

Abstract
Combinations of group A and B streptogramins (i.e., dalfopristin and quinupristin) are “last-resort” antibiotics for the treatment of infections caused by Gram-positive pathogens, including methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus faecium. Resistance to streptogramins has arisen via multiple mechanisms, including the deactivation of the group A component by the large family of virginiamycin O-acetyltransferase (Vat) enzymes. Despite the structural elucidation performed for the VatD acetyltransferase, which provided a general molecular framework for activity, a detailed characterization of the essential catalytic and antibiotic substrate-binding determinants in Vat enzymes is still lacking. We have determined the crystal structure of S. aureus VatA in apo, virginiamycin M1- and acetyl-coenzyme A (CoA)-bound forms and provide an extensive mutagenesis and functional analysis of the structural determinants required for catalysis and streptogramin A recognition. Based on an updated genomic survey across the Vat enzyme family, we identified key conserved residues critical for VatA activity that are not part of the O-acetylation catalytic apparatus. Exploiting such constraints of the Vat active site may lead to the development of streptogramin A compounds that evade inactivation by Vat enzymes while retaining binding to their ribosomal target.
INTRODUCTION
Due to resistance, the treatment of infections caused by Gram-positive bacteria, such as Staphylococcus aureus and Enterococcus spp., is increasingly difficult (1). This necessitates the search for alternative chemical entities with antimicrobial activity or the modification of existing scaffolds to be less prone to known resistance mechanisms. Streptogramins, comprising a mixture of two structurally distinct classes (group A and group B, i.e., dalfopristin and quinupristin; Fig. 1A) were thought to represent a useful class of antibiotics against vancomycin-resistant enterococci (VRE) (2). These compounds are synthetically modified variants of natural molecules produced by Streptomyces pristinaespiralis; in combination, they are bactericidal and inhibit protein synthesis via their synergistic binding to distinct but overlapping sites on the 50S ribosomal subunit (3).
FIG 1.

Overall structure of VatA. (A) Chemical structures of streptogramin A compounds virginiamycin M1 and dalfopristin. The O-18 arrows indicate the sites of O-acetylation. (B) Top, domain architecture (cap, LβH, α domain, C-terminal/CT) of the three chains of trimeric VatA. Bottom, structure of Vat, colored according to the top domain architecture diagram. The ternary complex shown was modeled by the superposition of the crystal structures of the binary complexes of VatA-virginiamycin M1 and VatA-acetyl-CoA. Virginiamycin M1 and acetyl-CoA are shown in a stick representation. Yellow stars indicate the locations of the catalytic centers. The subscripts after the domain names, acetyl-CoA ligands, or termini (N, N terminus; C, C terminus) indicate the protein chain. (C) Superposition of VatA and VatD structures (8, 9). The VatA chains are colored in red, blue, and green, and all chains of VatD are colored dark gray.
A molecular understanding of the mechanisms of resistance may prolong the usefulness of established antimicrobials. In the context of alleviating enzyme-mediated resistance, two main approaches are employed. The inhibition of the resistance-conferring enzymes by compounds that are not necessarily antimicrobial in nature has been shown to increase the effectiveness of the main antimicrobial (4). In the other approach, reduced susceptibility to modification/inactivation of the established antimicrobial is achieved via chemical modification of its structure. The chemical modification approach is more widely applied and has been employed, for example, in the development of broad-spectrum β-lactam antibiotics that are poorer substrates for β-lactamase (5). Structure-guided studies of antibiotic-modifying enzymes allow a focus on such chemical modification and drug development by identifying the regions of the active site that are functionally critical.
In this vein, a structural understanding of streptogramin resistance mechanisms may provide the necessary clues to improve these compounds. There are currently four established mechanisms of resistance to streptogramins (6). O-Acetylation by Virginiamycin acetyltransferase (Vat) enzymes is sufficient to cause resistance to the streptogramin A-B combination.
The Vat-catalyzed acetylation of the O18 of streptogramin A compounds decreases their affinity for their ribosomal binding site by disrupting multiple critical drug target contacts formed by this group (3). Vat enzymes comprise a sequence-related family (average 60% sequence identity at the protein level) and are encoded by the plasmid-borne genes vat(A), vat(B), and vat(C) from S. aureus, vat(D)/sat(A), vat(E)/sat(G), and vat(H) from Enterococcus faecium, and the vat(F) chromosomal gene from the Gram-negative bacterium Yersinia enterocolitica. Putative vat genes have also been identified in environmental microbial species (7).
In spite of the identification of numerous and widespread Vat enzymes in clinical isolates, little is known about the molecular mechanisms of their activity. The first insights into the molecular characteristics of the Vat enzyme family were based on structural data obtained for VatD (8, 9). The VatD structure features a left-handed β-helix (LβH) fold (10) that is closely related to the fold of xenobiotic acetyltransferases (XAT), such as chloramphenicol acetyltransferase (CAT) (11). The VatD enzyme forms a triangular-shaped homotrimer, with three catalytic centers to accommodate the acetyl-coenzyme A (CoA) and virginiamycin M1-dalfopristin cosubstrates, located at each of the interchain interfaces. These studies established that the O-acetylation reaction is dependent on a conserved histidine residue thought to act as a catalytic base. Due to the significant sequence conservation of Vat enzymes, the general fold, locations of the catalytic centers, and enzymatic reaction are all expected to be conserved. However, the streptogramin A specificity of Vat enzymes and the roles of other active-site residues in the O-acetylation reaction remain unexplored.
We conducted a detailed structural and functional characterization of VatA from S. aureus (12). This identified several active-site residues essential in the catalytic mechanism and for substrate recognition. Thus, these results highlighted key regions of the streptogramin A scaffold critical for recognition by Vat enzymes that might be exploited in the development of next-generation compounds of this class.
MATERIALS AND METHODS
Antibiotics.
Virginiamycin M1 and dalfopristin were obtained as a gift from Sanofi-Aventis.
Cloning, protein expression, and purification.
