

Abstract
The in vitro antimalarial activities of artemisone and artemisone entrapped in Pheroid vesicles were compared, as was their ability to induce dormancy in Plasmodium falciparum. There was no increase in the activity of artemisone entrapped in Pheroid vesicles against multidrug-resistant P. falciparum lines. Artemisone induced the formation of dormant ring stages similar to dihydroartemisinin. Thus, the Pheroid delivery system neither improved the activity of artemisone nor prevented the induction of dormant rings.
TEXT
Since 2006, artemisinin-based combination treatment (ACT) has been the cornerstone of malaria chemotherapy (1). However, high treatment failure rates with artesunate-mefloquine (AS-MQ) (2) and dihydroartemisinin (DHA)-piperaquine (3) in western Cambodia highlight the urgent need for effective ACTs until more potent replacement drugs can be developed.
Although artemisinin and its derivatives are the most potent and rapidly acting drugs for the treatment of Plasmodium falciparum malaria (4), this drug class is associated with high recrudescence. Artemisone (AMS), a new derivative, has potent antiplasmodial activity, good oral bioavailability, and metabolic stability (5) and is well tolerated in humans (6), with an effective curative dose approximately one-third that of artesunate (7). However, use of artemisone alone, as with the other artemisinins, also leads to recrudescence in nonhuman primates (8). A plausible explanation for recrudescence is drug-induced quiescence or dormancy that protects ring-stage parasites against artemisinin exposure (9, 10). The artemisinin-treated ring stages of P. falciparum thereby enter a temporary growth arrest (11, 12), wherein they survive drug treatment, resuming normal growth once drug pressure is removed (13–15).
Formulations involving liposomes and self-emulsifying drug delivery systems enhance the efficacy of anti-infective agents, including antimalarial drugs, such as artemisinins (16–19). A Pheroid delivery system has been shown to increase the in vitro antimalarial activities of azithromycin, mefloquine, and quinine significantly (20, 21). Entrapment of artemisone in Pheroid vesicles also has been shown to enhance blood artemisone concentrations in mice (22) and primates (23). We investigated the effect of a Pheroid formulation on the antimalarial activity of artemisone and on dormancy in vitro. If this formulation prevents the induction of dormancy in vitro, it may circumvent recrudescences occurring following artemisinin treatment.
Chloroquine diphosphate (CQ) and MQ (Sigma-Aldrich, St. Louis, MO), atovaquone (ATQ) (GlaxoSmithKline, Middlesex, United Kingdom), DHA, and AS (DK Pharma, Hanoi, Vietnam) were used. Artemisone and its metabolite M1 were obtained from the Hong Kong University of Science and Technology. The active M1 metabolite is formed via dehydrogenation in the thiomorpholine-dioxide moiety of artemisone (5, 6). The artemisone-entrapped Pheroid vesicles (AMS-Phe) were prepared by adding 30 mM artemisone to a pro-Pheroid formulation (i.e., oil phase only) containing 4.9% polyethylene glycol 400 (PEG-400) instead of 20%, vitamin F ethyl ester, PEGylated ricinoleic acid (Kolliphor), and α-tocopherol as previously described (23). This oil phase was saturated with nitrous oxide. The Pheroid vesicles (microparticles and nanoparticles) are spontaneously formed, entrapping the artemisone when the aqueous RPMI 1640 (low para-aminobenzoic acid/low folic acid [LPLF]) culture medium is added to the oil phase. The AMS-Phe dilutions were prepared in hypoxanthine-free RPMI 1640 (LPLF) complete medium containing 10% human plasma, followed by culture medium containing drug-free pro-Pheroid concentration of 0.004% in each well. There was no difference in in vitro antimalarial activity when artemisone was prepared in the pro-Pheroid formulation and entrapped in Pheroid vesicles (data not shown).
