

Abstract
Resistance to the antibacterial antifolate trimethoprim (TMP) is increasing in members of the family Enterobacteriaceae, driving the design of next-generation antifolates effective against these Gram-negative pathogens. The propargyl-linked antifolates are potent inhibitors of dihydrofolate reductases (DHFR) from several TMP-sensitive and -resistant species, including Klebsiella pneumoniae. Recently, we have determined that these antifolates inhibit the growth of strains of K. pneumoniae, some with MIC values of 1 μg/ml. In order to further the design of potent and selective antifolates against members of the Enterobacteriaceae, we determined the first crystal structures of K. pneumoniae DHFR bound to two of the propargyl-linked antifolates. These structures highlight that interactions with Leu 28, Ile 50, Ile 94, and Leu 54 are necessary for potency; comparison with structures of human DHFR bound to the same inhibitors reveal differences in residues (N64E, P61G, F31L, and V115I) and loop conformations (residues 49 to 53) that may be exploited for selectivity.
INTRODUCTION
Infections caused by Gram-negative bacteria are a significant health care burden yet are effectively treated with only a limited number of antibacterial agents. The innate outer membrane of Gram-negative bacteria that presents a natural barrier to the penetration of many known antibiotics and acquired resistance including efflux pumps, drug-modifying enzymes, and plasmid-acquired gene duplication restricts the number of effective agents and exacerbates the ability to treat these infections (1, 2). The increasing prevalence and resistance create a pressing need for new effective antibacterials that target Gram-negative bacteria.
Klebsiella pneumoniae and Escherichia coli, Gram-negative bacteria belonging to the Enterobacteriaceae family, are the most common causes of Gram-negative bacteremia (3, 4). Members of the family Enterobacteriaceae typically cause infections of the urinary tract as well as pneumonia, especially in immune-suppressed patients (3, 4), and are additionally a significant cause of bloodstream infections (5). Resistance in Enterobacteriaceae infections is a serious and expanding problem. Enterobacteriaceae can harbor plasmids encoding resistance to multiple antibiotics, including fluoroquinolones, cephalosporins, and carbapenems (6, 7). Often, K. pneumoniae exhibits an increased prevalence of resistant strains (8–10) over E. coli. The therapeutic options for highly resistant K. pneumoniae are extremely limited; colistin, an older drug with significant toxicity, remains the therapy of choice (2, 11).
Dihydrofolate reductase (DHFR), an essential enzyme for all cells, has been a validated target since the development of methotrexate and trimethoprim in the 1950s (12–14). Despite the fact that DHFR is a highly evolutionarily conserved enzyme across both prokaryotic and eukaryotic organisms, there exist exploitable structural differences that allow for the creation of potent and selective inhibitors of the bacterial enzymes. Trimethoprim-sulfamethoxazole (TMP-SMX) (Bactrim) that selectively targets DHFR and dihydropteroate synthase, respectively, in both Gram-positive and Gram-negative bacteria is the therapy of choice for uncomplicated urinary tract infections (UTIs) (15). However, resistance to TMP-SMX is increasing worldwide in strains of Enterobacteriaceae and now reaches 14 to 30% (4, 16, 17). Resistance generally occurs through the acquisition of a TMP-insensitive DHFR (18–20). A more broadly acting DHFR inhibitor should be achievable by potently inhibiting both wild-type and resistant forms of the enzyme.
We have been developing a series of propargyl-linked antifolates (PLAs) that are potent inhibitors of DHFR from multiple species. One of the strengths of this series is the flexibility and versatility to target TMP-resistant species, regardless of whether that resistance occurs natively (21–23) or via acquired mutations (24, 25). We have achieved success in targeting both wild-type and resistant forms of DHFR from the Gram-positive bacteria, Staphylococcus aureus (25) and aim to apply those design principles to the Gram-negative bacteria, Klebsiella pneumoniae. Excitingly, several of the latest PLAs exhibit MIC values for K. pneumoniae that are ∼1 μg/ml. With these compounds in hand, we determined the structure of the wild-type K. pneumoniae DHFR in order to enable a structure-based approach to the design, synthesis, and evaluation of a broadly effective PLA against K. pneumoniae. Comparisons with previously determined structures of human DHFR bound to the same compounds (26) specifically guide compound design for enhanced selectivity.
MATERIALS AND METHODS
Purification of Klebsiella pneumoniae DHFR.
Gene synthesis and cloning of K. pneumoniae DHFR (KpDHFR) into the pET41a(+) vector were carried out by GenScript resulting in a KpDHFR-HisTag pET41a(+) plasmid. As all experiments in this work use the histidine-tagged (HisTag) protein, it will be referred to as KpDHFR. Escherichia coli BL21(DE3) cells (Novagen) were transformed with the KpDHFR pET41a(+) plasmid. Expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an optical density at 600 nm (OD600) of 0.8 to 1.0, followed by growth for 6 h at 30°C. Cells were harvested and lysed using B-PER reagent (Thermo Scientific) with 1 mM phenylmethylsulfonyl fluoride (PMSF) and 100 U DNase. Soluble lysate was filtered with a 0.45-μm filter and added to a nickel column (Ni-nitrilotriacetic acid [Ni-NTA] agarose; Qiagen) equilibrated with buffer (20 mM Tris [pH 8.0], 0.01 M dithiothreitol [DTT], 10% [vol/vol] glycerol). KpDHFR was eluted by adding increasing amounts of a buffer containing 20 mM Tris (pH 8.0), 200 mM KCl, 0.25 M imidazole, 0.01 M DTT, and 10% (vol/vol) glycerol to the column. Fractions containing KpDHFR were identified by SDS-PAGE, pooled, and concentrated using a Centricon centrifugal filter unit (Millipore). The protein sample was then added to an S200 column equilibrated with buffer (20 mM Tris [pH 7.5], 1 mM DTT, 10% [vol/vol]) glycerol, 1 mM PMSF). Aliquots of purified protein were flash frozen at a concentration of 15 mg/ml with liquid nitrogen and stored at −80°C.
Propargyl-linked antifolates (PLAs) compound 1 (22), compound 2 (21), and compounds 3 to 5 (25) were synthesized and characterized as previously reported.
Enzyme inhibition assay.
Enzyme inhibition assays were performed as previously described (26). Briefly, the assay measures the rate of NADPH oxidation over time at 340 nm with limiting concentrations of enzyme and excess concentrations of NADPH (100 μM) and dihydrofolate (DHF) (100 μM). The assay was carried out in buffer containing 20 mM N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (TES) (pH 7.0), 50 mM KCl, 0.5 mM EDTA, 10 mM β-mercaptoethanol, and 1 mg/ml bovine serum albumin and performed at room temperature. Inhibitors were incubated with the enzyme and NADPH for 5 min prior to the addition of DHF before the reaction is monitored. Measurements were repeated at least three times, with averaged 50% inhibitory concentrations (IC50s) reported along with standard deviations.
MICs.
MICs were determined using the microdilution broth assay based on Clinical and Laboratory Standards Institute (CLSI) conditions and K. pneumoniae strain 10031 (ATCC). Specifically, an inoculum of 1 × 105 CFU/ml was incubated with compound in Isosensitest broth (Oxoid) for 16 h at 37°C. Growth was monitored at A600 using the alamarBlue assay; the MIC is defined as the lowest concentration of inhibitor to completely inhibit growth.
Crystallization.
Pure KpDHFR (15 mg/ml), 1 mM NADPH, and 1 mM inhibitor (compound 3 or 4) were mixed and incubated on ice for 3 h before being concentrated to 24 mg/ml. The KpDHFR/NADPH/compound 4 crystals were grown by adding protein and reservoir solution (100 mM cacodylate [pH 7.4], 150 mM sodium acetate, 25% [wt/vol] polyethylene glycol 8000 [PEG 8000]) in a 1:2 ratio in a 10-μl sitting drop over a 1,000-μl reservoir with an additional 1 mM (final concentration) l-glutathione (Hampton Research) nonvolatile additive that was included after mixing protein and reservoir. KpDHFR/NADPH/compound 3 crystals were grown by adding protein and reservoir solution (100 mM cacodylate [pH 7.4], 125 mM sodium acetate, 25% [wt/vol] PEG 8000, 11 mM calcium chloride) in a 1:2 ratio in a 10-μl sitting drop with an additional 11 mM (final concentration) betaine hydrochloride (Hampton Research) nonvolatile additive. In both cases, crystals grew between 1 and 5 days at 4°C. The crystals were harvested at 4°C and transferred into increasing amounts of ethylene glycol from 4 to 12% (vol/vol) with 2% (wt/vol) sucrose. The crystals were then flash frozen in liquid nitrogen. The final freezing solution for the KpDHFR/NADPH/compound 3 crystal contained 8% (vol/vol) ethylene glycol and 2% (wt/vol) sucrose. Diffraction data were collected at Brookhaven National Laboratory using the X4C beamline. Processing of the KpDHFR/NADPH/compound 4 and KpDHFR/NADPH/compound 3 data sets was performed with HKL2000 and Rigaku Structure Studio Process, respectively. Diffraction statistics are listed in Table 2.
TABLE 2.
Diffraction data and refinement statistics
| Parametera | Value(s) for the following: |
|
|---|---|---|
| KpDHFR/NADPH/compound 4 | KpDHFR/NADPH/compound 3 | |
| PDB ID or accession no. | 4OR7 | 4OSG |
| Space group | P3221 | P1 |
| No. of monomers in asymmetric unit | 1 | 4 |
| Unit cell | ||
| a, b, c (Å) | 61.161, 61.161, 105.426 | 36.12, 74.21, 82.53 |
| α, β, γ (°) | 90, 90, 120 | 67.94, 77.70, 75.92 |
| Resolution (range) (Å) | 37.4–1.76 (1.81–1.76) | 39.3–2.7 (2.77–2.70) |
| Completeness (%) (last shell) | 99.99 (100) | 94.18 (94.96) |
| No. of unique reflections | 25,725 (2,538) | 19,765 (2,017) |
| Redundancy | 8.2 (8.3) | 1.67 (1.66) |
| Rsym (last shell) | 0.072 (0.430) | 0.049 (0.331) |
| <I/σ> (last shell) | 13.3 (1.6) | 13.4 (2.1) |
| R-factor/Rfree | 0.2103/0.2504 | 0.2390/0.2483 |
| No. of atoms (protein, ligands, solvent) | 1,407, 73, 233 | 5,056, 330, 13 |
| RMS deviation bond lengths (Å), angles (°) | 0.013, 2.13 | 0.013, 2.22 |
| Avg B-factor for protein (Å2) | 17.6 | 86.9 |
| Avg B-factor for ligand (Å2) | 18.8 | 93.1 |
| Avg B-factor for solvent molecules (Å2) | 29.8 | 62.6 |
| Residues in most favored regions (%) | 91.7 | 90.8 |
| Residues in additional allowed regions (%) | 8.3 | 9.2 |
| Residues in generously allowed regions (%) | 0 | 0 |
| Residues in disallowed regions (%) | 0 | 0 |
RMS, root mean square.