The full-length vat(A) sequence was cloned in plasmid pET28a. The sequence for residues 7 to 219 of VatA was subcloned in the Gateway-compliant ligation-independent cloning vector p15Tv-LIC (13) leading to an N-terminal His6-tagged protein with a tobacco etch virus (TEV) protease cleavage site between the tag and residue 7 of VatA. The protein was expressed and purified as described for APH(4)-Ia (14). The molecular weights in solution were verified by size exclusion chromatography by loading 10 to 30 μl of 60 and 30 mg/ml VatA and VatD protein, respectively, onto a Superdex 200 10/300 GL size exclusion column (GE Healthcare) using the running buffer (0.3 M NaCl, 50 mM HEPES [pH 7.5], 0.5 mM tris[2-carboxyethyl]phosphine).
Mutagenesis.
Mutations in vat(A) were introduced using the QuikChange mutagenesis kit (Stratagene) with oligonucleotide pairs (see Table S1 in the supplemental material).
Crystallization.
VatA (apo) was crystallized at room temperature using the hanging drop method, with 1 μl of 60 mg/ml protein solution mixed with 1 μl of reservoir solution (2 M ammonium sulfate, 0.2 M potassium-sodium tartrate, and 0.1 M trisodium citrate dihydrate [pH 5.6]). This crystal was cryoprotected with reservoir solution supplemented with 14% glycerol into a liquid nitrogen stream and then annealed into Paratone oil. VatA (acetyl-CoA [AcCoA] complex) was crystallized by soaking pregrown VatA (apo) crystals into a drop containing reservoir solution (2 M ammonium sulfate, 0.1 M HEPES [pH 7.5], 2% [wt/vol] polyethylene glycol 400 [PEG 400]) supplemented with 2.5 mM acetyl-CoA. This crystal was cryoprotected with reservoir solution supplemented with 25% ethylene glycol into a liquid nitrogen stream. VatA (virginiamycin M1 complex) underwent cocrystallization using the hanging drop method, in which 5 mg of virginiamycin M1 in solid phase was added to 1 μl of 60 mg/ml protein solution mixed with 1 μl of reservoir solution (2 M ammonium sulfate, 0.1 M HEPES [pH 7.5], and 2% [wt/vol] PEG 400). This crystal was cryoprotected with reservoir solution supplemented with 20% ethylene glycol in a liquid nitrogen stream.
X-ray diffraction data collection and structural determination.
For VatA (apo), diffraction data at 100 K were collected at the Structural Genomics Consortium on a Rigaku MicroMax-007HF rotating anode source with a Rigaku Saturn A200 CCD. For VatA (acetyl-CoA complex) and VatA (virginiamycin M1 complex), diffraction data at 100 K were collected at the Advanced Photon Source (APS), Argonne National Laboratory, Life Sciences Collaborative Access Team beamline 21-ID-F, fitted with a MarMosaic 225 CCD. All X-ray data were reduced with HKL-3000 (15). The VatA (apo) structure was solved by molecular replacement using the structure of VatD (PDB accession no. 1MR7 [9]) and Phenix.phaser (16). Refinement was performed using Phenix.refine and Coot (17), with translation-libration-screw-rotation (TLS) parametrization (the TLS groups were residues 7 to 84, 85 to 113, and 114 to 217). Geometry was verified using the Phenix (16) and Coot validation tools plus the Protein Data Bank (PDB) Adit server. The structures have good backbone geometry (as verified by the Research Collaboratory for Structural Bioinformatics PDB validation server) with the following percentage of residues in the most favored, additional allowed, generously allowed, and disallowed regions, respectively, of the Ramachandran plots: VatA (apo), 88.1, 10.8, 1.1, and 0%; VatA (virginiamycin M1 complex), 88.9, 10.0, 1.1, and 0%; and VatA (acetyl-CoA complex), 91.5, 7.6, 0.9, and 0%. The presence of virginiamycin M1 and acetyl-CoA was validated using omit maps: all atoms of the ligand were deleted, followed by simulated annealing (Cartesian) using Phenix.refine, with default parameters, followed by model building into residual positive Fo–Fc density. The occupancy values for virginiamycin M1 and acetyl-CoA were refined. B factors were refined as anisotropic for VatA atoms and isotropic for nonprotein atoms. All inspections of electron density were carried out using Coot. The average B factor and bond angle/bond length root mean square deviation (RMSD) values were calculated using Phenix.
Structural analysis.
Structural superpositions were performed using PyMOL (30). Interactions between the protein and substrates were identified using Coot and PyMOL. Crystallographic and noncrystallographic protein-protein interfaces were determined using the PDBePISA server (18). Interactions between VatA and virginiamycin M1 were chosen as those ≤4.1 Å in distance.
Sequence analysis.
BLAST searches of the NCBI nonredundant (nr) database were conducted using the VatD sequence as a query, and hits of >55% identity over the full length of hit and query sequences were retained. These sequences were aligned with the MUSCLE algorithm within the Jalview package (19). Representative sequences were chosen for alignment in Fig. 2A (Bacillaceae sequences with >90% identity to another sequence were omitted for clarity), with visualization using Jalview, while all sequences were used for phylogenetic reconstruction in Fig. 2B. Phylogenetic reconstruction was done with the maximum likelihood algorithm in MEGA (20) after selection of the best substitution model (Whelan and Goldman [WAG], Gamma distribution of sites), including 500 bootstrap replicates, and the tree was visualized with FigTree (http://tree.bio.ed.ac.uk/software/figtree). For the phylogenetic analysis, the outgroup sequences of two CAT enzymes, xenobiotic acetyltransferase from Pseudomonas aeruginosa (PDB accession no. 1XAT [11]) and chloramphenicol acetyltransferase (PDB accession no. 3EEV), plus perosamine N-acetyltransferase PerB (PDB accession no. 4EA9 [21]) were chosen to represent LβH fold-containing enzymes to which streptogramin A acetyltransferases are structurally related but that are not expected to be active against streptogramin A. These sequences were added to the MUSCLE-derived alignment by structure-based alignment with MUSTANG (22).
FIG 2.