The activities of the drugs against a sensitive P. falciparum D6 line (from Sierra Leone) and five multidrug-resistant lines, W2 (from Indochina), 7G8 (from Brazil), and TM93-1088, TM91-C235, and TM90-C2B (all from Thailand), were assessed using the [3H]hypoxanthine growth inhibition assay (24). Briefly, the assays (in 96-well plates) were initiated when the majority of parasites (>90%) were at the early trophozoite (ring) stage. Parasite cultures (100 μl/well) at 1% initial parasitemia and 2% hematocrit in hypoxanthine-free RPMI 1640 (LPLF) medium were exposed to 10 2-fold serial dilutions of the compounds for 48 h, with [3H]hypoxanthine (0.2 μCi/well) added 24 h after the beginning of the experiment. Three independent experiments were carried out for each compound, with each assay performed in triplicate. The mean 50% (IC50) and 90% (IC90) inhibitory concentrations are presented in Table 1.
TABLE 1.
In vitro antimalarial activities of artemisone and standard drugs against six Plasmodium falciparum linesa
| Drug | IC50 (nM) for: |
|||||
|---|---|---|---|---|---|---|
| D6 | W2 | 7G8 | TM93-C1088 | TM91-C235 | TM-C2B | |
| AMS | 1.0 ± 0.4 | 1.3 ± 0.5 | 0.8 ± 0.1 | 0.7 ± 0.2 | 1.1 ± 0.5 | 1.1 ± 0.4 |
| AMS-Phe | 0.9 ± 0.7 | 1.3 ± 1.0 | 1.0 ± 0.4 | 0.8 ± 0.1 | 1.4 ± 0.7 | 1.1 ± 0.6 |
| M1 | 4.7 ± 0.2 | 6.6 ± 0.4 | 2.6 ± 0.8 | 2.5 ± 0.4 | 5.0 ± 0.6 | 8.6 ± 8.2 |
| DHA | 1.7 ± 0.4 | 2.2 ± 0.8 | 1.3 ± 0.2 | 1.4 ± 0.4 | 2.3 ± 0.7 | 2.0 ± 0.7 |
| CQ | 16 ± 2 | 195 ± 70 | 84 ± 18 | 360 ± 38 | 70 ± 12 | 95 ± 23 |
| MQ | ND | ND | 5.4 ± 1.7 | 16 ± 5.0 | 107 ± 41 | 130 ± 51 |
| ATQ | ND | ND | 3.1 ± 0.9 | 18,830 ± 5,102 | 2.2 ± 0.7 | 31,850 ± 6,833 |
| AS | ND | 3.0 ± 1.6 | 1.5 ± 0.2 | 1.4 ± 0.1 | 2.9 ± 1.4 | 2.5 ± 0.5 |
ND, not determined. W2 is chloroquine resistant, D6 is chloroquine sensitive, 7G8 is chloroquine resistant, TM90-C2B and TM93-C1088 are atovaquone and chloroquine resistant, and TM91-C235 is chloroquine and mefloquine resistant. CQ, chloroquine; MQ, mefloquine; ATQ, atovaquone; AS, artesunate; DHA, dihydroartemisinin; M1, metabolite of artemisone; AMS, artemisone; AMS-Phe, artemisone entrapped in Pheroid vesicles. Values represent the means ± SD from three independent experiments carried out in triplicate. There were no significant differences between AMS and AMS-Phe (P > 0.05) using Student's unpaired t test.
Artemisone had the lowest IC50s and was equally potent against both the drug-sensitive and -resistant P. falciparum lines (Table 1). The range of IC50s for artemisone was approximately 2-fold lower than those of artesunate and dihydroartemisinin. This compares with the 2.4-fold-greater antimalarial activity of artemisone reported by Marfurt (25), but the difference was markedly less than that established by Vivas and colleagues (26). The IC50s for chloroquine, mefloquine, and atovaquone were in good agreement with previous results (12, 27–29). The artemisone metabolite M1 was 3- to 8-fold less active than artemisone but still highly potent, with IC50s ranging from 2.5 to 8.6 nM, which compares well with the value of 4.2 ± 1.3 nM reported against the K1 strain of P. falciparum (6). Artemisone entrapped in Pheroid vesicles was no more active than artemisone in the six P. falciparum lines (P > 0.05) (Table 1).
In order to assess the effect of artemisone on dormancy, synchronous cultures of the Plasmodium strain W2 (ring stages > 95%) with an initial parasitemia of 1% and hematocrit of 4% were exposed to dihydroartemisinin (700 nM; ∼200× IC90) as described by Teuscher et al. (9) and to mefloquine (4,230 nM; ∼100× IC90). Concentrations of artemisone, artemisone entrapped in the Pheroid vesicles, and metabolite M1 of 200, 400, and 800 nM were evaluated. These concentrations cover the maximum plasma concentrations of artemisone and its metabolite measured in healthy subjects (6). The exposure period for all drugs was 6 h. Control drug-free cultures were evaluated in parallel. After exposure for 6 h, the drugs were removed, medium was changed, and each culture (10 ml) was divided into two equal aliquots. When the parasites in the control culture had reached the late trophozoite-schizont stage (∼33 to 36 h), one set of cultures was exposed to 5% d-sorbitol for 5 min to ensure removal of those parasites that had not become dormant but continued to grow. Thin and thick blood films were prepared daily from each culture, stained with Giemsa stain, and examined by light microscopy to determine the parasitemia. Two independent experiments were performed. Culture medium was replaced, and fresh human erythrocytes were added to cultures every 7 days.
The control parasites progressed from rings to trophozoites by 24 h, to schizonts by ∼33 to 36 h, and after schizogony to rings again by 40 h after commencing the experiment. Parasite growth was arrested at the ring stage following a single 6-h exposure to dihydroartemisinin, artemisone, artemisone entrapped in the Pheroid vesicles, and metabolite M1. Morphologically abnormal rings (rings that possessed smaller nuclei and condensed, rounded cytoplasm compared to untreated parasites) and no trophozoites were observed 48 h after commencement of the experiment (Fig. 1). Dormant rings were also seen and had blue-stained cytoplasm with red nuclei, as previously described (9, 12). Unlike the artemisinin derivatives, exposure of rings to mefloquine arrested parasite development at the late ring to early trophozoites stage (Fig. 1).
FIG 1.
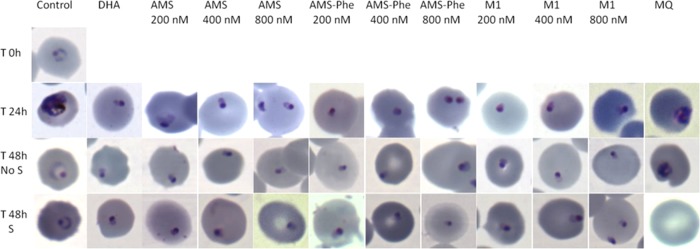
Images of Plasmodium falciparum W2 control parasites and W2 parasites exposed to dihydroartemisinin (DHA), artemisone (AMS), artemisone entrapped in Pheroid vesicles (AMS-Phe), artemisone metabolite M1, and mefloquine (MQ). T, time (h); No S, cultures not treated with sorbitol; S, cultures treated with sorbitol on day 2.
In dihydroartemisinin, artemisone, artemisone entrapped in the Pheroid vesicles, and metabolite cultures that were not exposed to sorbitol, growing parasites (trophozoites) were first detected on day 3 after drug treatment and with a parasitemia of approximately 1% by day 6 (Fig. 2). With sorbitol treatment that selectively kills all late parasite stages, growing parasites were first detected on day 5.0 ± 1.4 (mean ± standard deviation [SD]), and the time to reach 1% parasitemia was 9.0 ± 1.4 days. This delay in recovery indicated that there was a small number of parasites that were unaffected by the artemisinin derivatives. Artemisone and M1 induced dormancy but not in a concentration-dependent manner, since cultures treated with different concentrations of these drugs reached 1% parasitemia within the same time period. As with artemisone, M1, dihydroartemisinin, and artemisone entrapped in the Pheroid vesicles induced dormant parasites, resulting in parasite recovery. No growing parasites were observed in the mefloquine-treated cultures during the 30-day follow-up period. Although artemisone entrapped in Pheroid vesicles increased in vivo drug concentrations in blood (22, 23), it is unlikely that this Pheroid delivery system will prevent recrudescence.
FIG 2.
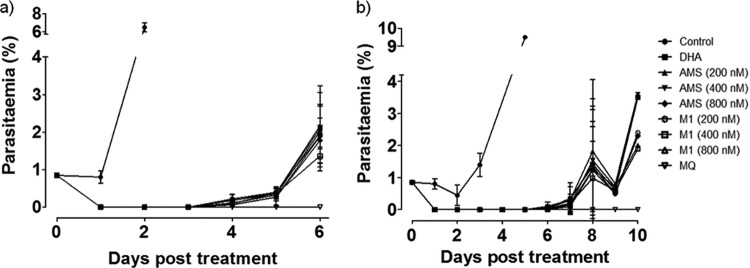
The effect of dihydroartemisinin (DHA), artemisone (AMS), artemisone entrapped in Pheroid vesicles (AMS-Phe), metabolite M1, and mefloquine (MQ) against the Plasmodium falciparum W2 line without (a) or with (b) sorbitol treatment on day 2. Values represent the mean parasitemia ± SD (%) from two independent experiments.
In conclusion, although artemisone is the most active artemisinin derivative against P. falciparum in vitro, it induces dormant ring-stage parasites, and if it is used alone, recrudescence can be expected to occur. Artemisone either entrapped in Pheroid vesicles or alone showed similar in vitro activities, and neither formulation prevented the induction of dormancy in ring-stage parasites. The future of artemisone lies in the selection of a suitable partner drug that can prevent the induction and/or recovery of dormant parasites.
ACKNOWLEDGMENTS
We thank Kerryn Rowcliffe for in vitro cultivation of the P. falciparum strains and the Australian Red Cross Blood Service (Brisbane) for providing human erythrocytes and plasma.
We acknowledge financial support from the Technology Innovation Agency and strategic funds from North-West University.
The opinions expressed herein are ours and do not necessarily reflect those of the Australian Defense Force and/or extant Australian Defense Force policy.
Footnotes
Published ahead of print 6 October 2014
REFERENCES
- 1.WHO. 2010. World malaria report 2010. WHO, Geneva, Switzerland: http://whqlibdoc.who.int/publications/2010/9789241500470_eng.pdf. Accessed 4 January 2011. [Google Scholar]
- 2.Rogers WO, Sem R, Tero T, Chim P, Lim P, Muth S, Socheat D, Ariey F, Wongsrichanalai C. 2009. Failure of artesunate-mefloquine combination therapy for uncomplicated Plasmodium falciparum malaria in southern Cambodia. Malar. J. 8:10. 10.1186/1475-2875-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leang R, Barrette A, Bouth DM, Menard D, Abdur R, Duong S, Ringwald P. 2013. Efficacy of dihydroartemisinin-piperaquine for treatment of uncomplicated Plasmodium falciparum and Plasmodium vivax in Cambodia, 2008 to 2010. Antimicrob. Agents Chemother. 57:818–826. 10.1128/AAC.00686-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White NJ. 2008. Qinghaosu (artemisinin): the price of success. Science 320:330–334. 10.1126/science.1155165. [DOI] [PubMed] [Google Scholar]
- 5.Haynes RK, Fugmann B, Stetter J, Rieckmann K, Heilmann H, Chan H, Cheung M, Lam W, Wong H, Croft SL, Vivas L, Rattray L, Stewart L, Peters W, Robinson BL, Edstein MD, Kotecka B, Kyle DE, Beckermann B, Gerisch M, Radtke M, Schmuck G, Steinke W, Wollborn U, Schmeer K, Römer A. 2006. Artemisone—a highly active antimalarial drug of the artemisinin class. Angew. Chem. Int. Ed. Engl. 45:2082–2088. 10.1002/anie.200503071. [DOI] [PubMed] [Google Scholar]
- 6.Nagelschmitz J, Voith B, Wensing G, Roemer A, Fugmann B, Haynes RK, Kotecka BM, Rieckmann KH, Edstein MD. 2008. First assessment in humans of the safety, tolerability, pharmacokinetics, and ex vivo pharmacodynamic antimalarial activity of the new artemisinin derivative artemisone. Antimicrob. Agents Chemother. 52:3085–3091. 10.1128/AAC.01585-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krudsood S, Wilairatana P, Chalermrut KW, Leowattana W, Voith B, Hampel B, Looareesuwan S. 2005. Artemifone, a new anti-malarial artemisinin derivative: open pilot trial to investigate the antiparasitic activity of bay 44-9585 in patients with uncomplicated P. falciparum malaria, p 142.. In Medicine and health in the tropics. Proceedings of the XVI International Congress for Tropical Medicine and Malaria, Marseilles, France. [Google Scholar]
- 8.Obaldia N, Kotecka BM, Edstein MD, Haynes RK, Fugmann B, Kyle DE, Rieckmann KH. 2009. Evaluation of artemisone combinations in Aotus monkeys infected with Plasmodium falciparum. Antimicrob. Agents Chemother. 53:3592–3594. 10.1128/AAC.00471-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teuscher F, Gatton ML, Chen N, Peters J, Kyle DE, Cheng Q. 2010. Artemisinin-induced dormancy in Plasmodium falciparum: duration, recovery rates, and implications in treatment failure. J. Infect. Dis. 202:1362–1368. 10.1086/656476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Codd A, Teuscher F, Kyle DE, Cheng Q, Gatton ML. 2011. Artemisinin-induced parasite dormancy: a plausible mechanism for treatment failure. Malar. J. 10:56–61. 10.1186/1475-2875-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mok S, Imwong M, Mackinnon MJ, Sim J, Ramaodoss R, Yi P, Mayxay M, Chotivanich K, KekYee L, Russel B, Socheat D, Newton PN, Day NPJ, White NJ, Preiser PR, Nosten F, Dondorp AM, Bozdech Z. 2011. Artemisinin resistance in Plasmodium falciparum is associated with an altered temporal pattern of transcription. BMC Genomics 12:391. 10.1186/1471-2164-12-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tucker MS, Mutka T, Sparks K, Patel J, Kyle DE. 2012. Phenotypic and genotypic analysis of in vitro-selected artemisinin-resistant progeny of Plasmodium falciparum. Antimicrob. Agents Chemother. 56:302–314. 10.1128/AAC.05540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kyle DE, Webster HK. 1996. Postantibiotic effect of quinine and dihydroartemisinin on Plasmodium falciparum in vitro: implications for a mechanism of recrudescence, abstr 0-22-6 Abstr. 14th Int. Congr. Tropic. Med. Malar. [Google Scholar]
- 14.Hoshen MB, Na-Bangchang K, Stein WD, Ginsburg H. 2000. Mathematical modelling of the chemotherapy of Plasmodium falciparum malaria with artesunate: postulation of ‘dormancy', a partial cytostatic effect of the drug, and its implication for treatment regimens. Parasitology 121:237–246. 10.1017/S0031182099006332. [DOI] [PubMed] [Google Scholar]
- 15.LaCrue AN, Scheel M, Kennedy K, Kumar N, Kyle DE. 2011. Effects of artesunate on parasite recrudescence and dormancy in the rodent malaria model Plasmodium vinckei. PLoS One 6:1–12. 10.1371/journal.pone.0026689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isacchi B, Arrigucci S, la Marca G, Bergonzi MC, Vannucchi MG, Novelli A, Bilia AR. 2011. Conventional and long-circulating liposomes of artemisinin: preparation, characterization, and pharmacokinetic profile in mice. J. Liposome Res. 21:237–244. 10.3109/08982104.2010.539185. [DOI] [PubMed] [Google Scholar]
- 17.Isacchi B, Bergonzi MC, Grazioso M, Righeschi C, Pietretti A, Severini C, Bilia AR. 2012. Artemisinin and artemisinin plus curcumin liposomal formulations: enhanced antimalarial efficacy against Plasmodium berghei-infected mice. Eur. J. Pharm. Biopharm. 80:528–534. 10.1016/j.ejpb.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 18.Memvanga PB, Préat V. 2012. Formulation design and in vivo antimalarial evaluation of lipid-based drug delivery systems for oral delivery of β-arteether. Eur. J. Pharm. Biopharm. 82:112–119. 10.1016/j.ejpb.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Chimanuka B, Gabriëls M, Detaevernier M, Plaizier-Vercammen J. 2002. Preparation of beta-artemether liposomes, their HPLC-UV evaluation and relevance for clearing recrudescent parasitaemia in Plasmodium chabaudi malaria-infected mice. J. Pharm. Biomed. Anal. 28:13–22. 10.1016/S0731-7085(01)00611-2. [DOI] [PubMed] [Google Scholar]
- 20.Du Plessis LH, van Niekerk AC, Maritz MM, Kotze AF. 2012. In vitro activity of Pheroid vesicles containing antibiotics against Plasmodium falciparum. J. Antibiot. 65:609–614. 10.1038/ja.2012.89. [DOI] [PubMed] [Google Scholar]
- 21.du Plessis LH, Helena C, van Huysteen E, Wiesner L, Kotzé AF. 2014. Formulation and evaluation of Pheroid vesicles containing mefloquine for the treatment of malaria. J. Pharm. Pharmacol. 66:14–22. 10.1111/jphp.12147. [DOI] [PubMed] [Google Scholar]
- 22.Steyn JD, Wiesner L, du Plessis LH, Grobler AF, Smith PJ, Chan W, Haynes RK, Kotzé AF. 2011. Absorption of the novel artemisinin derivatives artemisone and artemiside: potential application of Pheroid technology. Int. J. Pharm. 414:260–266. 10.1016/j.ijpharm.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 23.Grobler L, Grobler A, Haynes RK, Masimirembwa C, Thelingwani S, Steenkamp P, Steyn HS. 2014. The effect of the Pheroid delivery system on the in vitro metabolism and in vivo pharmacokinetics of artemisone. Expert. Opin. Drug Metab. Toxicol. 10:313–325. 10.1517/17425255.2014.885503. [DOI] [PubMed] [Google Scholar]
- 24.Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. 1979. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 16:710–718. 10.1128/AAC.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marfurt J, Chalfein F, Prayoga P, Wabiser F, Wirjanata G, Sebayang B, Piera KA, Wittlin S, Haynes RK, Möhrle JJ, Anstey NM, Kenangalem E, Price RN. 2012. Comparative ex vivo activity of novel endoperoxides in multidrug-resistant plasmodium falciparum and P. vivax.Antimicrob. Agents Chemother. 56:5258–5263. 10.1128/AAC.00283-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vivas L, Rattray L, Stewart LB, Robinson BL, Fugmann B, Haynes RK, Peters W, Croft SL. 2007. Antimalarial efficacy and drug interactions of the novel semi-synthetic endoperoxide artemisone in vitro and in vivo. J. Antimicrob. Chemother. 59:658–658. 10.1093/jac/dkl563. [DOI] [PubMed] [Google Scholar]
- 27.Korsinczky M, Chen N, Kotecka B, Saul A, Rieckmann K, Cheng Q. 2000. Mutations in Plasmodium falciparum cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob. Agents Chemother. 44:2100–2108. 10.1128/AAC.44.8.2100-2108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winter RW, Kelly JX, Smilkstein MJ, Dodean R, Hinrichs D, Riscoe MK. 2008. Antimalarial quinolones: synthesis, potency, and mechanistic studies. Exp. Parasitol. 118:487–497. 10.1016/j.exppara.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fivelman QL, Adagu IS, Warhurst DC. 2004. Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob. Agents Chemother. 48:4097–4102. 10.1128/AAC.48.11.4097-4102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]