Structure determination.
Both data sets were initially evaluated with Xtriage in the Phenix-1.8.2-1309 (27) package. The Xiage software revealed that the KpDHFR/NADPH/compound 3 crystal possesses a pseudomerohedral twin occupying approximately 44% of the data set and following operations h, h-k, and h-l, the data were also observed to be anisotropic. These two factors were taken into account during refinement using Phenix. The default setting for determination of the Rfree data set (10%) within Phenix was used; however, Phenix does not account for twinning in Rfree calculations. Structures were determined using molecular replacement using Phaser within the Phenix package. The probe model used for molecular replacement for the KpDHFR/NADPH/compound 4 structure was Escherichia coli dihydrofolate reductase (PDB identifier [ID] or accession no. 1DDR [28]). The refined structure of KpDHFR/NADPH/compound 4 was then used as the probe model in molecular replacement attempts for the KpDHFR/NADPH/compound 3 structure. Prodrg (29) was used to produce cif and pdb files for the inhibitors. The model was refined using NCS and secondary structure restraints with the program, Phenix. Coot (30) was used in the course of model building and density interpretation. There is one molecule of KpDHFR bound to NADPH and compound 4 per asymmetric unit in the KpDHFR/NADPH/compound 4 crystal and four molecules of KpDHFR bound to NADPH and compound 3 in the KpDHFR/NADPH/compound 3 crystal.
RESULTS
Propargyl-linked antifolates inhibit K. pneumoniae.
For several years, we have been optimizing the propargyl-linked antifolates to inhibit the growth of Gram-positive bacteria by inhibiting the DHFR enzymes from these bacteria (24, 25, 31, 32). Relatively hydrophobic biphenyl PLAs (compounds 1 and 2) potently inhibit methicillin-resistant Staphylococcus aureus and Streptococcus pyogenes, but they do not inhibit the growth of Gram-negative species such as Klebsiella pneumoniae, despite apparent inhibition of the K. pneumoniae DHFR enzyme (Table 1). For example, compounds 1 and 2 have IC50s of 15 and 58 nM, respectively, but they have MIC values of >50 μg/ml and 5 μg/ml, respectively (Table 1). However, as the compounds incorporated basic nitrogen atoms in the C-ring and increased polarity, several inhibited the KpDHFR enzyme as well as the growth of K. pneumoniae with MIC values of ∼1 μg/ml. For example, compounds 3 and 5 with a pyridyl C-ring exhibit MIC values of 1.25 or 1.56 μg/ml, respectively (Table 1). The cellular selectivity of compounds 3 to 5 remains high, as they do not inhibit the growth of human cells at concentrations less than 80 μM.
TABLE 1.
Biological evaluation of propargyl-linked antifolates
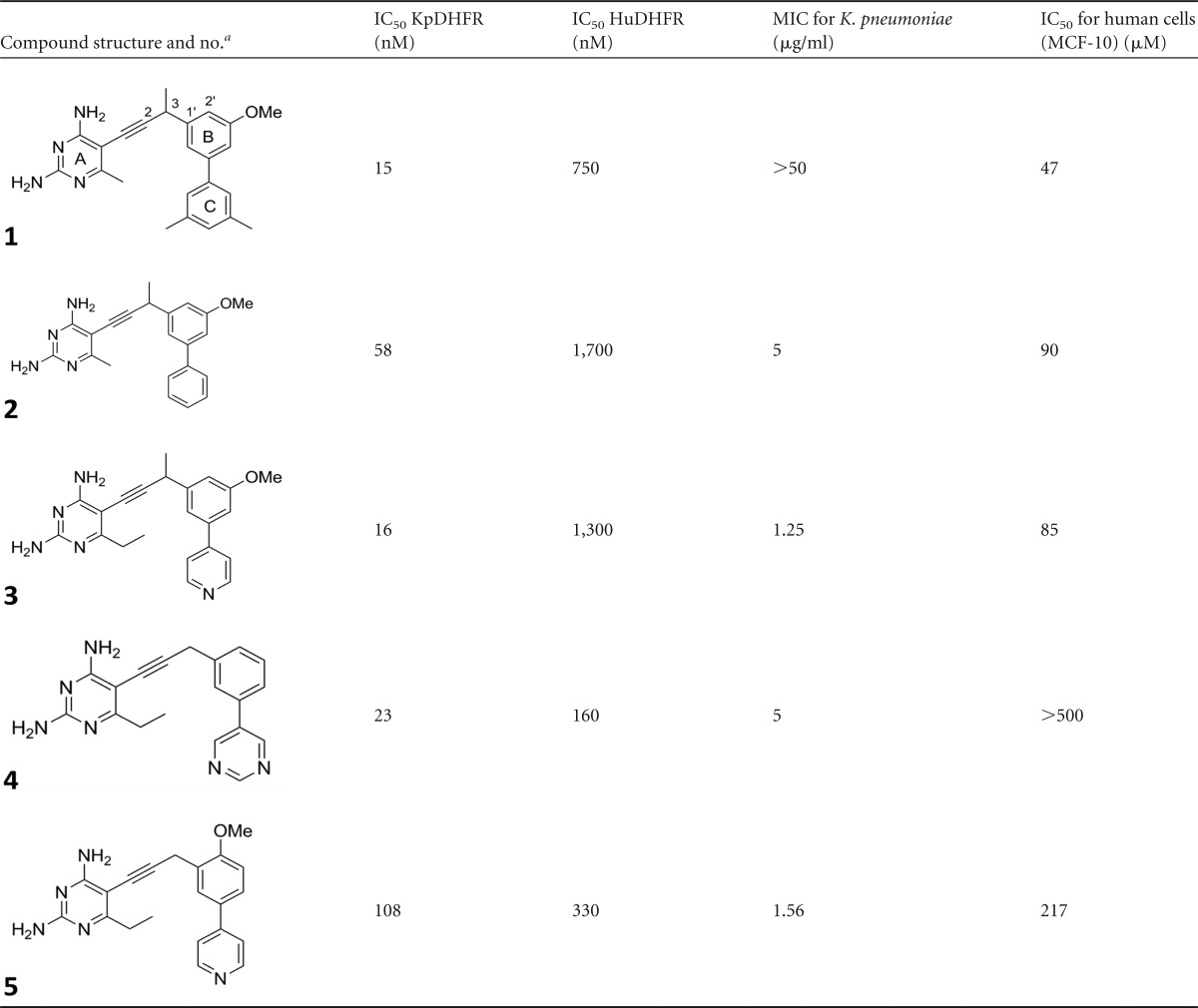
The compound numbers are shown in boldface type under the structures. The A, B, and C rings and positions are labeled in the structure for compound 1. Me, methyl.
Novel structure of Klebsiella pneumoniae DHFR.
In order to further develop the potency and selectivity of the PLA series against wild-type and resistant enzymes using a structure-based approach, we determined the first structure of K. pneumoniae DHFR bound to its cofactor NADPH and the PLA, compound 4. The protein was cocrystallized with compound 4 and NADPH using sitting drops incubated at 4°C; the crystals were then harvested and flash frozen for data collection. The crystal diffracted to a maximum resolution of 1.76 Å (Table 2). The KpDHFR/NADPH/compound 4 crystal belongs to space group P3121 and possesses one molecule of the ternary complex within the asymmetric unit. Similar to DHFR enzymes from other species, the enzyme has an eight-stranded β-sheet flanked by four α-helices (Fig. 1a). As KpDHFR and DHFR from E. coli share 92% sequence identity, the overall structures of the two enzymes are very similar. The acidic active site residue Asp 27 of KpDHFR forms hydrogen bonds with the nitrogen (N1) and 2-amino group in the pyrimidine ring (Fig. 1b; see Fig. S1a in the supplemental material). In addition, the 4-amino group in the same ring forms a hydrogen bond with the backbone carbonyl of Ile 94, and the 2-amino group forms a water-mediated hydrogen bond with Thr 113. The 6-ethyl substituent on the ring forms hydrophobic interactions with Leu 28, contributing to the anchoring capacity of this ring. Other interactions important to the binding of compound 4 in the active site include hydrophobic interactions between Ile 50 and the phenyl B-ring and a weak hydrogen bonding interaction (3.5 Å) between Arg 52 and the nitrogen in the pyrimidine C-ring. Additional hydrophobic interactions between the propargyl linker and Ile 50 as well as those between the pyrimidine ring and Leu 54 and Phe 31 also increase inhibitor affinity.
FIG 1.

K. pneumoniae DHFR bound to NADPH and propargyl-linked antifolates. (a) View of the overall structure of the sole protein chain in the KpDHFR/NADPH/compound 4 structure (NADPH in gray, compound 4 in magenta). (b) Active site of KpDHFR bound to NADPH (gray) and compound 4 (magenta). (c) Active site from chain A of the KpDHFR/NADPH/compound 3 structure (compound 3 in cyan). (d) Overlay of the active sites of chains A (cyan) and D (dark blue) in the KpDHFR/NADPH/compound 3 structure.
Upon reviewing the symmetry mates of the crystal, however, it is apparent that Asp 127 from a symmetry mate forms a hydrogen bond with Arg 52, which likely perturbs the placement of this residue. In fact, it may be possible that Arg 52 is closer to the pyrimidyl nitrogen and thus capable of forming a stronger hydrogen bond than that observed in the current structure. In order to gain a clearer view of the KpDHFR active site, a second crystal structure with KpDHFR/NADPH/compound 3, belonging to a different space group, P1, was determined (Table 2).
Structure of KpDHFR/NADPH/compound 3.
The crystal with KpDHFR/NADPH/compound 3 contains four molecules of the ternary complex in the asymmetric unit. While the maximum resolution of diffraction for this crystal was lower (2.7 Å as opposed to 1.76 Å) and the average B-factors for the overall protein are higher, two of the four molecules (chains A and D) possess active sites free of noncrystallography symmetry and symmetry mate perturbations, permitting an unbiased view of the active site residues. Also, it should be noted that the majority of the active site residues have B-factors lower than the average for the protein. Density for the diaminopyrimidine, propargyl linker, and C-ring of the ligand was observed in the first electron density maps after molecular replacement with the protein probe molecule and after a simulated annealing omit map calculation, lending support for the final placement of these groups (see Fig. S1b and S1d in the supplemental material). As the density for the B-ring was lower, interpretations of the ligand interactions are modulated with this consideration.
The KpDHFR/NADPH/compound 3 structure shows that compound 3 is anchored within the active site via the diaminopyrimidine ring as previously described for KpDHFR/NADPH/compound 4 (Fig. 1c). As in the structure with compound 4, the 6-ethyl group in the compound 3 molecule bound in chain D forms a hydrophobic interaction with Leu 28. In chain A, this interaction is absent, but the 2-amino group forms an additional hydrogen bond with Thr 113 (3.0 Å). While the anchoring hydrogen bonds in the pyrimidine rings remain intact for the inhibitor in chains A and D, the position of the pyrimidine ring in chain D is angled approximately 30° toward Ile 94 relative to that in chain A (see Fig. 1d for a superposition of the two conformations). The dihedral angle, beginning at the propargyl carbons at positions 2 and 3 and continuing through the carbons at positions 1 and 2 on the phenyl group (see notation in Table 1) differs in the two copies, resulting in the B and C rings being shifted relative to each other (Fig. 1d). The angular shift results in several stronger interactions between compound 3 and the residues in chain A relative to those in chain D: hydrophobic interactions between the propargyl methyl and Ile 94 (3.5 Å versus 4.1 Å) and Thr 46 (3.4 Å as opposed to 3.9 Å) as well as apparent hydrophobic interactions between the methoxy group on the B-ring with Met 42. Both conformations of compound 3 have notable hydrophobic interactions with Phe 31 and Leu 28.
The approximate binding energies of the two conformations of compound 3 were calculated using Flexidock (Flexidock within Sybyl-X 1.2; Tripos International, St. Louis, MO, USA). Overall, there appears to be an increase of ∼24% in binding affinity for the inhibitor orientation in chain A compared with that in chain D. The binding energy of compound 4 and that of compound 3 calculated from the orientation in chain A are essentially the same.
A comparison of the structures of KpDHFR/NADPH/compound 4 and chains A and D from KpDHFR/NADPH/compound 3 reveals some of the dynamics within the active site. In particular, the loop comprised of residues 49 to 53 appears to be flexible with distances of up to 1.1 Å between respective Ile 50 Cα atoms (Fig. 2c with human DHFR [HuDHFR]; see Fig. S2 in the supplemental material).
FIG 2.

Comparison of KpDHFR and human DHFR reveals residue changes and flexibility. (a) Comparison of the sole protein chain in the KpDHFR/NADPH/compound 4 structure (protein shown in purple and compound 4 in magenta) with chain A from the human DHFR/NADPH/compound 4 structure (both protein and ligand in salmon) with residue changes noted. (b) Comparison of chain A in the KpDHFR/NADPH/compound 3 structure (both shown in green) and chain A of the human DHFR/NADPH/compound 3 structure (shown in pink). (c) Residues in loop 49 to 53 (KpDHFR numbering) show flexibility in several KpDHFR structures (cyan, blue, and purple) as well as human DHFR structure (pink). Compound 4 is shown for reference. (d) The solvent-accessible surface areas of the KpDHFR (bright pink) active site taken from chain A from KpDHFR/NADPH/compound 3 and human DHFR (chain A from HuDHFR/NADPH/compound 3) active site (darker purple) show two lobes.
Comparison of HuDHFR and KpDHFR structures.
We previously reported structures of human DHFR bound to NADPH and compounds 3 and 4 (26), promoting a direct comparison of residues in the active sites that may be exploited for selectivity designs. There are clearly a number of amino acid residue variations between the two species: Asn 64 (HuDHFR) to Arg 52 (KpDHFR), Pro 61 (HuDHFR) to Gly 51 (KpDHFR), Phe 31 (HuDHFR) to Leu 28 (KpDHFR), and Val 115 (HuDHFR) to Ile 94 (KpDHFR) (Fig. 2a and b). Additionally, the flexible loop noted above (residues 49 to 53 in KpDHFR and residues 58 to 64 in HuDHFR), roughly corresponding to helix C (33), shows even greater conformational change when these structures are compared. For example, the distance between KpDHFR Gly 51 (chain A of the KpDHFR/NADPH/compound 3 structure) and the corresponding residue in HuDHFR, Pro 61, is 4.8 Å (Fig. 2c). A comparison of the solvent-accessible surface areas of HuDHFR and KpDHFR, generated from both DoGSiteScorer (http://dogsite.zbh.uni-hamburg.de/index.html) and the Sybyl docking suite (Flexidock) suggest that the KpDHFR and human DHFR active sites occupy the same general region of the protein but utilize two slightly different “pockets” of the site (Fig. 2d).
The inhibitor compound 4 binds KpDHFR selectively by 14.3-fold compared with HuDHFR. While the pyrimidine ring is shifted by 1 Å in the two structures, the largest difference is that the inhibitor binds in an opposite orientation within the HuDHFR binding site relative to that in the KpDHFR site with differences in dihedral angles as previously defined (see Fig. S3 in the supplemental material for detail): 81° (HuDHFR) and −125° (KpDHFR). The B-ring and C-ring are oriented in different directions within the active sites with the B-ring of compound 4 within KpDHFR pointed toward Ser 49 and Ile 50, while that in HuDHFR is oriented toward Leu 67 and Ile 60. The C-ring of compound 4 within KpDHFR is oriented toward Leu 28, while that in HuDHFR is oriented toward Ile 60/Pro 61 and Asn 64 (Fig. 2a). The resulting effect is that one of the nitrogen atoms in the pyrimidine ring of compound 4 approaches Arg 52 to form a very weak hydrogen bond (3.5 Å) while it is too far from the corresponding residue, Asn 64 in HuDHFR (4.6 Å). The 6-ethyl group forms hydrophobic interactions with Leu 28 (3.7 Å) in KpDHFR, but the same group is 4.4 Å away from Phe 31 in HuDHFR. Additionally, there are weaker interactions between the B-ring and Ile 60/Leu 67 in human DHFR relative to those in KpDHFR with Ile 50/Leu 54.
Comparing the interactions of compound 3 between HuDHFR and KpDHFR explains the greater potency (7-fold) of the ligand for KpDHFR. The orientation of compound 3 in chain A of KpDHFR enables the propargylic methyl group to form van der Waals interactions with Ile 94, which is not possible with the analogous residue (Val 115) in HuDHFR. Additionally, there are greater van der Waals interactions with Phe 31, Ile 50, and Leu 54 in KpDHFR relative to the analogous residues in HuDHFR (Fig. 2a).
DISCUSSION
The increase in resistance to currently available therapies for K. pneumoniae infections makes the development of new antibiotics critical. Over the past several years, strains of K. pneumoniae resistant to the clinically used antifolate trimethoprim have arisen; these strains predominantly carry a plasmid-encoded copy of resistant DHFR. We have been developing the propargyl-linked antifolates (PLAs) to be potent and selective inhibitors of wild-type and trimethoprim-resistant species of DHFR (23, 24, 34). Here, we show that the PLAs inhibit DHFR from K. pneumoniae and inhibit the growth of K. pneumoniae. In an effort to elucidate the basis of potency and selectivity evident in these initial compounds and to enrich future design, we determined two crystal structures bound to two PLAs in the KpDHFR active site. These structures may be used to understand the interactions of the ligands with the protein and to form the foundation for the design of inhibitors active against both wild-type and TMP-resistant forms of the enzyme.
The two structures of KpDHFR bound to NADPH and two PLAs; compounds 4 and 3 reveal several features critical for potency. In addition to the anchoring hydrogen bonds of the diaminopyrimidine ring, there are key hydrophobic interactions between the 6-ethyl position on the pyrimidine and Leu 28. The propargyl linker forms hydrophobic interactions with Ile 50, and the propargylic methyl substituent interacts with Ile 94 and Thr 46. The B-ring interacts with Ile 50 and Met 42, and the C-ring interacts with Leu 54 and Phe 31. The structures also reveal that loop residues 49 to 53 are quite flexible; this observation has been noted previously in molecular dynamics studies of other species of DHFR (35).
Selectivity over the human form of the enzyme is important for achieving low toxicity. A comparison of the structures of KpDHFR and human DHFR bound to the same ligands shows that there are a number of residue substitutions (N64E, P61G, F31L, and V115I) that provide excellent potential sites for garnering selectivity. For example, a hydrogen bond donor could be placed on the C-ring to form a selective interaction with KpDHFR Arg 52 (Asn 64 in HuDHFR), or a hydrophilic group could be added to the B-ring to interfere with the human Pro 61 residue without disrupting KpDHFR interactions. Additionally, flexibility in loop residues 49 to 53 may allow an alternate conformation of compound 4 in the active site of KpDHFR relative to human DHFR. The selection of such an alternate binding conformation of a ligand yields new opportunities for selective interactions. In summary, the critical points for selectivity appear to involve interactions between the 6-ethyl pyrimidyl group and Leu 28, the B-ring with Ile 50 and Leu 54, and the propargylic methyl group with Ile 94.
The expansion of antibacterial activity of the propargyl-linked antifolates into the Gram-negative pathogen K. pneumoniae presents an exciting opportunity to develop new compounds against wild-type and resistant bacterial strains. The high-resolution crystal structures presented here will guide the design of this class and potentially other antifolates toward the development of efficacious antibiotics.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge funding from the NIH (AI104841).
Nigel Priestley is a founder and CEO of Promiliad Biopharma. Jeremy Alverson is an employee of Promiliad Biopharma. Dennis Wright and Amy Anderson are inventors on a patent that describes the propargyl-linked antifolates and is licensed by Promiliad Biopharma.
Footnotes
Published ahead of print 6 October 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03555-14.
REFERENCES
- 1.Rice L. 2008. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 197:1079–1081. 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 2.Boucher H, Talbot G, Bradley J, Edwards J, Gilbert D, Rice L, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48:1–12. 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 3.Meatherall B, Gregson D, Ross T, Pitout J, Laupland K. 2009. Incidence, risk factors, and outcomes of Klebsiella pneumoniae bacteremia. Am. J. Med. 122:866–873. 10.1016/j.amjmed.2009.03.034. [DOI] [PubMed] [Google Scholar]
- 4.Tsay R, Siu L, Fung C, Chang F. 2002. Characteristics of bacteremia between community-acquired and nosocomial Klebsiella pneumoniae infection: risk factor for mortality and the impact of capsular serotypes as a herald for community-acquired infection. Arch. Intern. Med. 162:1021–1027. 10.1001/archinte.162.9.1021. [DOI] [PubMed] [Google Scholar]
- 5.Peleg A, Hooper D. 2010. Hospital-acquired infections due to gram-negative bacteria. N. Engl. J. Med. 362:1804–1813. 10.1056/NEJMra0904124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yigit H, Queenan A, Anderson G, Domenech-Sanchez A, Biddle J, Steward C, Alberti S, Bush K, Tenover F. 2001. Novel carbapenem-hydrolyzing beta-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob. Agents Chemother. 45:1151–1161. 10.1128/AAC.45.4.1151-1161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradford PA, Bratu S, Urban C, Visalli M, Mariano N, Landman D, Rahal JJ, Brooks S, Cebular S, Quale J. 2004. Emergence of carbapenem-resistant Klebsiella species possessing the class A carbapenem-hydrolyzing KPC-2 and inhibitor-resistant TEM-30 beta-lactamases in New York City. Clin. Infect. Dis. 39:55–60. 10.1086/421495. [DOI] [PubMed] [Google Scholar]
- 8.Bush K. 2010. Alarming β-lactamase-mediated resistance in multidrug-resistant Enterobacteriaceae. Curr. Opin. Microbiol. 13:558–564. 10.1016/j.mib.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Souli M, Galani I, Giamarellou H. 2009. Emergence of extensively drug-resistant and pandrug-resistant Gram-negative bacilli in Europe. Euro Surveill. 13(47):pii=19045 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19045. [PubMed] [Google Scholar]
- 10.European Centre for Disease Prevention and Control. 2012. Antimicrobial resistance surveillance in Europe 2011. Annual Report of the European Antimicrobial Resistance Surveillance Network (EARS-Net). European Centre for Disease Prevention and Control, Stockholm, Sweden. [Google Scholar]
- 11.Ho J, Tambyah P, Paterson D. 2010. Multiresistant Gram-negative infections: a global perspective. Curr. Opin. Infect. Dis. 23:546–553. 10.1097/QCO.0b013e32833f0d3e. [DOI] [PubMed] [Google Scholar]
- 12.Hawser S, Lociuro S, Islam K. 2006. Dihydrofolate reductase inhibitors as antibacterial agents. Biochem. Pharmacol. 71:941–948. 10.1016/j.bcp.2005.10.052. [DOI] [PubMed] [Google Scholar]
- 13.Purcell W, Ettinger D. 2003. Novel antifolate drugs. Curr. Oncol. Rep. 5:114–125. 10.1007/s11912-003-0098-3. [DOI] [PubMed] [Google Scholar]
- 14.Wright D, Anderson A. 2011. Antifolate agents: a patent review (2006–2010). Expert Opin. Ther. Pat. 21:1293–1308. 10.1517/13543776.2011.587804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hooton T. 2003. The current management strategies for community-acquired urinary tract infection. Infect. Dis. Clin. North Am. 17:303–332. 10.1016/S0891-5520(03)00004-7. [DOI] [PubMed] [Google Scholar]
- 16.Zhanel G, Karlowsky J, Harding G, Carrie A, Mazzulli T, Low D, The Canadian Urinary Isolate Study Group. Hoban D. 2000. A Canadian national surveillance study of urinary tract isolates from outpatients: comparison of the activities of trimethoprim-sulfamethoxazole, ampicillin, mecillinam, nitrofurantoin, and ciprofloxacin. Antimicrob. Agents Chemother. 44:1089–1092. 10.1128/AAC.44.4.1089-1092.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia Garcia M, Munoz Bellido J, Garcia Rodriguez J, Spanish Cooperative Group for the Study of Antimicrobial Susceptibility of Community Uropathogens 2007. In vitro susceptibility of community-acquired urinary tract pathogens to commonly used antimicrobial agents in Spain: a comparative multicenter study (2002–2004). J. Chemother. 19:263–270. 10.1179/joc.2007.19.3.263. [DOI] [PubMed] [Google Scholar]
- 18.Blahna M, Zalewski C, Reuer J, Kahlmeter G, Foxman B, Marrs C. 2006. The role of horizontal gene transfer in the spread of trimethoprim-sulfamethoxazole resistance among uropathogenic Escherichia coli in Europe and Canada. J. Antimicrob. Chemother. 57:666–672. 10.1093/jac/dkl020. [DOI] [PubMed] [Google Scholar]
- 19.Brolund A, Sundqvist M, Kahlmeter G, Grape M. 2010. Molecular characterisation of trimethoprim resistance in Escherichia coli and Klebsiella pneumoniae during a two-year intervention on trimethoprim use. PLoS One 5:e9233. 10.1371/journal.pone.0009233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seputiene V, Povilonis J, Ruzauskas M, Pavilonis A, Suziedeliene E. 2010. Prevalence of trimethoprim resistance genes in Escherichia coli isolates of human and animal origin in Lithuania. J. Med. Microbiol. 59:315–322. 10.1099/jmm.0.015008-0. [DOI] [PubMed] [Google Scholar]
- 21.Bolstad D, Bolstad E, Frey K, Wright D, Anderson A. 2008. A structure-based approach to the development of potent and selective inhibitors of dihydrofolate reductase from Cryptosporidium. J. Med. Chem. 51:6839–6852. 10.1021/jm8009124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J, Bolstad D, Smith A, Priestley N, Wright D, Anderson A. 2008. Structure-guided development of efficacious antifungal agents targeting Candida glabrata dihydrofolate reductase. Chem. Biol. 15:990–996. 10.1016/j.chembiol.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beierlein J, Frey K, Bolstad D, Pelphrey P, Joska T, Smith A, Priestley N, Wright D, Anderson A. 2008. Synthetic and crystallographic studies of a new inhibitor series targeting Bacillus anthracis dihydrofolate reductase. J. Med. Chem. 51:7532–7540. 10.1021/jm800776a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frey K, Liu J, Lombardo M, Bolstad D, Wright D, Anderson A. 2009. Crystal structures of wild-type and mutant methicillin-resistant Staphylococcus aureus dihydrofolate reductase reveal an alternative conformation of NADPH that may be linked to trimethoprim resistance. J. Mol. Biol. 387:1298–1308. 10.1016/j.jmb.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Viswanathan K, Frey K, Scocchera E, Martin B, Swain P, Alverson J, Priestley N, Anderson A, Wright D. 2012. Toward new therapeutics for skin and soft tissue infections: propargyl-linked antifolates are potent inhibitors of MRSA and Streptococcus pyogenes. PLoS One 7:e29434. 10.1371/journal.pone.0029434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamb K, G-Dayanandan N, Wright D, Anderson A. 2013. Elucidating features that drive the design of selective antifolates using crystal structures of human dihydrofolate reductase. Biochemistry 52:7318–7326. 10.1021/bi400852h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams P, Afonine P, Bunkóczi G, Chen V, Davis I, Echools N, Headd J, Hung L-W, Kapral G, Grosse-Kunstleve R, McCoy A, Moriarty N, Oeffner R, Read R, Richardson D, Richardson J, Terwilliger T, Zwart P. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dunbar J, Yennawar H, Banerjee S, Luo J, Farber G. 1997. The effects of denaturants on protein structure. Protein Sci. 6:1727–1733. 10.1002/pro.5560060813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schüttelkopf A, van Aalten D. 2004. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60:1355–1363. 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.Frey K, Lombardo M, Wright D, Anderson A. 2010. Towards the understanding of resistance mechanisms in clinically isolated trimethoprim-resistant, methicillin-resistant Staphylococcus aureus dihydrofolate reductase. J. Struct. Biol. 170:93–97. 10.1016/j.jsb.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frey K, Viswanathan K, Wright D, Anderson A. 2012. Prospectively screening novel antibacterial inhibitors of dihydrofolate reductase for mutational resistance. Antimicrob. Agents Chemother. 56:3556–3562. 10.1128/AAC.06263-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bolin J, Filman D, Matthews D, Hamlin R, Kraut J. 1982. Crystal structures of Escherichia coli and Lactobacillus casei dihydrofolate reductase refined at 1.7 A resolution. I. General features and binding of methotrexate. J. Biol. Chem. 257:13650–13662. [PubMed] [Google Scholar]
- 34.Zhou W, Scocchera E, Wright D, Anderson A. 2013. Antifolates as effective antimicrobial agents: new generations of trimethoprim analogs. Med. Chem. Comm. 4:908–915. 10.1039/c3md00104k. [DOI] [Google Scholar]
- 35.Paulsen J, Viswanathan K, Wright D, Anderson A. 2013. Structural analysis of the active sites of dihydrofolate reductase from two species of Candida uncovers ligand-induced conformational changes shared among species. Bioorg. Med. Chem. Lett. 23:1279–1284. 10.1016/j.bmcl.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.