

Abstract
BMS-791325 is a hepatitis C virus (HCV) inhibitor binding to the thumb domain of the NS5B RNA-dependent RNA polymerase. BMS-791325 is well characterized in genotype 1 (GT1) and exhibits good inhibitory activity (50% effective concentration [EC50], <10 nM) against hybrid replicons containing patient NS5B sequences from GT3a, -4a, and -5a while potency against GT2 is significantly reduced (J. A. Lemm et al., Antimicrob. Agents Chemother. 58:3485–3495, 2014, doi:http://dx.doi.org/10.1128/AAC.02495-13). BMS-791325 potency against GT6a hybrid replicons is more variable, with two of three hybrid clones having EC50s similar to that for GT1 while a third patient clone was ∼10 times less susceptible to BMS-791325. To characterize the resistance profile of BMS-791325 beyond GT1, curing studies were performed across GT1a and -3a to -6a and demonstrated that GT1a has the highest resistance barrier versus BMS-791325 while GT6a has the lowest. Selection of GT3 to -6 NS5B chimeric replicon cells at different concentrations of BMS-791325 revealed substitutions in the thumb domain of NS5B at residues 494 and 495 that conferred different levels of resistance to BMS-791325 but remained susceptible to NS5A or NS3 protease inhibitors. In addition, we demonstrate that the reduced potency of BMS-791325 against one GT6a patient is due to an A494 polymorphism present in ∼21% of sequences in the European HCV database. The results from this report suggest that BMS-791325 is a candidate for combination treatment of HCV GT3 to -6 chronic infections, and the resistance profiles identified will provide useful information for future clinical development.
INTRODUCTION
Hepatitis C virus (HCV) is a member of the Flaviviridae family with a positive-sense, single-strand RNA genome of approximately 9.6 kb in length. The HCV genome encodes a polyprotein that is processed into 10 different proteins: core, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B (1). The nonstructural proteins NS3 to NS5B are involved in replication of the viral genome, whereas the structural proteins (core, E1, and E2) are components of the viral particle (2, 3). HCV is classified into 6 major genotypes (GTs) with nucleotide sequence divergence of as much as 35%, each with multiple subtypes. Substantial regional differences exist in the global distribution of HCV genotypes. GT1a and -1b, which share approximately 88% genetic similarity (4, 5), are the predominant subtypes in the United States and Europe. In Japan, subtype 1b is responsible for up to 73% of cases of HCV infection (6). HCV subtypes 2a and 2b are relatively common in North America, Europe, and Japan, while HCV GT3a is particularly prevalent in intravenous drug abusers in Europe and the United States (7). GT4 to -6 are distributed less widely than GT1 to -3, with GT4 found mainly in Egypt and Africa, GT5 in South Africa, and GT6 in southeastern Asia (8).
Approximately 170 million people worldwide are infected with HCV, and persistent infection may lead to chronic hepatitis, cirrhosis, or hepatocellular carcinoma (9, 10). Treatment for HCV-infected patients often consists of a combination of pegylated alpha interferon (Peg-IFN-α) and ribavirin (RBV), which produces serious side effects and incomplete antiviral efficacy in many patients. Only ∼50% of the patients infected with HCV GT1 achieve a sustained viral response (SVR) upon treatment, although higher rates (∼80%) have been reported for patients infected with GT2 and GT3 (11–13). The new direct-acting antiviral agents (DAAs) telaprevir and boceprevir are NS3 protease inhibitors being used in combination with Peg-IFN-α and RBV that increase SVR rates and shorten the treatment duration for patients infected with GT1 only (14). The recently approved nucleoside inhibitor sofosbuvir, although it has pan-genotype coverage and can be used with RBV alone for some patients, should combine with RBV and Peg-IFN-α for GT1 and GT4 patients. The newly approved NS3 protease inhibitor simeprevir was prescribed in combination with Peg-IFN-α and RBV to treat GT1 patients, including those with liver disease (15). However, some participants experienced severe photosensitivity and had to be hospitalized (16). Thus, there is still an unmet medical need for more effective and broad-spectrum HCV therapies with good safety profiles.
The HCV RNA-dependent RNA polymerase (RdRp) is essential for viral replication and is an attractive target for the development of anti-HCV therapies. The structure of NS5B polymerase resembles a characteristic “right-hand” motif fold with finger, palm, and thumb domains (17). Two classes of NS5B polymerase inhibitors can be distinguished: nucleoside and nonnucleoside analogue inhibitors that bind to different allosteric sites. There are at least 4 distinct allosteric binding sites (thumb1, thumb2, palm1, and palm2) on the HCV polymerase which show no cross-resistance. BMS-791325 is a site I inhibitor binding to the thumb1 domain of NS5B polymerase. The error-prone nature of the RdRp contributes to the production of viral quasispecies, a population of highly genetically heterogeneous variants (18, 19). Since the high rate of viral replication and high mutation rate of the NS5B polymerase lead to rapid generation of drug-resistant mutants, emergence of resistant viruses is a major challenge in the development of successful antiviral therapies and combination therapy will be required.
Development of the replicon system was a significant breakthrough in HCV drug discovery and has been invaluable for the in vitro study of HCV replication (20). Since then, subgenomic replicons of several GTs (e.g., GT1a, -2a, -3a, -4a, and -6a) have been developed (21–26). In order to determine the antiviral activity of HCV polymerase inhibitors against various GTs, we have generated GT1a-H77c and 1b-Con1 shuttle replicons with unique restriction sites for cloning of patient-derived NS5Bs from other GTs (27). Using this tool, we have created a panel of replication-competent chimeric replicon cell lines with NS5B sequences derived from GT2 to -6 clinical samples for the evaluation of the antiviral spectrum of NS5B polymerase inhibitors.
