

Abstract
The dermatological disease molluscum contagiosum (MC) presents as lesions restricted solely to the skin. The poxvirus molluscum contagiosum virus (MCV) is responsible for this skin disease that is easily transmitted through casual contact among all populations, with greater frequency in children and immunosuppressed individuals. In addition, sexual transmission of MCV in adolescents and adults is a health concern. Although the skin lesions ultimately resolve in immunocompetent individuals, they can persist for extended periods, be painful, and result in scarring. Treatment is problematic, and there is no drug that specifically targets MCV. The inability of MCV to propagate in cell culture has impeded drug development. To overcome these barriers, we integrated three new developments. First, we identified a new MCV drug target (mD4) that is essential for processive DNA synthesis in vitro. Second, we discovered a small chemical compound that binds to mD4 and prevents DNA synthesis in vitro. Third, and most significant, we engineered a hybrid vaccinia virus (mD4-VV) in which the natural vaccinia D4 (vD4) gene is replaced by the mD4 target gene. This hybrid virus is dependent on mD4 for viral growth in culture and is inhibited by the small compound. This target system provides, for the first time, a platform and approach for the discovery and evaluation of new therapeutics that can be used to treat MC.
INTRODUCTION
Molluscum contagiosum (MC) is a skin disease caused by the poxvirus molluscum contagiosum virus (MCV). MC presents as skin lesions that can last from months to years before resolving (1). MC lesions occur in children and adults, particularly immunosuppressed individuals, and are restricted strictly to the skin. MCV is transmitted by direct skin-to-skin contact, sexual contact, autoinoculation from scratching lesions, and indirect inoculation from contaminated fomites (1–3). The lesions can be painful following treatments intended to reduce spread. The lesions are also psychologically distressful and even more so when they result in scarring. MC occurs in 2% to 10% of the worldwide population. In the USA, it constitutes about 1% of all diagnosed skin disorders and approaches 5% of diagnoses in children (2, 4). Significantly, in immunocompromised individuals, this infectious disease can be severe and protracted. Between 5% and 18% of HIV-infected patients have MC (2, 5). Often, severe MC disease in AIDS patients begins to resolve while they are treated with highly active antiretroviral therapy (HAART) (6). However, there have been documented cases of MC lesions developing soon after patients started HAART, suggesting that immune reconstitution inflammatory syndrome (IRIS) might play a role in the reemergence of MCV (7, 8).
The current treatments for MC usually employ physical therapy or chemical agents, which are not uniformly effective or safe, often fail to completely eliminate lesions, and may result in scarring (9). In addition, the broad-spectrum antiviral drug cidofovir, a dCMP analogue, has been used effectively as topical or intravenous medication for MC in immunocompromised patients, but with side effects, including inflammation, erosion, and pain for topical treatment and potential nephrotoxicity for systemic application (9). To date, there is not a single antiviral therapeutic that is licensed for the specific treatment of MC. The development of such an effective and safe treatment has been hampered mainly by the inability of MCV to propagate in culture (10, 11).
Processivity factors (PFs) are attractive antiviral therapeutic targets. The function of PFs is to tether DNA polymerases (Pols) to the template to enable the synthesis of extended strands. PFs are specific for their cognate DNA Pol and are essential for DNA synthesis (12, 13). As a case in point, Kaposi's sarcoma herpesvirus Pol (Pol-8) alone incorporates only three nucleotides, whereas in the presence of its PF (PF-8), it can incorporate many thousands of nucleotides (13). All DNA Pols, from phage to human, function with a single cognate PF. However, the prototypic poxvirus vaccinia virus (VV) is somewhat unusual in that a heterodimer comprising the A20 and D4 viral proteins constitutes the functional PF (14–18). D4, which can also function as a uracil DNA glycosylase repair enzyme, binds to its PF partner A20 but not to E9 Pol (14, 15, 18). A20, on the other hand, binds to both E9 and D4, suggesting that it serves, in part, as a bridge that indirectly connects D4 to E9.
In this study, we present three novel findings related to MCV therapeutic targeting. First, we cloned and expressed the MCV DNA Pol (mE9) and its cognate PF partners (mD4 and mA20) as potential drug targets. This MCV protein triad is functionally capable of processive DNA synthesis in vitro. Indeed, mD4, which has only 55% identity to the vaccinia protein, was completely able to substitute for vaccinia D4 (vD4) in DNA synthesis in vitro. Second, we demonstrated that a small chemical inhibitor (named compound 10) binds mD4 and blocks mD4-dependent processive DNA synthesis. Third, we engineered an mD4-VV hybrid virus and demonstrated that compound 10 blocks viral growth. This is of major significance, since the inability to propagate MCV in culture (10, 11) has hampered drug development. This viral targeted system provides a platform for the discovery and evaluation of new therapeutics that can be used to treat MC disease.
MATERIALS AND METHODS
Cell culture.
Rabbit kidney (RK) cells were maintained in minimal essential medium (MEM) supplemented with 10% fetal bovine serum and antibiotic/antimycotic mix (Invitrogen). The medium to grow the RK-D4R cells was further supplemented with 400 μg/ml of hygromycin B. African green monkey epithelial cells (BSC-1) were maintained in Dulbecco's minimal essential medium (DMEM) supplemented with 10% fetal bovine serum and 10 μg/ml of gentamicin.
Cloning of MCV E9L, A20R, and D4R.