Sequence analysis of Vat and closely related enzymes. (A) Multiple-sequence alignment. The sequence names are colored by groups as defined below, and the numbers refer to NCBI gi numbers. The domain architecture is indicated above the sequences. VatA residues that contacted an antibiotic (virginiamycin M1) are labeled. (B) Maximum likelihood phylogenetic tree constructed from alignment. The cluster 1 and 2 Vat enzymes are highlighted in red and blue, respectively. The bootstrap percentage is indicated at each node. The arrows refer to the tree root, and the dashed line indicates the branches for the outgroup sequences, which exceed the size of the page. The scale bar represents 0.2 substitutions per site.
Enzyme activity assay.
Due to limited solubility of virginiamycin M1 and dalfopristin in water, and since dimethyl sulfoxide interferes with the acetyltransferase assay (23), the compounds were solubilized in ethanol. A 35 mM stock solution of virginiamycin M1 was prepared in 80% ethanol, and a 100 mM stock solution of dalfopristin was prepared in 50% ethanol. The enzyme assays were performed as described previously (23), with the following modifications. The 50-μl reaction mixture contained 50 mM Tris-HCl (pH 8.0), 1 mM acetyl-CoA (AcCoA), and streptogramin A compound at the indicated concentrations. The enzymes were diluted in dilution buffer (50 mM Tris-HCl [pH 8.0], 500 mM sodium chloride), and 10 μl of enzyme was used to initiate the reactions, making the final concentration of sodium chloride 100 mM. The reactions were allowed to proceed for 10 min at 37°C and were stopped as described previously (23). Substrate saturation curves were produced by varying the concentration of virginiamycin M1 or dalfopristin (0 to 1 mM) while keeping the AcCoA concentration constant at 1 mM. The compounds were diluted from concentrated stock solutions to obtain solutions with a final concentration of ethanol at 5%. The final concentration of ethanol in each reaction mixture was 2%. In this assay, one unit of activity is the amount of enzyme that produces 1 μmol of CoA per min. The kinetic parameters for the wild-type (WT) and mutant enzymes were determined from substrate saturation curves of virginiamycin M1 or dalfopristin at 1 mM AcCoA. The concentration of enzyme used in each reaction mixture was 29 nM. The kinetic parameters were determined from plots of velocity versus various concentrations of streptogramin A compound by fitting data to a modified Hill equation: V = (Vo + [Vmax − Vo] [C]n)/(kn + [C]n) using Origin 8v1, as described previously. C is the concentration of streptogramin A compound, and k is equivalent to the S0.5, which is the concentration of substrate (streptogramin A compound) needed to reach half of the maximal velocity (Vmax).
Protein structure accession numbers.
The structures of VatA, VatA plus virginiamycin M1, and VatA plus acetyl-CoA have been deposited to the Protein Data Bank under the accession no. 4MYO, 4HUS, and 4HUR, respectively.
RESULTS
VatA crystal structure revealed a VatD-like fold but a distinct oligomeric form.
As a first insight into the molecular details of the function of S. aureus VatA, we determined the crystal structure in the apoenzyme form (Fig. 1B). The structure was solved to 2.7 Å by molecular replacement using the VatD structure (PDB accession no. 1MR7) as a model (Table 1). The asymmetric unit of the VatA crystal contained three protein chains marked by a 3-fold noncrystallographic symmetry axis through the center of the triangular trimer (Fig. 1B). The conformations of the main-chain residues comprising each chain in the VatA trimer were essentially identical (0.28 to 0.41 Å root mean square deviation [RMSD] in pairwise superpositions of 173 to 181 matching Cα atoms).
TABLE 1.
X-ray diffraction data collection and refinement statistics
| Characteristic | Data for liganda |
||
|---|---|---|---|
| None | Virginiamycin M1 | Acetyl-CoA | |
| PDB code | 4MYO | 4HUS | 4HUR |
| Data collection | |||
| Space group | C2221 | C2221 | C2221 |
| Cell dimensions (a, b, c) (Å) | 91.8, 184.0, 96.9 | 92.9, 184.5, 99.0 | 93.3, 184.7, 98.6 |
| Resolution (Å) | 25.0–2.70 | 30.0–2.02 | 30.0–2.30 |
| Rsymb | 0.078 (0.368) | 0.091 (0.507) | 0.079 (0.576) |
| I/σ (I) | 10.74 (2.01) | 19.0 (2.13) | 25.7 (2.64) |
| Completeness (%) | 100 (100) | 98.4 (94.0) | 99.1 (100) |
| Redundancy | 6.1 (6.1) | 5.0 (4.5) | 6.0 (5.8) |
| Refinement | |||
| Resolution (Å) | 24.67–2.70 | 29.18–2.15 | 29.39–2.36 |
| No. of reflections (working, test) | 22,955, 1,148 | 46,063, 1,677 | 34,991, 1,521 |
| R factor/free R factorc | 18.9/25.4 (25.0/31.5) | 16.8/20.2 (22.6/29.5) | 18.6/22.8 (21.9/25.7) |
| No. of refined atoms, molecules | |||
| Protein | 5,094, 3 | 5,105, 3 | 5,094, 3 |
| Substrate | NAd | 153, 3 | 38, 1 |
| Solvent | 17 | 127 | 158 |
| Water | 116 | 633 | 535 |
| B factors | |||
| Protein | 57.9 | 46.9 | 41.6 |
| Substrate | NA | 50.9 | 62.4 |
| Solvent | 72.1 | 72.8 | 67.8 |
| Water | 46.6 | 63.3 | 49.3 |
| RMSD | |||
| Bond length (Å) | 0.003 | 0.012 | 0.011 |
| Bond angle (°) | 0.691 | 1.382 | 1.238 |
The numbers in parentheses indicate the values for the outer shells of the data.
Rsym = ΣhΣi|Ii(h) − 〈I(h)〉/ΣhΣiIi(h), where Ii(h) and 〈I(h)〉 are the ith and mean measurement of the intensity of reflection h.
R = Σ|Fpobs − Fpcalc|/ΣFpobs, where Fpobs and Fpcalc are the observed and calculated structure factor amplitudes, respectively.
NA, not applicable.
The overall structure of VatA closely resembled that of VatD (RMSD, 0.67 Å over 198 matching Cα atoms, with 62% sequence identity) (Fig. 1C). The domain composition also matched that of VatD, in that VatA contained three main domains (Fig. 1B): a central LβH fold (residues 22 to 171), an insert to the LβH fold that formed a small domain comprising two α-helices (α-domain, residues 82 to 115) and a C-terminal domain comprising three α-helices (CT domain, residues 172 to 217). The N-terminal residues 6 to 21 formed a loop that adopted the same conformation in each chain of the trimer; this region formed a cap over the α- and CT domains and approached the C terminus. The interface between chains to form the VatA trimer was spread across each of three domains, and the small α-helical domain was largely buried against the LβH and CT domain of the neighboring chain.
Analysis of the crystallographic packing of VatA suggested that VatA trimers interacted, thereby forming a hexamer. In line with this observation, size exclusion chromatography showed that VatA formed a hexamer under the solution conditions tested (see Fig. S1A in the supplemental material). In contrast, VatD formed only trimeric species. The VatA hexameric interface observed in the crystal packing was along a crystallographic 2-fold axis and placed one edge of the triangular structure on an interaction surface with the same surface of the trimer in the neighboring asymmetric unit (see Fig. S1B in the supplemental material). Of the 24 residues involved in the hexamerization interface of VatA, only 14 are identical between VatA and VatD, and these differences may explain the distinct oligomeric states of these homologues. Overall, these results showed that VatA and VatD are largely similar in structure, as expected from their significant sequence identity, but nonetheless, the functional species in solution are of distinct oligomeric states.
Structures of VatA in complex with virginiamycin M1 and acetyl-CoA identified residues involved in substrate recognition.
To gain a better understanding of the specific features of the active site of VatA and to identify residues important in the modification of streptogramin A compounds, we next solved the crystal structures of the VatA-virginiamycin M1 and VatA-acetyl-CoA complexes (to 2.36-Å and 2.15-Å resolution, respectively) (Fig. 1B and Table 1). Crystallization trials for the ternary complex of VatA-virginiamycin M1-CoA were also attempted but were unsuccessful. Each of the determined VatA complex structures superimposed with the apoenzyme structure, with RMSD values between 0.24 Å and 0.37 Å across 211 matching Cα atoms of each monomer, pointing to no significant conformational changes of the protein backbone upon cosubstrate binding.
Under the successful crystallization conditions, virginiamycin M1 was bound to only one active site of the VatA trimer (formed by chains A and C). Another active center (formed by chains A and B) was obstructed by the C terminus of a symmetry-related chain in the crystal, while additional electron density corresponding to a polyethylene glycol molecule present in the crystallization solution was found in the third active center of the trimer (formed by chains B and C).
The antibiotic-binding site was localized to a thin and deep wedge formed mainly by the α-domain that cupped the substrate molecule via a small α-helical hairpin (Fig. 3A). A neighboring chain contributed to the antibiotic-binding site through the N-terminal cap and one edge of an LβH domain. This substrate-binding region was conserved between VatA and VatD; the antibiotic molecules assumed nearly the same position, differing only by a translation of 0.8 Å (Fig. 4A).
FIG 3.

Active sites of VatA. (A) Streptogramin A binding site of VatA. VatA is shown in a surface representation and colored as in Fig. 1A. Virginiamycin M1 is shown in sticks. The white dashed box is the location of the acetyl-CoA-binding tunnel. (B) Details of interactions between VatA and virginiamycin M1. The dashes indicate hydrogen bonds. The electron density for virginiamycin M1 shown is a simulated omit Fo–Fc density contoured at 1.0 σ. (C) Acetyl-CoA binding site of VatA. VatA is shown in a surface representation and colored as in Fig. 1A. The yellow dashed box is the location of the virginiamycin M1 binding site. (D) Details of interactions between VatA and acetyl-CoA. The dashes indicate hydrogen bonds. The electron density for acetyl-CoA shown is a simulated omit Fo–Fc density contoured at 2.0 σ.
FIG 4.

Comparison of ligand binding to VatA and VatD. (A) Comparison of interactions between VatA and virginiamycin M1 (Virg. M1) with VatD (8, 9) and virginiamycin M1 or dalfopristin. Two views are shown, rotated 80°. The enzyme side chains are colored as follows: purple and green for chain A and chain C of VatA, gray for both chains A and B of VatD; side chains from the VatD-dalfopristin complex are not shown, as no major conformational differences are observed with the VatD-virginiamycin M1 complex. Virginiamycin M1 bound to VatA and VatD is colored in yellow and light gray, respectively, and dalfopristin bound to VatD is colored in black. (B) Comparison of acetyl-CoA binding to VatA (three copies per trimer, colored purple) and VatD (three copies per trimer [9], colored gray). The circle highlights the distinct conformation of acetyl and β-mercaptoethylamine groups. (C) Comparison of interactions between acetyl-CoA and VatA and VatD, colored the same as in panel A.
The virginiamycin M1 cosubstrate was held in place in the VatA active site by an extensive network of interactions contributed by 17 residues (Fig. 3B). The majority of these interactions were hydrophobic in nature, with extensive interactions provided by Leu23, Leu98, Leu113 of VatA, a stacking interaction of Tyr59, and one edge of Tyr57. In addition, four residues formed hydrogen bonds with the antibiotic, including the side chain of Asn20 from the N-terminal cap and virginiamycin M1 O-11, the side chain of Tyr42 from the LβH domain and O-18, the side chain of His87 from the α-domain and O-18, and between the side chain of His97 from the α-domain and O-27. His87 was also hydrogen bonded to the backbone carbonyl oxygen of Thr93, an interaction seen in other Vat and XAT enzymes (8, 9, 11). Finally, VatA Asp44 formed an electrostatic interaction with O-15 of the antibiotic molecule. The conformations of the abovementioned residues in the virginiamycin M1-bound structure were the same as in the VatA apoenzyme structure, except for those of six residues, Tyr59, Val61, Ala85, His87, His97, and Met107, which changed their rotamer conformations upon substrate binding.
The modeling of the dalfopristin molecule into the VatA antibiotic-binding site based on the VatA-virginiamycin M1 structure (Fig. 4A) did not reveal any significant steric clashes, except for an oxygen atom from the sulfonyl group that approached the side chain of Val61 and a close approach of Tyr59 with the O-27 atom. This analysis is in line with previous reports of VatA conferring resistance to the quinupristin-dalfopristin combination (24).