In this study, we evaluated the resistance barrier and also selected and analyzed the in vitro resistance profile of BMS-791325 in the major HCV genotypes using the NS5B chimeric replicon system. The correlation between replicon and clinical resistance development in GT1 (27, 28) helps to validate the replicon system and provide guidance for clinical resistance emerging in other genotypes. We also show that replicons resistant to BMS-791325 remain fully sensitive to other DAAs, such as NS3 protease or NS5A replication complex inhibitors, further supporting the use of BMS-791325 in combination therapy.
MATERIALS AND METHODS
Cell lines and HCV inhibitors.
Human hepatoma cells (Huh-7) were propagated in Dulbecco modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (HyClone) and 100 IU/ml penicillin and 100 μg/ml streptomycin (Invitrogen). Stable replicon cell lines were cultured using the same medium supplemented with 0.5 mg/ml G418 (Geneticin; Invitrogen). Genotype 1a (H77c) and 1b (Con1) replicon cells used in this study have been described previously (27, 29). BMS-791325 was synthesized at Bristol-Myers Squibb. Daclatasvir (DCV; NS5A replication complex inhibitor) and asunaprevir (ASV; NS3 protease inhibitor) were previously described (27–39).
Construction of HCV genotype 2 to 6 NS5B chimeric replicons.
HCV chimeric replicon clones which replaced the Con1 or H77c NS5B gene with clinical isolate sequences from GT2 to -6 were constructed. GT1a or -1b shuttle replicons were prepared by introducing unique SpeI-ClaI or SpeI-SnaB1 sites flanking the GT1a or -1b NS5B genes by site-directed mutagenesis using the QuikChange mutagenesis protocol (Stratagene). Restriction sites generated in the shuttle replicons have no effect on replication efficiency or compound susceptibility. The GT1 shuttle replicons contain amino acid substitutions E1202G in NS3 and S2204I in NS5A as shown in Fig. 1. Plasma samples of HCV patients infected with GT2b, -3a, -4a, and -5a were purchased from Boca Biolistics (Coconut Creek, FL, USA), while GT6 patient sera were obtained from SeraCare Life Science (Milford, MA, USA) or provided by Huy Trinh. RNA was isolated from HCV-infected patient sera using the QIAamp MinElute virus vacuum system according to the manufacturer's instructions. First-strand cDNA synthesis was performed with random hexamers using a Superscript III first-strand cDNA synthesis kit (Invitrogen Corporation, Carlsbad, CA). Patient-derived NS5Bs were amplified by nested PCR with Platinum Taq high-fidelity DNA polymerase (Invitrogen). Amplicons were subjected directly to sequence analysis and cloned into the GT1a shuttle replicon for GT3a and the GT1b shuttle replicon for GT2b to -6 using the in-fusion HD EcoDry cloning kit (Clontech, Mountain View, CA, USA). In addition, for HCV GT2b, -4a, and -5a, a patient-derived NS5B sequence known to generate a viable chimeric replicon (26) was synthesized by DNA 2.0 and cloned into the GT1b shuttle replicon.
FIG 1.
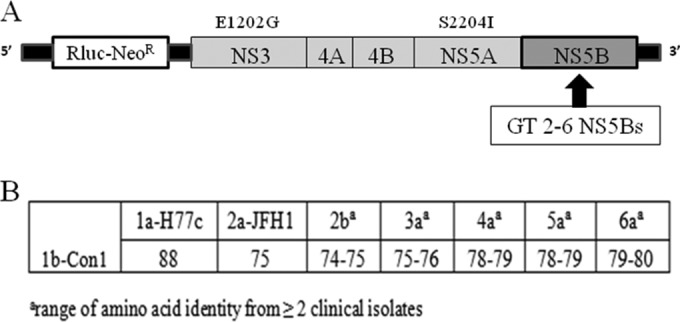
Construction of HCV GT2 to -6 NS5B chimeric replicons. (A) Schematic diagram of 1b-Con1 or 1a-H77c replicons with Renilla luciferase and neomycin resistance genes. GT2 to -6 chimeric replicons were constructed by replacing the entire NS5B coding region of the parent replicon with patient-derived NS5Bs from GT3a for the 1a-H77c backbone and GT2b, -4a, -5a, or -6a for the 1b-Con1 backbone. Each of the replicons contained engineered adaptive mutations of E1202G in NS3 and S2204I in NS5A. All NS5B chimeric replicon cell lines exhibited luciferase activities comparable to those of the GT1a/1b parent cell lines. (B) Percent NS5B amino acid identity of HCV GT2 to -6 patient samples to 1b-Con1.
Resistance selection.
HCV replicon cells were seeded in 10-cm tissue culture dishes (∼5 × 106 cells), allowed to attach overnight, and then maintained in tissue culture medium with 0.5 mg/ml G418 and the desired concentration of BMS-791325 in dimethyl sulfoxide (DMSO). Control cells were maintained with an equivalent percent volume of DMSO (0.1%). Cells were split or fed with fresh medium containing inhibitor twice a week for up to 4 to 5 weeks. Surviving colonies were expanded for resistance testing and sequence analysis.
Generation of chimeric replicons with selected mutants.
Total RNA was isolated from populations of resistant cells using either TRIzol or an RNeasy minikit (Qiagen, Valencia, CA) in accordance with the manufacturer's directions. First-strand cDNA synthesis was performed using 0.5 to 1 μg of total RNA and Superscript III reverse transcriptase with random hexamers. NS5B PCR products were generated, purified, and cloned using a TOPO TA cloning kit (Invitrogen). DNA was sequenced from ∼50 clones to establish the frequency and coincidence of mutations in a population. Sequence analysis and alignments were performed in Sequencher 4.6 software.