The MCV E9L (mE9), A20R (mA20), and D4R (mD4) genes were amplified by PCR from genomic DNA obtained from tissue samples (kindly provided by R. M. Buller, Saint Louis University School of Medicine) using the primer sets E9L forward (5′-AGAAAGCTTGCCATGGAGATCCGGTGTCTCAA) and reverse (5′-TCTCTCTAGACTAGTTCGAGAAGACGGGGCGCAC), A20R forward (5′-GAGAAAGCTTGCCATGGCCAAGGAGCCCGAT) and reverse (5′-TCTCTCTAGATTACTTCTCGGCGCTGGAC), and D4R forward (5′-GAGAAAGCTTGCCATGGTGCGCGAGCGCGCGCT) and reverse (5′-TCTCTCTAGAGGGGTACGAAGCCCT). PCR was performed using Herculase-enhanced DNA Pol according to the manufacturer's recommendation (Agilent Technologies, Inc.), with the addition of 1 M betaine and 5% dimethyl sulfoxide (DMSO) for each reaction. Cloning into pcDNA3.1(+) plasmid (Invitrogen) was accomplished using the HindIII (forward primer, underlined) and XbaI (reverse primer, underlined) sites. The Kozak sequence (GCCATGG) was included within the forward primers to allow for proper initiation of translation. All inserts were confirmed by DNA sequencing. The VV E9L (vE9), A20R (vA20), and D4R (vD4) genes were cloned as described previously (14).
Protein expression and purification.
The MCV and VV E9, A20, and D4 [35S]Cys/Met-labeled proteins, respectively, were generated from recombinant pcDNA3.1 using the transcription/translation system (TNT) T7-coupled reticulocyte lysate system (Promega). Labeled proteins were separated on SDS gel and visualized by autoradiography. To express N-terminally 6His-tagged mD4, the mD4R gene was amplified by PCR using the primers 5′mD4 (5′-AGACATATGCTGCGCGAGCGCGCGCTG) and 3′mD4 (5′-AGAGGATCCTAAAGGGGTACGAAGCCCTG) and cloned into the NdeI and BamHI sites of an Escherichia coli expression vector pET-15b (Novagen). The 6His-mD4 protein was expressed in the E. coli Rosetta (DE3) strain (EMD Millipore) by induction with 0.6 mM isopropyl-β-d-thiogalactopyranoside (IPTG) overnight at 17°C. Total protein was extracted from the cells by treatment with 0.25 mg/ml lysozyme for 1 h at 4°C in phosphate buffer (50 mM phosphate buffer, pH 7.1, 400 mM NaCl, 5% glycerol) containing 0.1% Triton X-100, 0.1% Tween 20, and 0.5 mM phenylmethylsulfonyl fluoride (PMSF), followed by 3× freeze/thaw and sonication. After centrifugation, the supernatant was loaded onto a Talon cobalt resin column (Clontech), followed by a thorough wash with the above-described phosphate buffer containing 20 mM imidazole. The 6His-mD4 protein was eluted with 200 mM imidazole and then further purified by Superdex 200 gel filtration with 25 mM phosphate buffer (pH 6.8) containing 200 mM NaCl and 5% glycerol. The in vitro-translated and E. coli-expressed proteins were used for processive DNA synthesis assays as described below.
Processive DNA synthesis assays.
Processive DNA synthesis was assessed by the rapid plate assay and the M13 assay. The rapid plate assay was performed as previously described (19–22). Briefly, a 5′-biotinylated 100-nucleotide template that contains adenines at only its 5′ distal end was annealed with a 15-nucleotide primer to its 3′ end and attached to streptavidin-coated 96-well plates (Roche Applied Science). DNA synthesis was carried out in a 50-μl reaction mixture containing 100 mM (NH)2SO4, 20 mM Tris-HCl (pH 7.5), 3 mM MgCl2, 0.1 mM EDTA, 0.5 mM dithiothreitol (DTT), 2% glycerol, 40 μg/ml bovine serum albumin (BSA), 5 μM dATP, 5 μM dCTP, 5 μM dGTP, 1 μM digoxigenin-11-dUTP, and E9/A20/D4 proteins. The TNT reticulocyte lysate or in vitro-translated luciferase was used as a negative control. After incubation at 37°C for 30 min, the plate was washed extensively with phosphate-buffered saline (PBS). The wells were then incubated with anti-digoxigenin-peroxidase antibody (Roche) for 1 h at 37°C, followed by washing with PBS. The substrate 2,2′-azino-bis(3-ethylbenzthiazoline)-sulfonate (Roche) was added, and the plates were gently rocked to allow color development. DNA synthesis was quantified by measuring the absorbance of each reaction at 405 nm with a microplate reader (Tecan). Experiments were conducted in triplicate and independently repeated at least twice.
The M13 assay was conducted as described previously (14). Briefly, the reaction mixture (50 μl) contained 45 fmol of primed M13mp18 single-stranded DNA, 100 mM (NH4)2SO4, 10 mM Tris-HCl pH 7.5, 8 mM MgCl2, 0.1 mM EDTA, 5 mM DTT, 40 μg/ml BSA, 4% glycerol, 60 μM dATP, 60 μM dGTP, 60 μM dTTP, and 20 μM [α-32P]dCTP. After incubation at 37°C for 1 h, the reaction was stopped by adding 6× loading dye containing 200 mM NaOH. The products were fractionated on a 1.3% alkaline agarose gel and visualized by autoradiography.
Protein pulldown assay.
A total of 1.5 μg purified 6His-mD4 was incubated with 10 μl in vitro-translated 35S-labeled A20, 15 μl Talon cobalt beads (pretreated with 5% BSA), and 300 μl PBS-NP buffer (PBS with 0.2% NP-40) for 2 h at 4°C. The beads were washed four times in PBS-NP buffer (15 min each). Pulled-down proteins were separated on SDS gel and visualized by autoradiography.
Thermal shift assay.
The thermal shift (differential scanning fluorimetry) assay was performed as previously described (23). Briefly, 5 μM purified 6His-mD4 was mixed with compounds in thin-wall PCR 96-well plates at 20 μl total volume containing 25 mM phosphate buffer (pH 6.8), 0.2 M NaCl, 2.5% glycerol, 2% DMSO, 0.005% (wt/vol) Triton X-100, and 1× Sypro orange. Fluorescence intensities were monitored using the Applied Biosystems 7500 fast real-time PCR system (Carlsbad, CA) at 582 nm from 25 to 80°C at a rate of 1°C/min. To generate the melting temperature (Tm), protein melting curves were plotted on the GraphPad Prism and fitted to the Boltzmann sigmoidal model. Thermal shift (ΔTm) is the difference between the 2% DMSO mock treatment and the inhibitor treatment. All experiments were duplicated and repeated independently.
Construction, isolation, and characterization of mD4-VV hybrid virus and vD4-VV rescue virus.
The vaccinia virus vD4-ZG lacking a functional D4R gene (VVΔD4R), a rabbit kidney cell line stably expressing the vaccinia D4 protein (RK-D4R), and plasmid pER, which contains D4R sequences flanked by D3R and D5R sequences, were originally engineered by G. W. Holzer and F. G. Falkner (24) and were provided to us by B. Moss.
To construct a plasmid to generate the mD4-VV hybrid virus, the mD4R gene was amplified using the primers 5′mD4-EcoRI (5′-GTGGAATTCAATGCTGCGCGAGCGCGCGCTG) and 3′mD4-HindIII (5′-GAGAAGCTTCTAAAGGGGTACGAAGCCCTG) and subcloned into the EcoRI and HindIII sites of the pER plasmid. This replaces the vD4R with the mD4R gene but with the derived clone containing two start ATG codons (in bold type, 5′-TATAATGAATTCAATGCTG), of which the first is from the vD4R gene. To remove the vD4R start codon, site-directed mutagenesis was performed using a pair of complementary primers (5′-AAAGGTATCTAATTTGATATAATaAAGCCATGCTGCGCGAGCGCG, in which the mutated nucleotide is lowercased and underlined nucleotides are mD4 sequences) to generate plasmid pER-mD4(ATG), which contains only the mD4R start codon and thus encodes wild-type mD4.
mD4-VV hybrid virus and vD4-VV rescue virus were generated by homologous recombination of pER-mD4(ATG) and pER, respectively, by transfection of plasmids into RK-D4R cells infected with parental virus VVΔD4R as previously described by others (16). After 48 h, cells were harvested and the virus released by freeze-thawing and sonication. Recombinant viruses were then isolated by infecting BSC-1 cells with the virus lysates from RK-D4R cells and successive plaque purifications of large plaques. The recombinant viruses were confirmed by PCR and sequencing. Compared to the mD4R sequence (VP0038088) found at www.poxvirus.org, the mD4R sequence in the recombinant mD4R-VV hybrid virus (plaque 224a1-1) was an identical match. Compared to the D4R WR sequence (VP0042547) found at www.poxvirus.org, the vD4R sequence in the recombinant vD4-VV rescue virus (plaque 225a1) contained amino acid Asn instead of Lys at residue 150. This base change was also present in the starting plasmid pER.
Virus growth kinetics were measured on confluent BSC-1 cells in wells of 24-well plates infected at a multiplicity of infection (MOI) of 0.05 PFU/cell with mD4-VV hybrid virus and vD4-VV rescue virus in quadruplicate. At various time points, media and cells were harvested, and virus titers were determined.
Viral plaque reduction and cytotoxicity assays.
The viral plaque reduction assay was performed using BSC-1 cells as previously described (23) in triplicate and independently repeated for compound 10. Briefly, cells were infected by adsorbing virus at 80 PFU/well in 100 μl of growth medium for 1 h in a 48-well plate, followed by 16 h of treatment with compounds. Cells were stained and plaques counted under a dissecting microscope, and data were plotted on the GraphPad Prism. Cytotoxicity on BSC-1 cells was assessed by the lactate dehydrogenase assay (LDH) as previously reported (23).
Dot blot hybridization.
BSC-1 cells were grown to confluence by seeding 1.2 × 105 cells/well in a 24-well plate and incubated at 37°C overnight. The cells were infected by adsorbing virus (∼1 MOI) in 200 μl of growth medium for 1 h, followed by treatment with 65 μM of compound 10. Cells were then collected at various time points. Viral DNA was extracted with 20 mM Tris buffer (pH 7.5) containing 20 mM EDTA, 0.5% (wt/vol) SDS, and 0.5 mg/ml proteinase K and used for dot blot hybridization as described previously (25). 32P-labeled vA20 DNA was used as a probe.
RESULTS
Cloning and expression of the DNA Pol and PF of MCV.
Based on sequence homology to vaccinia virus (VV) (26) (see Table S1 in the supplemental material), we predicted that the MCV PF (mA20 and mD4) should enable the cognate mE9 DNA Pol to synthesize DNA processively. In order to verify this assumption, we first cloned these three MCV genes from a clinical isolate obtained from an individual with MC (see Fig. S1 in the supplemental material for the orientations and genomic positions of the coding regions for mE9, mA20, and mD4). The complete coding region of each MCV gene was amplified with primers that contained a translational Kozak sequence and HindIII and XbaI restrictions sites for insertion into pcDNA3.1(+). All the clones were validated by DNA sequencing. When transcribed and translated in vitro, each of the cloned templates was able to generate a protein product of the predicted size (Fig. 1). These MCV proteins are similar in size to their corresponding counterparts of vaccinia virus (Fig. 1).
FIG 1.

In vitro translation of MCV and VV polymerases and processivity factors. The E9 polymerases and the processivity factors D4 and A20 from the molluscum contagiosum and vaccinia viruses were translated in vitro from cloned plasmids and labeled with [35S]Cys/Met. Proteins were fractionated on an SDS gel and visualized by autoradiography. Arrows indicate full-length proteins. Note that lanes from the original autoradiogram were rearranged for convenient comparison.
mE9, mD4, and mA20 perform processive DNA synthesis in the rapid plate assay.
In order to test for processive DNA synthesis of the MCV proteins (Fig. 1), we employed the rapid plate assay (RPA), which measures nucleotide incorporation (19, 20). Briefly, the RPA consists of a 100-nucleotide template with a biotin moiety on its 5′ end and a 15-nucleotide primer annealed to its 3′ end. The annealed primer template is attached to streptavidin-coated wells of a 96-well plate. The additions of DNA Pol and PF cause incorporation of dNTPs and dig-dUTP that is recognized by digoxigenin (DIG) antibody coupled to horseradish peroxidase (HRP) for colorimetric quantitation. This RPA has been successfully used to determine the strict requirement of all three members of the VV protein triad (vE9, vA20, vD4) in processive DNA synthesis (14). Indeed, omission of any member of this protein triad precludes processive DNA synthesis (14) (Fig. 2, compare bars 1 and 2). As shown in Fig. 2, the MCV mE9, mA20, and mD4 triad exhibited the same activity as the corresponding VV proteins (compare bars 2 and 3).
FIG 2.

mD4 can substitute for vD4 in processive DNA synthesis. DNA synthesis was conducted with homologous and heterologous combinations of the E9 polymerases and the A20 and D4 processivity factors from VV and MCV using the rapid plate assay. DNA synthesis was quantitated by the incorporation of dig-dUTP, which was detected by peroxidase-conjugated DIG antibody (optical density [OD] at 405 nm). The background OD value is arbitrarily set to 1. The data represent the means + SD from at least two independent experiments in triplicate.
mD4 fully substitutes for vD4 to enable processive DNA synthesis in the rapid plate assay.
We next inquired whether any of the triad proteins from MCV can substitute for their corresponding proteins from VV. As shown in Fig. 2, mE9 and mA20 can each only poorly substitute for their VV counterparts to enable processive DNA synthesis (Fig. 2, bars 4 and 5). In strong contrast, mD4 is completely capable of substituting for vD4 (Fig. 2, bar 6 versus bars 2 and 3).
mD4 physically associates with mA20 or vA20.
The VV processivity proteins vD4 and vA20 form a heterodimeric complex and are both required to enable DNA Pol to conduct extended DNA strand synthesis (14, 15, 18). It was thus important to demonstrate that the functional ability of mD4 to substitute for vD4 in processivity (Fig. 2) is related to its ability to bind vA20. Toward this end, 6His-tagged mD4 (6His-mD4) produced in E. coli was purified to homogeneity (Fig. 3A) for use in pulldown assays. As shown in Fig. 3B, 6His-mD4 was able to pull down in vitro-translated mA20 and vA20. This result strongly supports the premise that the ability of mD4 to functionally substitute for vD4 involves the formation of the mD4/vA20 heterocomplex.
FIG 3.
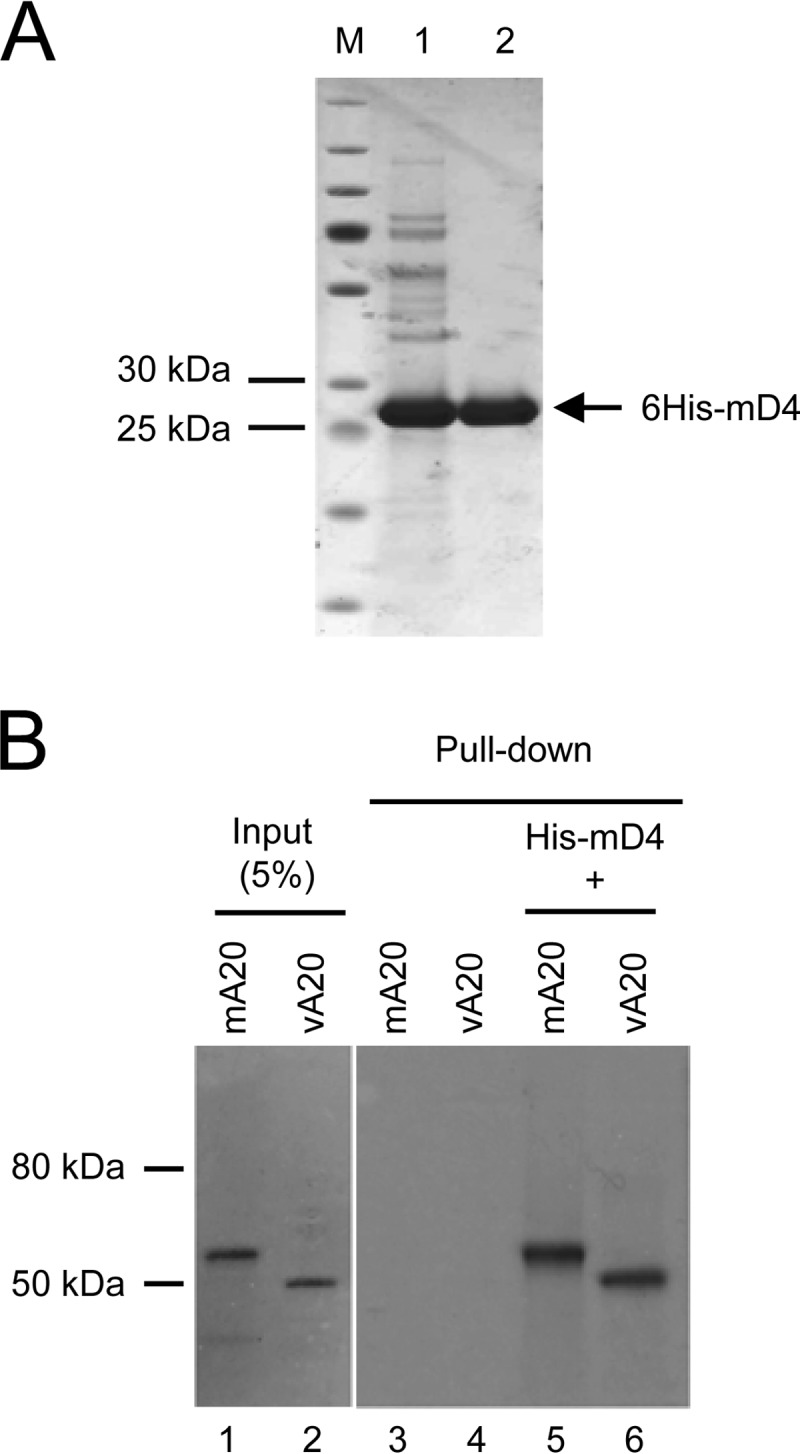
Purified mD4 physically interacts with A20 from both MCV and VV. (A) Expression and purification of His-tagged mD4 from bacteria. N-terminal 6× His-tagged mD4 (6His-mD4) was induced in E. coli and purified using cobalt metal-affinity resins (lane 1), followed by gel filtration chromatography on Superdex 200 (lane 2). Lane M indicates size markers. (B) Pulldown assay. Purified 6His-mD4 was incubated with in vitro-translated 35S-labeled mA20 or vA20 and pulled down by cobalt resins. Pulled-down proteins were separated on SDS gel and visualized by autoradiography. The input represents 5% of the radiolabeled proteins used for the pulldown assay.
mD4 enables long-chain processive DNA synthesis in the M13 assay.
The RPA (Fig. 2) is a reliable screening technology for evaluating processivity (19, 20), but it is limited to the maximum incorporation of nucleotides. In order to confirm that mD4 can completely substitute for vD4 in long-chain processive DNA synthesis, we employed a more rigorous assay using M13 single-strand DNA (7,249 nucleotides) as the template. In this assay, the M13 template was annealed to a 30-nucleotide primer and incubated with dNTPs in the presence of vE9, vA20, and either vD4 or 6His-mD4. As shown in Fig. 4, neither 6His-mD4 alone (lane 1) or vE9 alone (lane 4) nor the negative control luciferase (lane 5) was capable of generating newly synthesized DNA from the M13 template. In contrast, the 6His-mD4, vA20, and vE9 triad was able to synthesize the 7,249-nucleotide full-length strand (lane 2). As a positive control, the VV triad (vE9, vD4, vA20) was also shown to produce a full-length M13 product (lane 3). Of note, the assay is not quantitative for signal strength, since the synthetic source of each D4 protein was different, i.e., bacterial His-mD4 and in vitro-translated vD4. These data clearly demonstrate that mD4 is capable of substituting for vD4 in processive DNA synthesis.
FIG 4.

Purified mD4 is functional in processive DNA synthesis. Full-length M13 DNA (7,249 nucleotides) was annealed to a primer (left) and used as the template for DNA synthesis with in vitro-translated vE9, vA20, and either purified 6His-mD4 or in vitro-translated vD4. The newly synthesized DNA products were fractionated on a 1.3% alkaline agarose gel and visualized by autoradiography (right). The 6His-mD4-dependent mixed triad (lane 2) was able to synthesize the 7,249-nucleotide full-length M13 DNA, as did the VV triad (lane 3), which served as a positive control.
Compound 10 binds mD4 and blocks mD4-dependent processive DNA synthesis in vitro.
From a previous high-throughput screening that examined the disruption of interaction between vD4 and the N-terminal domain of vA20, we discovered five small chemical compounds that can bind vD4 and inhibit VV DNA synthesis and infection (23). The fact that mD4 can substitute for its VV counterpart vD4 (Fig. 2 and 4) prompted us to inquire whether mD4 can also be targeted by these five compounds. To examine this possibility, we first employed the Thermofluor assay (differential scanning fluorimetry). This assay can detect the binding of a small molecule to a protein by observing an increase in thermal stability (Tm). We found that one of the five small molecules, compound 10 (4-({[2-(benzyloxy)phenyl]methyl}amino)phenol), was clearly able to bind mD4. As seen in Fig. 5A, purified 6His-mD4 exhibited a Tm shift (ΔTm, 2.2°C) in the presence of 10 μM compound 10 and an even greater Tm shift (ΔTm, 3.3°C) in the presence of 40 μM compound 10. These results suggest that compound 10 binds to a conserved region of vD4 and mD4.
FIG 5.

Compound 10 binds mD4 and inhibits mD4-dependent processive DNA synthesis. (A) Molecular formula of compound 10 (top, Cmpd 10). Thermal shift of 6His-mD4 in the presence of compound 10 (bottom). Thermal shift (ΔTm) is the difference between DMSO mock treatment and compound 10. Note that the mean thermal shift of 6His-mD4 increases as the concentration of compound 10 is elevated from 10 μM (ΔTm, 2.2 ± 0.2°C [mean ± SD]) to 40 μM (ΔTm, 3.3 ± 0.3°C). The data were obtained from two independent experiments. (B) Inhibition of mD4-dependent processive DNA synthesis by compound 10. The rapid plate assay was used to quantitate DNA synthesis conducted by purified 6His-mD4 and in vitro-translated vA20 and vE9 in the presence of increasing concentrations of compound 10. The mean IC50 of compound 10 was 28 μM, from two independent assays.
We next inquired whether the binding of compound 10 to mD4 would inhibit its essential role as a PF in DNA synthesis. To examine this possibility, we employed the RPA that can quantitatively evaluate the potency (50% inhibitory concentration [IC50]) of small chemical inhibitors for their abilities to block processive DNA synthesis (19–23, 27). In order to be consistent with the M13 assay (Fig. 4), we used translated vE9, vA20, and bacterial 6His-mD4. As shown in Fig. 5B, compound 10 was able to block DNA synthesis with an IC50 of 28 μM. Taken together, these results indicate that compound 10 binds mD4 to disable it from functioning in processive DNA synthesis.
Constructing an mD4-VV hybrid virus to assay compounds that target mD4.
We next wanted to evaluate whether compound 10 is capable of blocking mD4 function in the context of a viral infection. However, the major impediment in discovering drugs that specifically target a critical function of MCV has been the inability to propagate this virus in all cell culture systems attempted thus far (10, 11). We have now circumvented this obstacle by engineering a vaccinia hybrid virus (mD4-VV) in which vD4 is replaced by mD4 (Fig. 6). The rationale for electing this approach was our demonstration that mD4 can completely substitute for vD4 in processive DNA synthesis (Fig. 2, 4, and 5). A depiction of how the mD4-VV virus was engineered is shown in Fig. 6A. Briefly, the mD4-VV hybrid virus was engineered from the VV D4 deletion mutant virus VVΔD4R (16, 24). The VVΔD4R virus (WR strain) can only be propagated on a complementing vD4-expressing rabbit kidney (RK) cell line (24) and not in the normal RK (or BSC-1) cells. We introduced mD4 into VVΔD4R by homologous recombination (16), as shown in Fig. 6A. The new mD4-VV hybrid virus was isolated by several rounds of plaque purification in BSC-1 cells. The amplified stock of mD4-VV hybrid virus was sequenced to verify that it encodes the complete and fully intact mD4 gene. As a positive control, we recombined the vD4 back into VVΔD4R to generate a rescue virus, vD4-VV. Figure 6B reveals that multistep growth curves for mD4-VV hybrid virus and the positive-control vD4-VV rescue virus are nearly identical when propagated in BSC-1 cells. Also, in BSC-1 cells, the mD4-VV hybrid virus produced plaques of similar size to those of the positive control virus vD4-VV (data not shown).
FIG 6.

Construction of a vaccinia hybrid virus containing mD4 of MCV. (A) Schematic of the construction of mD4-VV hybrid virus. The starting VV has vD4 deleted (VVΔD4R). Cloned mD4 with vD4 flanking sequences was transfected into RK-D4R helper cells infected with VVΔD4R, in which the natural vD4 gene had been deleted. The mD4-VV hybrid virus was obtained through homologous recombination and plaque purified, and the mD4 insert was verified by DNA sequencing. As a control, the vD4-VV rescue virus was similarly generated by using a vD4 plasmid. The boxes labeled F are vD4-flanking sequences. (B) Growth curves of mD4-VV and vD4-VV. BSC-1 cells were infected with mD4-VV and vD4-VV viruses (MOI, ∼0.05 PFU/cell). At indicated time points, media and cells were harvested, and virus titers were determined.
The mD4 inhibitor compound 10 blocks infection of the mD4-VV hybrid virus.
We next tested if compound 10 is able to inhibit infection by the mD4-hybrid virus. Indeed, as seen in the plaque reduction assay shown in Fig. 7A, compound 10 effectively blocked mD4-VV infection of BSC-1 cells with a 50% effective concentration (EC50) of 14 μM, which is comparable to its IC50 of 28 μM in inhibiting processive DNA synthesis (Fig. 5B) and far below its 50% cytotoxic concentration (CC50, 175.4 μM) on BSC-1 cells as previously determined (23). In comparison, an EC50 of 11 μM was obtained for compound 10 in inhibiting vD4-VV rescue virus (Fig. 7A). In contrast, the antiherpesvirus drug acyclovir had no inhibitory effect, even at high concentrations (data not shown).
FIG 7.

Compound 10 inhibits mD4-VV hybrid virus replication. (A) Plaque reduction assay. Confluent BSC-1 cells were infected with mD4-VV hybrid virus or vD4-VV rescue virus (80 PFU/well) in the presence of increasing concentrations of compound 10. After 16 h, cells were stained with crystal violet, and plaques were counted. The mean EC50 values of compound 10 obtained from two independent experiments were 14 ± 1 μM and 11 ± 1 μM (means ± SD) for mD4-VV and vD4-VV, respectively. (B) Dot blot hybridization. Confluent BSC-1 cells were infected with mD4-VV or vD4-VV (∼1 MOI) in the absence or presence of 65 μM compound 10. Cells were collected at indicated time points, and viral genomic DNA was extracted. Dot blot hybridization was then performed using 32P-labeled vA20 DNA as a probe. The relative viral DNA levels are shown below each panel. None, no infection.
To confirm that compound 10 blocks mD4-VV replication via inhibiting viral processive DNA synthesis, we evaluated its effect on viral DNA accumulation throughout an infection time course. As shown in Fig. 7B (upper row), treatment of BSC-1-infected cells with 65 μM compound 10 completely prevented mD4-VV genomic DNA accumulation at each time point of infection. In contrast, in the absence of compound 10 (DMSO alone), viral DNA was observed to accumulate over time as expected. A similar result was obtained with the control vD4-VV rescue virus (Fig. 7B, bottom row). These data clearly demonstrate that compound 10 inhibits viral DNA replication.
DISCUSSION
There is no licensed antiviral drug available that specifically treats molluscum contagiosum (MC), a common viral skin infection that is prevalent in children and adults. Any attempts to identify compounds that might block MCV infection have been impeded by the inability to propagate this poxvirus in cell culture (10, 11). In this study, we devised a two-step strategy to circumvent this roadblock. For the first step, we identified a novel and essential MCV target gene that is inhibited in vitro by a small chemical compound. Specifically, we cloned the mD4 gene of MCV that is essential for processive DNA synthesis. We then showed that compound 10, previously demonstrated to be efficacious in blocking vaccinia poxvirus vD4, was capable of both binding mD4 and blocking mD4-dependent processive DNA synthesis, as determined by in vitro assays. For the second step, we constructed a vaccinia hybrid virus (mD4-VV) that substitutes the mD4 target gene for the natural vD4 gene. mD4-VV exhibited the same growth characteristics as the vD4-rescued vaccinia virus. Significantly, we showed that compound 10 does indeed block the mD4-VV hybrid virus from replicating in cells. The fact that mD4 is required by the hybrid virus to be infectious and that mD4 is a physical and functional target of compound 10 predicts strongly that compound 10 will inhibit natural MCV infection. Importantly, this study provides, for the first time, a systematic path for discovering drugs that specifically target MCV replication and infection.
Of the MCV processivity triad, only mD4 substituted for its vD4 counterpart in the in vitro processive DNA synthesis assay, whereas A20 and E9 substituted poorly (Fig. 2). This may be related to the fact that mD4 exhibits a greater chemical identity to its vaccinia virus counterpart (55%) than does its processivity partner A20 (30%) (see Table S1 in the supplemental material). Significantly, purified mD4 can physically associate with both mA20 and vA20, as seen by pulldown assays, further validating the conservation of this poxvirus protein (Fig. 3B). Most important, compound 10 binds and inhibits mD4 function, as shown here, as well as vD4 (23). This suggests that compound 10 was able to bind to the conserved region of D4 that is involved in interacting with A20. Recently, the cocrystal structure of vD4 with the vA20 N-terminal domain revealed that the C-terminal residues 167 to 180 and 191 to 206 of vD4 are in direct contact with vA20 (28). Point mutations of Arg167 and Pro173 in this region of vD4 were able to perturb formation of the D4/A20 complex (28). Sequence comparison of mD4 and vD4 indicates that the C-terminal region (residues 167 to 206, including Arg167 and Pro173) is conserved in D4 (67.5% identity) (see Fig. S2A in the supplemental material). Future studies will address if mutations in the conserved C-terminal region of mD4, including the corresponding Arg164 and Pro180, can also perturb interaction between mD4 and vA20 or binding of compound 10 to mD4. It is notable that the predicted structure of mD4 superimposes directly onto the known crystal structure of vD4 (29) (see Fig. S2B in the supplemental material). This may largely explain why both mD4 and vD4 are targeted by compound 10. Since D4 proteins of other poxviruses share high sequence identity with vD4 (∼99%), compound 10 is anticipated to have broad-spectrum antipoxviral activity. Notably, four other compounds that exhibited binding to vD4 by thermal shift (23) failed to bind mD4 (this study, not shown), which may be accounted for by differences in their protein composition and local conformations. The mD4-VV viral hybrid system should permit identification of additional compounds that directly bind and inhibit multiple regions of mD4, thus enabling the development of new therapeutics that specifically target MCV for treatment of MC.
In summary, we have cloned and identified a novel antiviral target, the PF mD4, that can be inhibited by a small chemical compound to prevent the viral Pol from synthesizing extended strands of DNA. The type of MCV hybrid system described here can be exploited for discovering additional viral targets and compounds that will block infection. Compounds that can target specific MCV genes in this surrogate virus system are potential candidates to move forward in drug development.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant 5U01-A1082211 to R.P.R. and partially supported through a pilot project grant from the Penn Skin Disease Research Center, an NIH-funded program (P30 AR057217), to S.N.I.
Footnotes
Published ahead of print 29 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03660-14.
REFERENCES
- 1.Isaacs SN. 2013. Molluscum contagiosum. In Hirsch MS, Levy ML, Rosen T. (ed), UpToDate. Wolters Kluwer, Waltham, MA. [Google Scholar]
- 2.Tyring SK. 2003. Molluscum contagiosum: the importance of early diagnosis and treatment. Am. J. Obstet. Gynecol. 189(Suppl):S12–S16. 10.1067/S0002-9378(03)00793-2. [DOI] [PubMed] [Google Scholar]
- 3.Braue A, Ross G, Varigos G, Kelly H. 2005. Epidemiology and impact of childhood molluscum contagiosum: a case series and critical review of the literature. Pediatr. Dermatol. 22:287–294. 10.1111/j.1525-1470.2005.22401.x. [DOI] [PubMed] [Google Scholar]
- 4.Gottlieb SL, Myskowski PL. 1994. Molluscum contagiosum. Int. J. Dermatol. 33:453–461. 10.1111/j.1365-4362.1994.tb02853.x. [DOI] [PubMed] [Google Scholar]
- 5.Osinusi O, Greisman L, Menajovsky J, Pinczewski J, Ghosh M. 2011. A patient with AIDS with fungating lesions of the face and scalp. Clin. Infect. Dis. 52:1029–1030. 10.1093/cid/cir030. [DOI] [PubMed] [Google Scholar]
- 6.Calista D, Boschini A, Landi G. 1999. Resolution of disseminated molluscum contagiosum with highly active anti-retroviral therapy (HAART) in patients with AIDS. Eur. J. Dermatol. 9:211–213. [PubMed] [Google Scholar]
- 7.Bachmeyer C, Moguelet P, Baud F, Lescure FX. 2009. Efflorescence of facial molluscum contagiosum as a manifestation of immune reconstitution inflammatory syndrome in a patient with AIDS. Eur. J. Dermatol. 19:257–258. 10.1684/ejd.2009.0622. [DOI] [PubMed] [Google Scholar]
- 8.Pereira B, Fernandes C, Nachiambo E, Catarino MC, Rodrigues A, Cardoso J. 2007. Exuberant molluscum contagiosum as a manifestation of the immune reconstitution inflammatory syndrome. Dermatol. Online J. 13:6. [PubMed] [Google Scholar]
- 9.Chen X, Anstey AV, Bugert JJ. 2013. Molluscum contagiosum virus infection. Lancet Infect. Dis. 13:877–888. 10.1016/S1473-3099(13)70109-9. [DOI] [PubMed] [Google Scholar]
- 10.Bugert JJ, Melquiot N, Kehm R. 2001. Molluscum contagiosum virus expresses late genes in primary human fibroblasts but does not produce infectious progeny. Virus Genes 22:27–33. 10.1023/A:1008126217725. [DOI] [PubMed] [Google Scholar]
- 11.Sherwani S, Blythe N, Farleigh L, Bugert JJ. 2012. New method for the assessment of molluscum contagiosum virus infectivity. Methods Mol. Biol. 890:135–146. 10.1007/978-1-61779-876-4_8. [DOI] [PubMed] [Google Scholar]
- 12.Jeruzalmi D, O'Donnell M, Kuriyan J. 2002. Clamp loaders and sliding clamps. Curr. Opin. Struct. Biol. 12:217–224. 10.1016/S0959-440X(02)00313-5. [DOI] [PubMed] [Google Scholar]
- 13.Lin K, Dai CY, Ricciardi RP. 1998. Cloning and functional analysis of Kaposi's sarcoma-associated herpesvirus DNA polymerase and its processivity factor. J. Virol. 72:6228–6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Druck Shudofsky AM, Silverman JE, Chattopadhyay D, Ricciardi RP. 2010. Vaccinia virus D4 mutants defective in processive DNA synthesis retain binding to A20 and DNA. J. Virol. 84:12325–12335. 10.1128/JVI.01435-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stanitsa ES, Arps L, Traktman P. 2006. Vaccinia virus uracil DNA glycosylase interacts with the A20 protein to form a heterodimeric processivity factor for the viral DNA polymerase. J. Biol. Chem. 281:3439–3451. 10.1074/jbc.M511239200. [DOI] [PubMed] [Google Scholar]
- 16.De Silva FS, Moss B. 2003. Vaccinia virus uracil DNA glycosylase has an essential role in DNA synthesis that is independent of its glycosylase activity: catalytic site mutations reduce virulence but not virus replication in cultured cells. J. Virol. 77:159–166. 10.1128/JVI.77.1.159-166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boyle KA, Stanitsa ES, Greseth MD, Lindgren JK, Traktman P. 2011. Evaluation of the role of the vaccinia virus uracil DNA glycosylase and A20 proteins as intrinsic components of the DNA polymerase holoenzyme. J. Biol. Chem. 286:24702–24713. 10.1074/jbc.M111.222216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishii K, Moss B. 2002. Mapping interaction sites of the A20R protein component of the vaccinia virus DNA replication complex. Virology 303:232–239. 10.1006/viro.2002.1721. [DOI] [PubMed] [Google Scholar]
- 19.Lin K, Ricciardi RP. 2000. A rapid plate assay for the screening of inhibitors against herpesvirus DNA polymerases and processivity factors. J. Virol. Methods 88:219–225. 10.1016/S0166-0934(00)00190-7. [DOI] [PubMed] [Google Scholar]
- 20.Ricciardi RP, Lin K, Chen X, Dorjsuren D, Shoemaker R, Sei S. 2005. Rapid screening of chemical inhibitors that block processive DNA synthesis of herpesviruses: potential application to high-throughput screening. Methods Mol. Biol. 292:481–492. [DOI] [PubMed] [Google Scholar]
- 21.Ciustea M, Silverman JEY, Druck Shudofsky AM, Ricciardi RP. 2008. Identification of inhibitors of vaccinia virus DNA synthesis by high-throughput screening. J. Med. Chem. 51:6563–6570. 10.1021/jm800366g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silverman JEY, Ciustea M, Druck Shudofsky AM, Bender F, Shoemaker RH, Ricciardi RP. 2008. Identification of polymerase and processivity inhibitors of vaccinia DNA synthesis using a stepwise screening approach. Antiviral Res. 80:114–123. 10.1016/j.antiviral.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nuth M, Huang L, Saw YL, Schormann N, Chattopadhyay D, Ricciardi RP. 2011. Identification of inhibitors that block vaccinia virus infection by targeting the DNA synthesis processivity factor D4. J. Med. Chem. 54:3260–3267. 10.1021/jm101554k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holzer GW, Falkner FG. 1997. Construction of a vaccinia virus deficient in the essential DNA repair enzyme uracil DNA glycosylase by a complementing cell line. J. Virol. 71:4997–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Earl PL, Moss B. 2001. Characterization of recombinant vaccinia viruses and their products. Curr. Protoc. Mol. Biol. Chapter 16:Unit 16.18. 10.1002/0471142727.mb1618s43. [DOI] [PubMed] [Google Scholar]
- 26.Senkevich TG, Koonin EV, Bugert JJ, Darai G, Moss B. 1997. The genome of molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology 233:19–42. 10.1006/viro.1997.8607. [DOI] [PubMed] [Google Scholar]
- 27.Nuth M, Guan H, Zhukovskaya N, Saw YL, Ricciardi RP. 2013. Design of potent poxvirus inhibitors of the heterodimeric processivity factor required for viral replication. J. Med. Chem. 56:3235–3246. 10.1021/jm301735k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Contesto-Richefeu C, Tarbouriech N, Brazzolotto X, Betzi S, Morelli X, Burmeister WP, Iseni F. 2014. Crystal structure of the vaccinia virus DNA polymerase holoenzyme subunit D4 in complex with the A20 N-terminal domain. PLoS Pathog. 10:e1003978. 10.1371/journal.ppat.1003978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schormann N, Grigorian A, Samal A, Krishnan R, DeLucas L, Chattopadhyay D. 2007. Crystal structure of vaccinia virus uracil-DNA glycosylase reveals dimeric assembly. BMC Struct. Biol. 7:45. 10.1186/1472-6807-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.