In the VatA-acetyl-CoA binary complex, one acetyl group donor molecule bound to each chain in the trimer of the enzyme (Fig. 1B). The position of acetyl-CoA bound to VatA was similar to its position bound to VatD, P. aeruginosa XAT (PaXAT) (11), and other N-acetyltransferases, such as PerB (21). Accordingly, acetyl-CoA adopted a bent conformation, with the glycosidic linkage oriented parallel to the extended pantothenic acid region. This acetyl-CoA conformation was identical for all three binding sites of the VatA trimer. The carbonyl oxygen of the acetyl group pointed toward the VatA antibiotic-binding site, while the methyl group pointed toward the enzyme core, accommodated in a pleat in a central β-strand of the VatA LβH domain formed by the backbone atoms of Ser73, Ile74, and Gly75 (Fig. 3D). The carbonyl oxygen was hydrogen bonded to Tyr42 and His87. The remainder of the acetyl-CoA molecule formed numerous interactions with the enzyme, resulting in 49% of the surface of the cosubstrate molecule shielded from solvent (Fig. 3C). The 5′-diphosphate and ribose moieties of acetyl-CoA were less buried, and the 3′-phosphate was nearly completely exposed to solvent. The diphosphate negative charges were balanced by interactions with Lys116 and Arg170 side chains.
The superposition of the two VatA cosubstrate complex structures demonstrated that the acetylation site (O-18) of virginiamycin M1 was oriented toward acetyl-CoA and the presumed catalytic center (Fig. 3C). The antibiotic-binding site was connected to a long tunnel that linked one face of the trimer to the other. The tunnel was formed by two edges of the LβH domain, one from each neighboring chain in the trimer. This tunnel formed the acetyl-CoA-binding site (Fig. 3C). The catalytic center was found where the tunnel emerged to meet the wedge-shaped antibiotic-binding site.
Overall, our structural analysis pointed to a strong similarity between VatA and the previously characterized VatD enzyme, particularly with respect to the active-site architecture and to interactions with cosubstrates. This observation suggested that the elucidation of specific roles of VatA active-site residues would infer general features of the active center of this class of antibiotic-modifying enzymes.
Sequence conservation analysis of Vat enzymes exposed a conserved active center signature beyond the catalytic center.
In order to assess the plasticity and sequence conservation of the VatA active site, we undertook a detailed phylogenetic and structure-guided sequence analysis of the active site among Vat family representatives. We selected seven functionally characterized Vat enzymes and their sequence homologues in GenBank (>55% sequence identity, identifying 91 potential Vat enzymes), aligned them (Fig. 2A), and performed a phylogenetic reconstruction (Fig. 2B).
The phylogenetic reconstruction revealed that Vat enzymes separate into two clusters: cluster 1 included VatA, VatC, VatD, and VatH, and cluster 2 included VatB and VatE (Fig. 2B). An inspection of the sequence alignment (Fig. 2A) suggested that the two clusters were defined primarily by their sequence divergence in regions corresponding to the VatA cap and CT domains, while the LβH and α-domain sequences were more conserved.
Next, we correlated the virginiamycin M1-interacting residues of VatA identified through structural analysis with the multiple-sequence alignment. Of the 17 VatA residues that encompassed the virginiamycin M1 binding site (Fig. 3B), seven (Tyr42, Tyr59, His87, Thr93, Pro95, Pro108, and Leu113) were totally conserved across all Vat enzyme sequences. Five residues were highly conserved within their amino acid class across all Vat enzymes (Leu23, Val61, Ile62, Ser73, and Leu98). The remaining five substrate-interacting residues were conserved only in subsets of Vat enzymes, with Asn20, Asp44, and Tyr55 conserved exclusively in cluster 1 representatives.
Based on this analysis, we postulated that Vat enzymes possess an extended set of conserved active-site residues essential for cosubstrate binding that may not be directly involved in catalysis.
Mutational analysis-verified amino acids in the VatA active site far from the catalytic apparatus are critical for activity.
To clarify which VatA residues are critical for activity, we probed the individual roles of 14 active-site residues by mutagenesis, followed by testing of the mutants for acetyltransferase activity against streptogramin A substrates (virginiamycin M1 and dalfopristin) (Fig. 5). The VatA mutants that retained activity were further characterized to determine the full set of kinetic parameters (Table 2).
FIG 5.
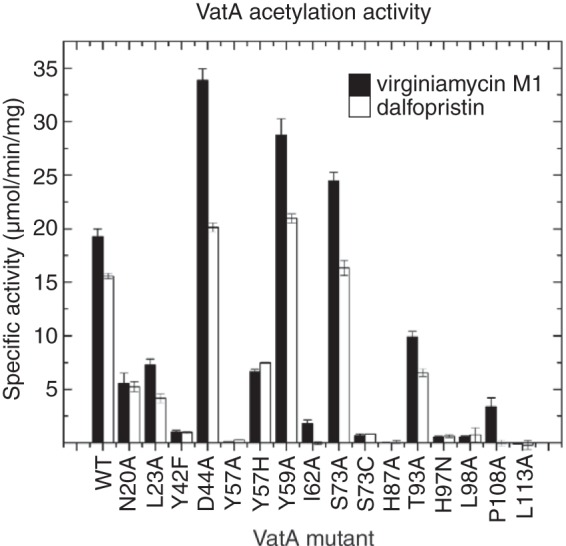
Acetylation activity of VatA and mutants against streptogramin A compounds. Shown is a comparison of the acetylation activity of VatA WT and mutants toward virginiamycin M1 (black bars) and dalfopristin (white bars). The specific activity of each enzyme is the average from the data of three independent experiments.
TABLE 2.
Kinetic and mutational analysis of VatA
| Antibiotic WT/mutants | Vmax (mean ± SD) (μmol min−1 mg−1) | S0.5 (mean ± SD) (μM) | No. (mean ± SD) | kcat (s−1) | Catalytic efficiency (M−1 s−1) |
|---|---|---|---|---|---|
| Virginiamycin M1 | |||||
| WT | 21.3 ± 1.4 | 124 ± 18 | 1.49 ± 0.23 | 8.84 | 7.13 × 104 |
| N20Aa | 6.44 ± 0.27 | 61.7 ± 7.2 | 1.56 ± 0.22 | 2.67 | 4.33 × 104 |
| L23A | 10.6 ± 0.43 | 66.7 ± 5.5 | 1.69 ± 0.16 | 4.40 | 6.60 × 104 |
| D44Ab | 64.2 ± 7.8 | 657 ± 157 | 1.07 ± 0.08 | 26.6 | 4.05 × 104 |
| Y57H | 8.34 ± 0.39 | 138 ± 14 | 3.38 ± 0.85 | 3.46 | 2.51 × 104 |
| Y59A | 28.0 ± 1.6 | 210 ± 26 | 1.45 ± 0.15 | 11.6 | 5.52 × 104 |
| I62A | NDc | ND | ND | ND | ND |
| S73A | 21.5 ± 1.4 | 151 ± 23 | 1.41 ± 0.19 | 8.92 | 5.91 × 104 |
| T93A | 12.9 ± 0.8 | 102 ± 14 | 1.41 ± 0.16 | 5.35 | 5.25 × 104 |
| P108A | 3.93 ± 0.37 | 170 ± 26 | 1.88 ± 0.34 | 1.63 | 9.59 × 103 |
| Dalfopristin | |||||
| WT | 24.1 ± 1.4 | 315 ± 35 | 1.23 ± 0.08 | 10.0 | 3.17 × 104 |
| N20A | 5.46 ± 0.49 | 317 ± 44 | 1.74 ± 0.26 | 2.27 | 7.16 × 103 |
| L23A | 17.8 ± 3.3 | 462 ± 141 | 1.43 ± 0.27 | 7.39 | 1.60 × 104 |
| D44A | ND | ND | ND | ND | ND |
| Y57H | 6.26 ± 0.53 | 212 ± 34 | 1.69 ± 0.29 | 2.60 | 1.23 × 104 |
| Y59A | 18.7 ± 0.8 | 160 ± 16 | 1.47 ± 0.14 | 7.76 | 4.85 × 104 |
| S73A | 22.7 ± 1.6 | 346 ± 47 | 1.26 ± 0.10 | 9.42 | 2.72 × 104 |
Substrate inhibition was seen at concentrations of >0.4 mM virginiamycin M1.
The large errors associated with the kinetic parameters for the D44A mutant are due to the fact that complete saturation was not achieved.
ND, the kinetic parameters were unable to be determined because the curves did not reach saturation.
According to our results, the VatA Tyr42Phe, His87Ala, and Leu113Ala variants showed a severe loss of activity, suggesting that these conserved residues are critical for VatA activity. The Pro108Ala variant was inactive against dalfopristin while retaining only ∼15% activity against virginiamycin M1, consistent with its conservation across both clusters of Vat enzymes. The substitution of residues Tyr57, Ile62, and Leu98, corresponding to positions occupied exclusively by hydrophobic residues in cluster 1 and 2 enzymes, also had dramatic effects on VatA activity. Unexpectedly, the His97Asn mutation was detrimental for VatA activity, despite the fact that several Vat enzymes possess an asparagine residue at this position.
Mapping our mutagenesis results onto the VatA-virginiamycin M1 complex structure showed that functionally critical residues were distributed across the VatA active site (Fig. 6). In particular, Ile62, Pro108, and Leu113 were spatially clustered in a region approximately 12 Å from the virginiamycin M1 acetylation site (O-18). These observations support the hypothesis that appropriate substrate positioning is critical for Vat enzyme activity, and this is achieved by multiple highly conserved residues spread around the streptogramin A binding site that are not expected to be directly involved in catalysis. Thus, the development of alternative streptogramin A compounds featuring additional chemical groups perturbing interactions with these conserved residues may make such compounds less prone, or resistant, to acetylation by this enzyme class.
FIG 6.

(A) Critical contacts between Vat enzymes and streptogramin A. Side chains from the VatA active site and virginiamycin M1 atoms are colored according to combined sequence and mutational analysis: darker blue, most critical for acetylation reaction, fully conserved; lighter blue, also important for acetylation reaction, highly conserved within amino acid class; red, critical for acetylation reaction, conserved within Vat enzyme clusters only; gray, positions tolerant to mutation, not well conserved. The hatched residues were not mutated. The distance labels are the distances from those particular side chain atoms to the acetylation site of virginiamycin M1 (O-18). (B) Putative catalytic mechanism of VatA.
DISCUSSION
Streptogramins used to hold promise as a treatment for infections caused by multidrug-resistant Gram-positive bacteria. The spread of resistance mechanisms, such as O-acetylation, by Vat enzymes reduced the effectiveness of these compounds. This, along with the introduction of other classes of antimicrobials, led to almost a complete removal of this class of antibiotics from clinical use.
Much of the ongoing antimicrobial research is devoted to the modification and/or synthetic tailoring of antimicrobial scaffolds to reduce their susceptibility to resistance. In line with this effort, we undertook a detailed structural and functional characterization of S. aureus VatA acetyltransferase to expand our understanding of Vat active-site features and to provide the necessary molecular information for the development of less-modification-prone streptogramin compounds.
Our structural data showed that the VatA enzyme shares the general fold and antibiotic substrate recognition mode with the previously characterized VatD, suggesting that these molecular features are common among Vat enzymes. However, we showed that the VatA enzyme likely functions as a hexamer or a dimer of trimers, which contrasts with the oligomeric state of VatD as a trimer. This difference is reflected in the poor conservation of residues contributing to the VatA hexamerization interface in the VatD enzyme. The functional implications of this difference remain to be clarified, since we did not observe cooperative effects in our kinetic studies of VatA.
Guided by substrate-bound crystal structures, we established that the acetylation reaction of VatA, and likely other Vat enzymes, relied on a key histidine (His87 in VatA) corresponding to His82 of VatD (8, 9) and the catalytic residue in LβH fold N-acetyltransferases. The highly conserved Tyr42 is also critical for the catalytic mechanism; the interaction between Tyr42 and virginiamycin M1 O-18 observed in the VatA-virginiamycin M1 complex structure suggests that this residue plays a role in positioning the streptogramin A O-18 atom for catalysis. Also, the positioning of the Tyr42 side chain suggests this interaction may stabilize the oxyanion tetrahedral intermediate in the catalytic cycle (Fig. 6B presents a putative mechanism for this enzyme).
An analysis of the VatA-virginiamycin M1 complex demonstrated that the substrate O-27, C-28, C-31, and C-35 atoms participate in multiple interactions with enzyme residues. VatA Pro108 and Leu113, taking part in interactions with this region of the compound, were distantly located from the antibiotic modification site but were nevertheless highly conserved across the VatA enzyme family. The positions of these amino acid side chains in clasping the distal end of the virginiamycin M1 ring suggested that these residues are important for orienting the antibiotic substrate into the position for acetylation. We thus propose that modifying the streptogramin A scaffold (specifically, the N-25–O-27 region) to block these interactions would decrease or completely abrogate Vat enzyme activity against these molecules. Consistent with this notion, the same region in this antibiotic molecule was previously identified as an avenue for the development of acetylation-resilient derivatives (9). As further validation of the importance of VatA Pro108 and Leu113 in the binding affinity for streptogramin A substrates, a recent study demonstrated that a derivative of virginiamycin M1 with a modification in this region (saturation of the C-28–C-29 bond) that is expected to interact with these two residues showed altered binding affinity to VatA (25).
Mutagenesis studies demonstrated that VatA activity against streptogramin A substrates was dramatically reduced by the individual alteration of an additional set of four well-conserved residues (Leu23, Ser73, Thr93, and Leu98) that were arrayed around the virginiamycin M1 ring. In particular, VatA was sensitive to the nature of the residue at position 73, as the Ser73Cys variant was inactive, while the Ser73Ala variant had increased activity relative to that of the wild type. This residue is closely positioned (within 4 Å) to the key catalytic His87, and therefore, mutations at this position may play a role in influencing the catalytic properties of that residue. Thus, these active-site residues appeared to be important for substrate binding and structural modifications of the drug scaffold to impact these interactions may also be exploited for blocking inactivation in a Vat family-wide manner.
The comprehensive survey of Vat enzyme sequences indicated that another five VatA residues (Asn20, Asp44, Tyr57, Tyr59, and His97) forming interactions with the antibiotic substrate were not conserved across the Vat enzyme family. While the mutagenic analysis showed that three of these residues (Asn20, Tyr57, and His97) were important for VatA activity, we suspect that the modification of the streptogramin A scaffold to block these interactions may not provide Vat family-wide protection against acetylation.
Phylogenetic analysis identified two clusters in the Vat enzyme family that may reflect divergent evolutionary histories among these enzymes. Given the distribution of experimentally characterized Vat enzymes, putative streptogramin A O-acetyltransferases, other putative acetyltransferases, and LβH fold-containing enzymes in the phylogenetic tree, we speculate that the enzymes found in the family Bacillaceae bacteria represent the protoresistance reservoir (26) of enzymes from which Vat enzymes may have evolved. Based on multiple-sequence alignment and the conservation of motifs defining the LβH fold, we predict that these Bacillaceae enzymes adopt similar folds and possess acetyltransferase activities. Further support for this hypothesis is provided by the presence of a cluster of homologue genes in the Paenibacillaceae family, which also share >60% sequence identity with VatA. This family of soil-inhabiting bacteria was identified as a source of glycopeptide antibiotic resistance elements (27).
The updated genome search also revealed the presence of putative vat genes in Enterococcus spp. beyond those that have been studied. These include two homologues from E. faecium, with one in each of clusters 1 and 2, and one representative in Enterococcus faecalis belonging to cluster 1 that might also represent streptogramin A resistance elements. The presence of an enzyme sequence from Clostridium papyrosolvens phylogenetically close to cluster 2 might explain the macrolide-lincosamide-streptogramin B (MLSB) resistance observed in this genus (28).
The bactericidal effects of streptogramin A and B compounds result from their interactions with the peptidyltransferase center of the ribosome. Based on an analysis of dalfopristin binding to 50S rRNA (3), these interactions do not involve the aforementioned C-28, C-31, and C-35 positions of the antibiotic molecule identified as potential modification sites that would disrupt recognition by Vat enzymes. Therefore, the modifications at these locations on the streptogramin A scaffold may not directly affect the ribosomal contacts of the compound, justifying further research in this direction.
An alternative approach to addressing the inactivation of streptogramin A compounds is the inhibition of Vat enzyme activity. The VatD structure was utilized in a virtual screening program for the discovery of the first small-molecule lead inhibitors of this antibiotic resistance enzyme class (29). Our structural analysis of the VatA enzyme would further inform such inhibitor screening efforts aimed at identifying Vat family-wide inhibitors.
In conclusion, our study provides further insights into the structures, mechanisms, and distribution of the Vat enzymes. Importantly, it produced a roadmap of functionally critical amino acid residues in the Vat enzyme active site for the rational development of novel acetylation-resilient streptogramin A compounds.
Supplementary Material
ACKNOWLEDGMENTS
The structures presented here were solved by the Center for Structural Genomics of Infectious Diseases (CSGID) (http://csgid.org); this project has been funded in whole or in part with U.S. federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contracts HHSN272200700058C (2007 to 27 September 2012) and HHSN272201200026C (starting 1 September 2012).
We thank Veronica Yim, Rosa Di Leo, Marina Kudritska, Olena Onopriyenko, and M. Krishnamoorthy for technical assistance, Aiping Dong at the Structural Genomics Consortium in Toronto and Zdzislaw Wawrzak and George Minasov at the Life Sciences Collaborative Access Team, Advanced Photon Source, for assistance in X-ray diffraction data collection and structure validation, and Djalal Meziane-Cherif for critical reading of the manuscript.
Footnotes
Published ahead of print 15 September 2014
For this virtual institution, see http://www.csgid.org/.
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03743-14.
REFERENCES
- 1.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48:1–12. 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Leclercq R, Courvalin P. 1998. Streptogramins: an answer to antibiotic resistance in Gram-positive bacteria. Lancet 352:591–592. 10.1016/S0140-6736(05)79570-2. [DOI] [PubMed] [Google Scholar]
- 3.Harms JM, Schlünzen F, Fucini P, Bartels H, Yonath A. 2004. Alterations at the peptidyl transferase centre of the ribosome induced by the synergistic action of the streptogramins dalfopristin and quinupristin. BMC Biol. 2:4. 10.1186/1741-7007-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalan L, Wright GD. 2011. Antibiotic adjuvants: multicomponent anti-infective strategies. Expert Rev. Mol. Med. 13:e5. 10.1017/S1462399410001766. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Shang X, Hu F, Lao X, Gao X, Zheng H, Yao W. 2013. β-Lactamase inhibitors: an update. Mini Rev. Med. Chem. 13:1846–1861. 10.2174/13895575113139990074. [DOI] [PubMed] [Google Scholar]
- 6.Roberts MC. 2002. Resistance to tetracycline, macrolide-lincosamide-streptogramin, trimethoprim, and sulfonamide drug classes. Mol. Biotechnol. 20:261–283. 10.1385/MB:20:3:261. [DOI] [PubMed] [Google Scholar]
- 7.D'Costa VM, Griffiths E, Wright GD. 2007. Expanding the soil antibiotic resistome: exploring environmental diversity. Curr. Opin. Microbiol. 10:481–489. 10.1016/j.mib.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 8.Sugantino M, Roderick SL. 2002. Crystal structure of Vat(D): an acetyltransferase that inactivates streptogramin antibiotics. Biochemistry 41:2209–2216. 10.1021/bi011991b. [DOI] [PubMed] [Google Scholar]
- 9.Kehoe LE, Snidwongse J, Courvalin P, Rafferty JB, Murray IA. 2003. Structural basis of Synercid (quinupristin-dalfopristin) resistance in Gram-positive bacterial pathogens. J. Biol. Chem. 278:29963–29970. 10.1074/jbc.M303766200. [DOI] [PubMed] [Google Scholar]
- 10.Raetz CR, Roderick SL. 1995. A left-handed parallel beta helix in the structure of UDP-N-acetylglucosamine acyltransferase. Science 270:997–1000. 10.1126/science.270.5238.997. [DOI] [PubMed] [Google Scholar]
- 11.Beaman TW, Sugantino M, Roderick SL. 1998. Structure of the hexapeptide xenobiotic acetyltransferase from Pseudomonas aeruginosa. Biochemistry 37:6689–6696. 10.1021/bi980106v. [DOI] [PubMed] [Google Scholar]
- 12.Allignet J, Loncle V, Simenel C, Delepierre M, el Solh N. 1993. Sequence of a staphylococcal gene, vat, encoding an acetyltransferase inactivating the A-type compounds of virginiamycin-like antibiotics. Gene 130:91–98. 10.1016/0378-1119(93)90350-C. [DOI] [PubMed] [Google Scholar]
- 13.Eschenfeldt WH, Lucy S, Millard CS, Joachimiak A, Mark ID. 2009. A family of LIC vectors for high-throughput cloning and purification of proteins. Methods Mol. Biol. 498:105–115. 10.1007/978-1-59745-196-3_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stogios PJ, Shakya T, Evdokimova E, Savchenko A, Wright GD. 2011. Structure and function of APH(4)-Ia, a hygromycin B resistance enzyme. J. Biol. Chem. 286:1966–1975. 10.1074/jbc.M110.194266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. 2006. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62:859–866. 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 16.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 18.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372:774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 19.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thoden JB, Reinhardt LA, Cook PD, Menden P, Cleland WW, Holden HM. 2012. Catalytic mechanism of perosamine N-acetyltransferase revealed by high-resolution X-ray crystallographic studies and kinetic analyses. Biochemistry 51:3433–3444. 10.1021/bi300197h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM. 2006. MUSTANG: a multiple structural alignment algorithm. Proteins 64:559–574. 10.1002/prot.20921. [DOI] [PubMed] [Google Scholar]
- 23.Kuhn ML, Majorek KA, Minor W, Anderson WF. 2013. Broad-substrate screen as a tool to identify substrates for bacterial Gcn5-related N-acetyltransferases with unknown substrate specificity. Protein Sci. 22:222–230. 10.1002/pro.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lina G, Quaglia A, Reverdy ME, Leclercq R, Vandenesch F, Etienne J. 1999. Distribution of genes encoding resistance to macrolides, lincosamides, and streptogramins among staphylococci. Antimicrob. Agents Chemother. 43:1062–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoang NH, Huong NL, Shrestha A, Sohng JK, Yoon YJ, Park JW. 2013. Regio-selectively reduced streptogramin A analogue, 5,6-dihydrovirginiamycin M1 exhibits improved potency against MRSA. Lett. Appl. Microbiol. 57:393–398. 10.1111/lam.12125. [DOI] [PubMed] [Google Scholar]
- 26.Morar M, Wright GD. 2010. The genomic enzymology of antibiotic resistance. Annu. Rev. Genet. 44:25–51. 10.1146/annurev-genet-102209-163517. [DOI] [PubMed] [Google Scholar]
- 27.Guardabassi L, Christensen H, Hasman H, Dalsgaard A. 2004. Members of the genera Paenibacillus and Rhodococcus harbor genes homologous to enterococcal glycopeptide resistance genes vanA and vanB. Antimicrob. Agents Chemother. 48:4915–4918. 10.1128/AAC.48.12.4915-4918.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt C, Löffler B, Ackermann G. 2007. Antimicrobial phenotypes and molecular basis in clinical strains of Clostridium difficile. Diagn. Microbiol. Infect. Dis. 59:1–5. 10.1016/j.diagmicrobio.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 29.Wang GF, Huang N, Meng ZH, Liu QH. 2007. Identification of novel inhibitors of the streptogramin group A acetyltransferase via virtual screening. Yao Xue Xue Bao. 42:47–53. 10.3321/j.issn:0513-4870.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 30.DeLano W. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA: http://www.pymol.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.