Mutations in the NS5B coding region were introduced into GT2 to -6 chimeric replicon clones using a QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions. Desired changes were confirmed by sequence analysis.
Transient replication assays.
Genotype 1a/1b replicons and GT2b, -4a, -5a, and -6a NS5B chimeric replicon clones were linearized by ScaI digestion; GT2a JFH1 or GT3a NS5B chimeric replicons were linearized with XbaI or HpaI digestion, respectively, and transcribed in vitro with a T7 RiboMAX Express large-scale RNA production system (Promega, Madison, WI) according to the manufacturer's directions. For transient replication assays, 5 to 10 μg of RNA was transfected into cured Huh-7 cells (∼2 × 106 cells/60-mm dish) with DMRIE-C reagent (Invitrogen). After 4 to 5 h, transfected cells were trypsinized and transferred into 96-well plates (104/well), dilutions of inhibitors in DMSO were added, and plates were incubated at 37°C for 72 h. Plates were washed with phosphate-buffered saline (PBS) and assayed with a Renilla luciferase assay system (Promega) according to the manufacturer's directions.
Generation of stable cell lines.
To generate stable replicon cell lines, transfected cells were switched to 0.3 mg/ml G418 medium 24 h after transfection and cultured continually in G418 medium. Colonies were picked after ∼3 weeks of selection and expanded. Luciferase activity was measured, and the cell line with the highest activity was used for antiviral assays.
HCV replicon luciferase assay.
To evaluate compound efficacy, HCV replicon cell lines were trypsinized and seeded into 96-well plates at 104 cells/well. Compounds serially diluted in DMSO or DMSO alone was added to individual wells to a final DMSO concentration of 0.5%. Cell plates were incubated at 37°C for 3 days, and Renilla luciferase activity was then assayed with EnduRen cell substrate (Promega) according to the manufacturer's directions. Plates were read on a TopCount NXT microplate scintillation and luminescence counter (Packard Instrument Company, Meriden, CT). The 50% effective concentration (EC50) was calculated as previously described (39).
Efficiency of replicon clearance from cultured cells.
HCV replicon cells (24,000 per 100-mm dish) were treated with BMS-791325 at a 300 nM concentration in cell growth medium for up to 10 days. Medium containing inhibitors was removed at different time points, and cells were maintained in growth medium containing 0.5 mg/ml G418. Medium was changed twice weekly for a period of ∼4 weeks, and then plates were washed and colonies were stained with 0.2% crystal violet.
RESULTS
In vitro antiviral activity of BMS-791325 against GT1 to -6 NS5B chimeric replicons.
BMS-791325 exhibits low-nanomolar potency against GT1a H77c and -1b Con1 strains, as well as chimeric replicons with NS5B sequences derived from GT1 clinical isolates (27). To evaluate BMS-791325 activity against additional genotypes, replication-competent NS5B chimeric replicons were generated from GT2 to -6 patient isolates (Fig. 1). The level of amino acid variation between clinical isolates within each GT ranged between 2 and 5%. In contrast, the divergence of NS5B sequences between GT1b Con1 and the clinical isolates ranged from 25 to 26% for GT2b, 24 to 25% for GT3a, 21 to 22% for GT4a and -5a, and 20 to 21% for GT6a as shown in Fig. 1. The antiviral activity of BMS-791325 against GT2 to -6 was evaluated using stable cell lines containing chimeric replicons with NS5B derived from various patient isolates (Table 1). As previously reported (27), significantly reduced susceptibility to BMS-791325 was observed for GT2a and -2b chimeric replicons (Table 1). In contrast, BMS-791325 exhibited nanomolar potency against GT3a, -4a, and -5a chimeric replicons generated from multiple patient isolates for each GT with EC50s ranging from 0.5 to 9.3 nM. BMS-791325 is highly active against GT6a chimeric replicons generated from two patient isolates (TT-003 and 752) that have a valine at residue 494 in NS5B with EC50s less than 10 nM, but an approximately 10-fold loss of potency (EC50, 72 nM) was observed from a 6a isolate (HN001) with a naturally occurring alanine at residue 494 in NS5B. This is consistent with the fact that a V494A substitution in GT1a confers low-level (∼2-fold) resistance to BMS-791325 (data not shown). To confirm that the alanine at position 494 was responsible for the reduced efficacy of BMS-791325, residue 494 was changed to a valine in GT6a isolate HN001 and this clone (6a-HN001-A494V) was evaluated for susceptibility to BMS-791325. Conversely, the valine at residue 494 of GT6a isolate 752 was changed to alanine to generate 6a-752-V494A and the effect of this change on BMS-791325 efficacy was determined. As shown in Fig. 2, the 6a-HN001-A494V replicon showed dramatically increased susceptibility to BMS-791325 (EC50, 7.3 nM) compared to the 6a-HN001-A494 clone with an EC50 of 72 nM. Likewise, BMS-791325 showed reduced potency on the 6a-752-V494A clone (EC50, 81 nM) compared to parent clone 6a-752-V494 (EC50, 9 nM). These data confirm that the alanine substitution at residue 494 of NS5B reduces susceptibility to BMS-791325 in GT6.
TABLE 1.
BMS-791325 activity against HCV genotype 1 to 6 chimeric replicons
| NS5B genotype of chimeric replicon | No. of isolates | EC50 (nM)a |
|
|---|---|---|---|
| Avg | Range | ||
| 1a | 4 | 5 | 2.1 to 7 |
| 1b | 2 | 7 | 4.5 to 9 |
| 2a | 2 | NAb | 76 to >500 |
| 2b | 2 | 925 | 450 to 1,400 |
| 3a | 6 | 4 | 1 to 5.6 |
| 4a | 3 | 6 | 2 to 9.3 |
| 5a | 4 | 3 | 0.5 to 5.5 |
| 6a | 3 | 30 | 9 to 71 |
EC50s are generated from at least two independent experiments for all cell lines.
NA, not available.
FIG 2.
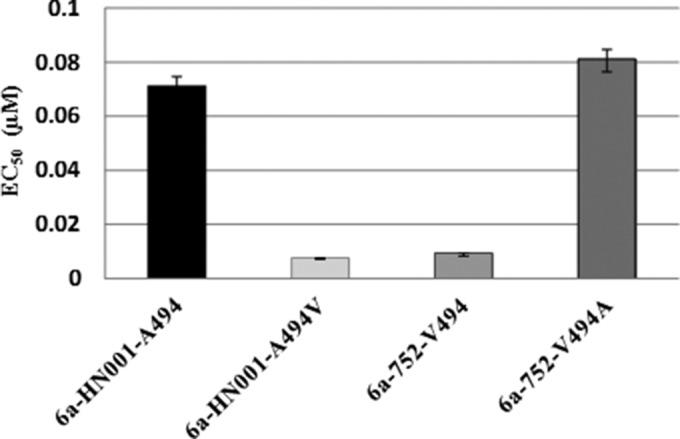
Phenotypic analysis of the NS5B A494V and V494A HCV GT6a replicons. Replicons containing substitutions of A494V in 6a-HN001 and V494A in 6a-752 were generated by site-directed mutagenesis. Stable cell lines were selected and tested for susceptibility to BMS-791325 in a 3-day replicon assay. Results are reported as average EC50 ± standard deviation from at least two independent experiments.
Clearance of HCV replicons by BMS-791325 treatment.
Although BMS-791325 exhibited efficacy against GT3 to -6, the resistance barrier of this compound against non-GT1 replicons was unknown. To evaluate the ability of the compound to eradicate HCV RNA from GT3 to -6 chimeric replicon cells, replicon clearance studies were performed using BMS-791325 at a 300 nM concentration for up to 10 days. Since BMS-791325 has demonstrated substantial antiviral effect against GT1 patients in the clinic (28), GT1a replicon cells were used as a control, and a 300 nM concentration was chosen as it is the average minimum concentration of drug (Cmin) observed at 24 h after a single 100-mg dose of BMS-791325 which resulted in a mean HCV RNA decline of approximately 1.3 log10 copies/ml in HCV GT1-infected patients (28). Consistent with the EC50 results, replicon clearance occurred efficiently in GT3a, -4a, and -5a chimeric replicons, similar to that observed in the GT1a replicon (Fig. 3A). Significant colony reduction occurred by day 5 for GT1a, -3a, -4a, and -5a, with continued reduction observed out to day 10. HCV RNA was not eliminated efficiently from GT2a and -2b replicon cells, consistent with the reduced potency of BMS-791325 against GT2. The GT6a-752 isolate showed susceptibility to BMS-791325 (EC50, ∼10 nM) similar to that of GT1, yet eradication of HCV RNA was less efficient against this hybrid, suggesting that resistance may be more easily generated in this clone.
FIG 3.

Resistance barrier of BMS-791325 on HCV GT1 to -6. Colony elimination studies were performed on GT1 to -6 wild-type replicons (A) or GT6a mutant replicons (B) using BMS-791325 monotherapy. NS5B chimeric replicon cells were treated with BMS-791325 (300 nM) for up to 10 days. Colonies were stained with crystal violet. Data shown are representative of results from two independent experiments.
Curing efficiencies of the GT6a isolates with different susceptibilities to BMS-791325 were also evaluated. Replicon clearance in the GT6a isolate HN001, with a naturally occurring alanine at residue 494 in NS5B, was much less efficient than that in GT6a isolate 752, which has a valine at the 494 position (Fig. 3B). This is consistent with the fact that BMS-791325 was ∼10-fold less potent on the GT6a-HN001 isolate than on the 752 isolate. Colony elimination was much more efficient against the 6a-HN001-A494V cell line, which contains valine at residue 494 and shows susceptibility to BMS-791325 similar to that of the 6a-752 clone (Fig. 3B). Likewise, the V494A substitution in the 6a-752 clone (6a-752-V494A) significantly reduced the ability of BMS-791325 to eradicate HCV RNA from this cell line, further highlighting the importance of this residue for BMS-791325 activity against GT6a. The A494 polymorphism observed in the GT6a-HN001 isolate is present in ∼21% of GT6a sequences in the European HCV database, with V494 conserved in the remaining 79%. Although a lower resistance barrier was observed with this A494 variant in colony elimination studies with BMS-791325, DAAs will not be used as monotherapy in the clinic but rather will be part of a combination regimen which may be able to overcome the lower HCV clearance efficiency resulting from this polymorphism.
Resistance selection with BMS-791325 across genotypes 3 to 6.
In order to characterize the resistance profile of BMS-791325 in GT3 to -6, NS5B chimeric replicon cells were selected with inhibitor at concentrations of 10× and 30× EC50s for up to 5 weeks until a resistant population emerged. GT1 subgenomic replicon cells were selected in parallel as a control. Selection at 30× EC50s in GT1a or -1b replicon cells yielded replicons with EC50s 37- to 42-fold higher than that of DMSO-treated cells (Table 2). Under the same selective pressure, resistance of GT3a or -6a selected cells was 46- or 22-fold higher, respectively, relative to wild-type (WT) EC50s. Higher levels of resistance were detected in GT5a selected cells, where a 91-fold increase in EC50 was observed. For GT4a, on the other hand, replicon cells survived only the 10× EC50 selection, yielding a 24-fold increase in BMS-791325 EC50 versus DMSO-treated cells. Multiple attempts to isolate GT4a cell lines using 30× selection were unsuccessful, even when increasing the concentration from 10× to 30× EC50 on the 10× selected cells. While the potency of BMS-791325 was decreased in the inhibitor-selected cells (EC50s, 129 to 342 nM) versus the DMSO-treated control cells (EC50s, 2 to 9 nM), no change in potency of DCV (EC50s, 0.006 to 0.018 nM for wild type and 0.005 to 0.028 nM for resistant replicons) or ASV (EC50s, 3 to 10 nM for WT and 3 to 22 nM for resistant replicons) was observed (Fig. 4), suggesting that these inhibitors display no cross-resistance with BMS-791325.
TABLE 2.
BMS-791325 resistance selection on HCV GT1 and GT3 to -6 NS5B replicon cells
| NS5B GT panel | Mean EC50 (nM) of parental cells ± SD | BMS-791325 selection concn (nM) | Mean EC50 (nM) of selected cells ± SD | Fold resistancea | NS5B amino acid substitution(s) |
|---|---|---|---|---|---|
| 1a-H77c | 3 ± 1 | 0b | 3 ± 1 | ||
| 90 | 110 ± 30 | 37 | P495A/S/L/T | ||
| 1b-Con1 | 6 ± 1 | 0b | 5 ± 1 | ||
| 180 | 210 ± 70 | 42 | P495A/S/L, V499A | ||
| 3a-Inx | 9 ± 2 | 0b | 8 ± 1 | ||
| 270 | 370 ± 71 | 46 | C494S, P495S | ||
| 4a-h2 | 6 ± 4 | 0b | 4 ± 1 | ||
| 60 | 95 ± 30 | 24 | V494A, P495A/L | ||
| 5a-h3 | 4 ± 1 | 0b | 5 ± 1 | ||
| 120 | 455 ± 49 | 91 | V494A, P495S/L/H | ||
| 6a-752 | 9 ± 3 | 0b | 12 ± 2 | ||
| 270 | 259 ± 23 | 22 | V494A |
Fold resistance = EC50 of BMS-791325-selected cells/EC50 of DMSO-selected cells of the same GT.
DMSO-treated cells.
FIG 4.
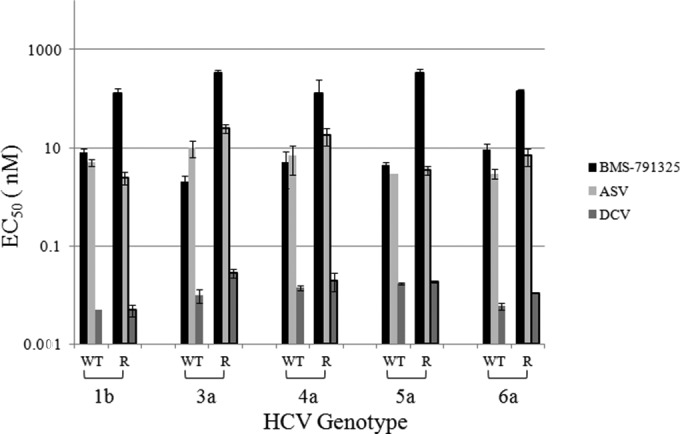
Potency of HCV NS3 protease (ASV) and NS5A replication complex (DCV) inhibitors on BMS-791325-resistant replicon cell lines. GT1 and GT3 to -6 NS5B wild-type (WT) and BMS-791325 resistant (R) replicon cell lines were tested for sensitivity to HCV inhibitors BMS-791325, ASV, and DCV. EC50s are the averages from at least 2 independent experiments, and error bars represent standard deviations.
Genotypic and phenotypic analysis of BMS-791325-selected variants.
To identify the resistant variants selected with BMS-791325, RNA was isolated from resistant and DMSO-treated cells and used to generate NS5B cDNA by reverse transcription-PCR (RT-PCR). Sequence analysis of NS5B revealed variations at residues 494, 495, and 499 in cDNA clones from BMS-791325-treated GT1 and -3 to -6 NS5B chimeric replicon cells. Similar to previous reports (27, 39), the major resistant substitutions selected in GT1 were at the P495 site: P495A, -S, -L, or -T for GT1a and P495A, -S, or -L for GT1b (Table 2). A V499A mutant was also selected in GT1b at a lower frequency (5%). Amino acid substitutions identified in GT3 to -6 NS5Bs are as follows: GT3a-C494S or -P495S; GT4a-V494A or -P495A or -L; GT5a-V494A, -P495S, -L, or -H; and GT6a-V494A (Table 2). In order to characterize the variants identified in BMS-791325-resistant cells, selected mutants were engineered into the wild-type replicons by site-directed mutagenesis and tested in transient replication assays. In order to assess the impact on BMS-791325 potency, replicon stable cell lines were established for some mutants that showed poor replication in a transient assay. Mutants with mutations at the P495 position in GT1 conferred 15- to 64-fold resistance to BMS-791325 with the following rank in order of resistance: P495S > P495T > P495L > P495A in GT1a and P495S/L > P495A in GT1b (Table 3). Mutants that conferred a higher level of resistance had poor replication fitness. The V499A substitution in GT1b gave only a negligible shift in potency (1.6-fold) but reduced replication efficiency approximately 50% compared to the WT replicon (Table 3). In GT3a, NS5B residue 494 is cysteine rather than valine, which is present in the other genotypes. C494S was the predominant substitution identified in GT3a selected cells (present at 76%), but replicons with this change did not replicate in transient assays; however, a stable cell line which conferred 34-fold resistance to BMS-791325 was selected (Table 3). A P495S substitution was also observed in GT3a selected cells, although at a lower frequency (24%). Replication of the P495S mutant could not be detected in transient replication assays, and several attempts to isolate stable cell lines of this mutant were unsuccessful. For GT4a, P495A and V494A mutants were analyzed in transient replication assays (Table 3). P495A conferred modest levels of resistance to BMS-791325 with an increase in EC50 25-fold above the wild-type control level while the V494A mutation had only a slight impact on BMS-791325 potency (3-fold). Reduced replication fitness was observed for both mutants, ∼30 to 46% relative to WT in transient assays. The GT4a-P495L mutant did not replicate in transient assays, and a stable cell line could not be selected for evaluation. Amino acid substitutions selected in GT5a replicon cells were mainly detected at residue 495, with P495S, -L, and -H variants observed (9/23/6 clones out of 39 cDNA clones, respectively). A V494A substitution was also identified in 1 out of 39 clones which was not linked with substitution at the P495 site. The GT5a selected variants were not active in transient replication assays, and we were not able to select stable cell lines for the P495S/L or V494A variants. However, the P495H mutant replicon cells survived and conferred a high level of resistance to BMS-791325, with a 167-fold increase in EC50 over that of the wild type. V494A was the only variant selected in GT6a replicon cells. Although the GT6a V494A mutant was not active in a transient assay, a stable cell line was successfully isolated, and it conferred 17-fold resistance to BMS-791325. Interestingly, this is the same substitution observed in the GT6a isolate HN001, which was ∼10-fold less susceptible to BMS-791325.
TABLE 3.
Resistance profile of BMS-791325 in genotypes 1a, 1b, 3a, 4a, 5a, and 6a
| NS5B genotype | Variant | Mean BMS-791325 EC50 (nM) ± SD | Fold resistancea | Replication efficiencyb |
|---|---|---|---|---|
| 1a-H77c | WT | 4 ± 1.1 | 1 | |
| P495A | 58 ± 5 | 15 | 0.6–0.8 | |
| P495S | 253 ± 36 | 64 | 0.16–0.18 | |
| P495L | 133 ± 24 | 32 | 0.25–0.3 | |
| P495T | 142 ± 34 | 36 | 0.07–0.14 | |
| 1b-Con1 | WT | 7 ± 1.4 | 1 | |
| P495A | 144 ± 33 | 20 | 0.38–0.45 | |
| P495S | 378 ± 58 | 54 | 0.06–0.07 | |
| P495L | 368 ± 111 | 53 | 0.08–0.14 | |
| V499A | 11.5 ± 0.7 | 1.6 | 0.42–0.55 | |
| 3a-Inx | WT | 5 ± 1 | ||
| C494S | 170 ± 14 | 34 | Cell linec | |
| P495S | NDd | No replication | ||
| 4a-h2 | WT | 4.5 ± 2.1 | 1 | |
| V494A | 15.5 ± 3.5 | 3.4 | 0.33–0.46 | |
| P495A | 111 ± 41 | 25 | 0.3–0.34 | |
| P495L | ND | No replication | ||
| 5a-h3 | WT | 4 ± 1 | ||
| V494A | ND | No replication | ||
| P495H | 670 ± 30 | 167 | Cell linec | |
| P495L | ND | No replication | ||
| P495S | ND | No replication | ||
| 6a-752 | WT | 10.6 ± 3.5 | ||
| V494A | 179 ± 9.5 | 17 | Cell linec |
Against WT.
Replication efficiency relative to wild type in a transient replication assay.
Stable cell lines selected.
ND, not determined.
DISCUSSION
BMS-791325 is a HCV polymerase inhibitor that showed substantial antiviral activity in a single ascending dose (SAD) study in GT1-infected patients (28) and is currently in phase 3 studies. To gain insight into GT coverage, we utilized NS5B chimeric replicons to assess the efficacy and resistance profile of BMS-791325 in non-GT1 subtypes. A chimeric replicon approach was successfully employed to analyze NS5B GT2 to -6 subtypes in the absence of full-length replicons with several tactics attempted to generate replication-competent GT2 to -6 NS5B chimeric replicon cell lines. The first approach used a 2a-JFH1 backbone to generate chimeras since efficient replication of this replicon did not require adaptive mutations. However, we were able only to select an active 2a-J6 NS5B chimeric replicon cell line with no replication observed for other genotypes. We then employed both GT1a and -1b backbones and were able to successfully select active stable cell lines for GT2b, -4a, -5a, and -6a NS5B chimeric replicons by using the 1b-Con1 backbone and for GT3a NS5B chimeric replicons using the 1a-H77c backbone.
We evaluated the efficacy of BMS-791325 in nongenotype1 replicons by determining the EC50s against GT2b, -3a, -4a, -5a, and -6a NS5B chimeras from multiple clinical isolates for each genotype. The fold shift in antiviral potency in GT2b versus GT1a-H77c ranged from 150- to greater than 350-fold increases in EC50. Lack of BMS-791325 susceptibility in GT2b could possibly be attributed to the I392 and A494 polymorphisms, residues that interact with the inhibitor. L392I and V494A variants in GT1b conferred low-level resistance to BMS-791325 with 7-fold-reduced potency for L392I (27) and ∼2- to 3-fold-reduced potency observed for the V494A replicon (data not shown). Structural analysis suggests that genotype variants result in a different shape of the inhibitor binding site that is responsible for the resistance of GT2b polymerase to site 1 inhibitors (40). Antiviral activity was achieved for BMS-791325 in GT3a, -4a, -5a, and -6a isolates with a valine at residue 494, with EC50s of less than 10 nM. However, reduced potency was observed for BMS-791325 on a GT6a isolate (HN001) with a naturally occurring A494 variant in NS5B, which represents about 21% of patients infected with GT6a HCV, according to the European HCV database (Table 4). Susceptibility to BMS-791325 was significantly improved (∼10-fold-lower EC50) with an A494V site-directed mutant in this 6a isolate. As expected, the 6a-752 V494A site-directed mutant, which changed valine to alanine, showed reduced susceptibility to BMS-791325 with an EC50 increase of 9-fold (Fig. 2). These data suggest that residue 494 plays an important role in determining BMS-791325 susceptibility in GT6a. Evaluation of BMS-791325 GT coverage by colony elimination assays showed good agreement with the in vitro replicon assay with efficient HCV clearance observed in the GT6a-752 variant compared to 6a isolate HN001 with the A494 polymorphism. Combination therapy with other DAAs will be needed to overcome the reduced clearance efficiency in the A494-containing GT6a isolates.
TABLE 4.
Prevalence of polymorphisms at NS5B positions important for BMS-791325 resistance
| Genotype (no.)a | Residue(s)/polymorphism(s) at NS5B position (% of sequences): |
||
|---|---|---|---|
| 494 | 495 | 499 | |
| 1a (384) | V (99.7), A (0.3) | P (100) | A (97.1), T (2.3), V (0.5) |
| 1b (763) | V (100) | P (100) | V (85.7), A (11), T (3), I (0.3) |
| 2a (31) | A (100) | P (100) | A (61.3), V (29), T (9.7) |
| 2b (27) | A (96.3), V (3.7) | P (100) | A (100) |
| 3a (44) | C (100) | P (100) | A (100) |
| 4a (25) | V (100) | P (100) | A (100) |
| 5a (4) | V (100) | P (100) | A (100) |
| 6a (22) | V (72.7), A (27.3) | P (95.5), L (4.5) | A (100) |
Numbers in parentheses are the numbers of GT1 to -6 NS5B sequences in the European HCV database with the indicated amino acid.
Due to the high mutation rate during HCV replication and error-prone nature of the NS5B RdRp, the rapid selection of drug-resistant variants upon treatment with antivirals is to be expected. The emergence of DAA drug resistance is one of the major challenges in the future of antiviral treatment regimens. P495 is the major resistance site identified from BMS-791325 selection in GT1 replicons. A good correlation was observed for BMS-791325-resistant mutants that were identified in vitro in a GT1a replicon and in clinical samples from the SAD study where P495S or -L was found in one patient at a single time point (T24) following a 3.5-log10 drop in viral RNA at a 900-mg dose of BMS-791325 (28). A V494A-P495S double mutant was also identified in 2 out of 150 clones during clonal analysis of this patient sample. Phenotypic analysis of the V494A-P495S-linked mutant was performed in a GT1a replicon, and although the replicon harboring the V494A-P495S double mutant is not viable in a transient replication assay, an active stable replicon cell line was selected with additional mutations F574S/A in NS5B identified from sequence analysis of the cell line. The F574S/A site-directed mutants did not confer resistance to BMS-791325 but significantly improved replication fitness compared to a WT GT1a replicon in a transient replication assay and appear to function as “compensatory” mutations that allow the replicon with the V494A-P495S double mutant to actively replicate in a transient replication assay (data not shown). The V494A-P495S-linked double mutant conferred a higher level of resistance (>100-fold) to BMS-791325 in the replicon than did P495S (64-fold) or V494A (∼2-fold) single mutants. Similar to GT1, resistance mutations selected with BMS-791325 were identified mainly at residue 494 or 495 for GT3 to -6 NS5B chimeric replicons. In GT3a selected cells, C494S and P495S mutants were identified in separate cDNA clones. V494A and P495A, -S, -L, or -H variants were selected in GT5a chimeric cell lines with BMS-791325, and again, the mutants were not linked based on clonal analysis. In GT4a, although the majority of V494A and P495A substitutions were present individually based on clonal analysis, there was a single linked double mutant found in one cDNA clone; however, we failed to select an active cell line with this clone. Interestingly, V494A was the only resistant mutant identified in the GT6a selection that resulted in substantial resistance to BMS-791325 in GT6a but very little resistance (2- to 3-fold) in GT1 or GT4a, possibly due to differences in the amino acid composition and the shape of the inhibitor binding pocket among NS5Bs across genotypes. The strong similarity in emerging resistant variants in GT1 between replicon in vitro studies and the recently reported SAD study from HCV-infected subjects validates the replicon system and helps to guide the clinical development of BMS-791325 in other genotypes.
Across HCV genotypes, the variation at NS5B residues that are important for BMS-791325 resistance development (residues 494, 495, and 499) was examined in sequences deposited in the European HCV database (Table 4). P495 is highly conserved (>95% conservation in sequence) across all genotypes, while residues 494 and 499 are less conserved. For the majority of sequences in the European HCV database, residue 494 is a valine for GT1 and GT4 to -6, a cysteine for GT3a, and an alanine for GT2. Although residue 494A in GT2 is likely to play a role in the resistance to BMS-791325, 494C found in GT3a did not change the inhibitor susceptibility. For residue 499, the majority of the sequences in the database are an alanine, except that it is a valine in GT1b. The V499A mutant in GT1b has only minimal impact on BMS-791325 potency (∼2-fold). Mutation of P495, a residue that locates on the thumb domain away from the active site, to A, L, or S confers resistance to BMS-791325, and the observation that replicons carrying P495 substitutions do not replicate efficiently suggests that this region of the molecule might play a key function during viral replication. The P495 site has been identified by X-ray crystallography as one of the key residues involved in the interaction with a noncatalytic GTP molecule on the NS5B surface (41). The binding sites for GTP and for the indole-based inhibitors are close in space but clearly distinct (42). Moreover, GTP binding is not associated with any protein conformational change (41), whereas the crystal structure NS5B-1b apoenzyme shows that allosteric inhibitors defined by P495 resistance binding require a displacement of the Δ1 finger loop from a thumb binding pocket. GTP stabilizes the thumb-finger interaction, while the inhibitor binding displaces the finger-tip domains and locks the enzyme in an inactive, “open” conformation. The polymerase Δ1 loop regulates the coordinated movement of the fingers and thumb during the polymerase reaction cycle (42). The movement of the Δ1 loop in the presence of P495-site inhibitors is also linked with trypsin hypersensitivity, and this is evidenced by the detection of trypsin cleavage products with changes in NS5B polymerase structure (43).
Given that HCV replicates at extremely high rates in humans (44) and given the error-prone nature of the RdRp, there is an increased potential for the selection of drug-induced resistance variants. Combination therapy that inhibits multiple targets will be required to achieve a sustained viral response. Replicon cell lines with NS5B variants exhibiting a high level of resistance to BMS-791325 remained fully sensitive to HCV NS5A and protease inhibitors, suggesting that the combination of the three could efficiently suppress the emergence of resistant replicons. This is in agreement with results from clinical studies where the all-oral, interferon-free, and ribavirin-free regimen of DCV, ASV, and BMS-791325 achieved high rates of SVR12 in patients with HCV GT1 infection (45). Data from this study suggest that BMS-791325 could be a component of a combination therapy approach in HCV GT3- to -6-infected patients, and results from the resistance analysis reported here provide guidance for resistance monitoring in future clinical trials.
ACKNOWLEDGMENTS
We thank Bernadette Kienzle and Xin Huang for assistance with NS5B sequence analysis. We thank Mark Cockett and Nicholas Meanwell for their support and leadership.
All authors are employees of Bristol-Myers Squibb and may hold stock and/or options in Bristol-Myers Squibb.
Footnotes
Published ahead of print 29 September 2014
REFERENCES
- 1.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 2.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5:453–463. 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 3.Bartenschlager R, Cosset F-L, Lohmann V. 2010. Hepatitis C virus replication cycle. J. Hepatol. 53:583–585. 10.1016/j.jhep.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 4.Simmonds P. 2004. Genetic diversity and evolution of hepatitis C virus—15 years on. J. Gen. Virol. 85:3173–3188. 10.1099/vir.0.80401-0. [DOI] [PubMed] [Google Scholar]
- 5.Zein NN. 2000. Clinical significance of hepatitis C virus genotypes. Clin. Microbiol. Rev. 13:223–235. 10.1128/CMR.13.2.223-235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takada N, Takase S, Takada A, Date T. 1993. Differences in the hepatitis C virus genotypes in different countries. J. Hepatol. 17:277–283. 10.1016/S0168-8278(05)80205-3. [DOI] [PubMed] [Google Scholar]
- 7.Pawlotsky JM, Taskiris L, Roudot-Thoraval F, Pellet C, Stuyver L, Duval J, Dhumeaux D. 1995. Relationship between hepatitis C virus genotypes and sources of infection in patients with chronic hepatitis C. J. Infect. Dis. 171:1607–1610. 10.1093/infdis/171.6.1607. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen MH, Keeffe EB. 2005. Prevalence and treatment of hepatitis C virus genotypes 4, 5, and 6. Clin. Gastroenterol. Hepatol. 3(10 Suppl 2):S97–S101. 10.1016/S1542-3565(05)00711-1. [DOI] [PubMed] [Google Scholar]
- 9.Thomas DL, Seeff LB. 2005. Nature history of hepatitis C. Clin. Liver Dis. 9:383–398. 10.1016/j.cld.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 10.Alter MJ. 2007. Epidemiology of hepatitis C virus infection. World J. Gastroenterol. 13:2436–2441. 10.3748/wjg.v13.i17.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Gonçales FL, Jr, Häussinger D, Diago M, Carosi G, Dhumeaux D, Craxi A, Lin A, Hoffman J, Yu J. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347:975–982. 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 12.Hadziyannis SJ, Sette H, Jr, Morgan TR, Balan V, Diago M, Marcellin P, Ramadori G, Bodenheimer H, Jr, Bernstein D, Rizzetto M, Zeuzem S, Pockros PJ, Lin A, Ackrill AM. 2004. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann. Intern. Med. 140:346–355. 10.7326/0003-4819-140-5-200403020-00010. [DOI] [PubMed] [Google Scholar]
- 13.Munir S, Saleem S, Idrees M, Tariq A, Butt S, Rauff B, Hussain A, Badar S, Naudhani M, Fatima Z, Ali M, Ali L, Akram M, Aftab M, Khubaib B, Awan Z. 2010. Hepatitis C treatments: current and future perspectives. Virol. J. 7:296. 10.1186/1743-422X-7-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hofmann WP, Sarrazin C, Zeuzem S. 2012. Current standards in the treatment of chronic hepatitis C. