac Kenzie W, Heilig C, Dooley K, Engle M, Nsubuga P, Phan H, Peloquin C, Dorman S, Tuberculosis Trials Consortium of the Centers for Disease Control and Prevention 2013. PKPD analysis of rifapentine in patients during intensive phase treatment for tuberculosis from Tuberculosis Trials Consortium studies 29 and 29X; abstr. 11 6th International Workshop on Clinical Pharmacology of Tuberculosis Drugs, Denver, CO, 9 September 2013. [Google Scholar]
- 14.Dorman SE, Goldberg S, Stout JE, Muzanyi G, Johnson JL, Weiner M, Bozeman L, Heilig CM, Feng PJ, Moro R, Narita M, Nahid P, Ray S, Bates E, Haile B, Nuermberger EL, Vernon A, Schluger NW, Tuberculosis Trials Consortium 2012. Substitution of rifapentine for rifampin during intensive phase treatment of pulmonary tuberculosis: study 29 of the Tuberculosis Trials Consortium. J. Infect. Dis. 10.1093/infdis/jis461. [DOI] [PubMed] [Google Scholar]
- 15.Dooley KE, Bliven-Sizemore EE, Weiner M, Lu Y, Nuermberger EL, Hubbard WC, Fuchs EJ, Melia MT, Burman WJ, Dorman SE. 2012. Safety and pharmacokinetics of escalating daily doses of the antituberculosis drug rifapentine in healthy volunteers. Clin. Pharmacol. Ther. 91:881–888. 10.1038/clpt.2011.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.U.S. Department of Health and Human Services, Center for Drug Evaluation and Research, Center for Veterinary Medicine. 2013. Guidance for industry: bioanalytical method validation. U.S. FDA, Rockville, MD: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidelines/UCM368107.pdf. [Google Scholar]
- 17.Matuszewski BK, Constanzer ML, Chavez-Eng CM. 1998. Matrix effect in quantitative LC/MS/MS analyses of biological fluids: a method for determination of finasteride in human plasma at picogram per milliliter concentrations. Anal. Chem. 70:882–889. 10.1021/ac971078+. [DOI] [PubMed] [Google Scholar]
- 18.Clinical and Laboratory Standards Institute. 2002. Method comparison and bias estimation using patient samples; approved guideline—2nd ed. CLSI document EP9-A2. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 19.Passing H, Bablok W. 1984. Comparison of several regression procedures for method comparison studies and determination of sample sizes. Application of linear regression procedures for method comparison studies in Clinical Chemistry, Part II. J. Clin. Chem. Clin. Biochem. 22:431–445. [DOI] [PubMed] [Google Scholar]
- 20.Bland JM, Altman DG. 1986. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet i:307–310. [PubMed] [Google Scholar]
- 21.Ting LS, Villeneuve E, Ensom MH. 2006. Beyond cyclosporine: a systematic review of limited sampling strategies for other immunosuppressants. Ther. Drug Monit. 28:419–430. 10.1097/01.ftd.0000211810.19935.44. [DOI] [PubMed] [Google Scholar]
- 22.Dooley K, Flexner C, Hackman J, Peloquin CA, Nuermberger E, Chaisson RE, Dorman SE. 2008. Repeated administration of high-dose intermittent rifapentine reduces rifapentine and moxifloxacin plasma concentrations. Antimicrob. Agents Chemother. 52:4037–4042. 10.1128/AAC.00554-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barfield M, Spooner N, Lad R, Parry S, Fowles S. 2008. Application of dried blood spots combined with HPLC-MS/MS for the quantification of acetaminophen in toxicokinetic studies. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 870:32–37. 10.1016/j.jchromb.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 24.Beaudette P, Bateman KP. 2004. Discovery stage pharmacokinetics using dried blood spots. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 809:153–158. 10.1016/j.jchromb.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 25.van Amsterdam P, Waldrop C. 2010. The application of dried blood spot sampling in global clinical trials. Bioanalysis 2:1783–1786. 10.4155/bio.10.158. [DOI] [PubMed] [Google Scholar]
- 26.Xu Y, Woolf EJ, Agrawal NG, Kothare P, Pucci V, Bateman KP. 2013. Merck's perspective on the implementation of dried blood spot technology in clinical drug development–why, when and how. Bioanalysis 5:341–350. 10.4155/bio.12.321. [DOI] [PubMed] [Google Scholar]
- 27.Spooner N, Lad R, Barfield M. 2009. Dried blood spots as a sample collection technique for the determination of pharmacokinetics in clinical studies: considerations for the validation of a quantitative bioanalytical method. Anal. Chem. 81:1557–1563. 10.1021/ac8022839. [DOI] [PubMed] [Google Scholar]
- 28.Capiau S, Stove VV, Lambert WE, Stove CP. 2013. Prediction of the hematocrit of dried blood spots via potassium measurement on a routine clinical chemistry analyzer. Anal. Chem. 85:404–410. 10.1021/ac303014b. [DOI] [PubMed] [Google Scholar]
- 29.Vu DH, Koster RA, Bolhuis MS, Greijdanus B, Altena RV, Nguyen DH, Brouwers JR, Uges DR, Alffenaar JW. 2014. Simultaneous determination of rifampicin, clarithromycin and their metabolites in dried blood spots using LC-MS/MS. Talanta 121:9–17. 10.1016/j.talanta.2013.12.043. [DOI] [PubMed] [Google Scholar]
- 30.Vu DH, Bolhuis MS, Koster RA, Greijdanus B, de Lange WC, van Altena R, Brouwers JR, Uges DR, Alffenaar JW. 2012. Dried blood spot analysis for therapeutic drug monitoring of linezolid in patients with multidrug-resistant tuberculosis. Antimicrob. Agents Chemother. 56:5758–5763. 10.1128/AAC.01054-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vu DH, Koster RA, Alffenaar JW, Brouwers JR, Uges DR. 2011. Determination of moxifloxacin in dried blood spots using LC-MS/MS and the impact of the hematocrit and blood volume. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 879:1063–1070. 10.1016/j.jchromb.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 32.Weiner M, Peloquin C, Burman W, Luo CC, Engle M, Prihoda TJ, Mac Kenzie WR, Bliven-Sizemore E, Johnson JL, Vernon A. 2010. Effects of tuberculosis, race, and human gene SLCO1B1 polymorphisms on rifampin concentrations. Antimicrob. Agents Chemother. 54:4192–4200. 10.1128/AAC.00353-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chigutsa E, Visser ME, Swart EC, Denti P, Pushpakom S, Egan D, Holford NH, Smith PJ, Maartens G, Owen A, McIlleron H. 2011. The SLCO1B1 rs4149032 polymorphism is highly prevalent in South Africans and is associated with reduced rifampin concentrations: dosing implications. Antimicrob. Agents Chemother. 55:4122–4127. 10.1128/AAC.01833-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boeree MJ, Plemper van Balen G, Aarnoutse RA. 2011. High-dose rifampicin: how do we proceed? Int. J. Tuberc. Lung Dis. 15:1133. 10.5588/ijtld.11.0198. [DOI] [PubMed] [Google Scholar]
- 35.Boeree M, Diacon AH, Dawson R, Venter A, duBois J, Narunsky K, Hoelscher M, Gillespie SH, Phillips PPJ, Aarnoutse RE. 2013. What is the “right” dose of rifampin? Abstr. 128, 20th Conference on Retroviruses and Opportunistic Infections, Atlanta, GA, 3 to 6 March 2013. [Google Scholar]
- 36.Allanson AL, Cotton MM, Tettey JN, Boyter AC. 2007. Determination of rifampicin in human plasma and blood spots by high performance liquid chromatography with UV detection: a potential method for therapeutic drug monitoring. J. Pharm. Biomed. Anal. 44:963–969. 10.1016/j.jpba.2007.04.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.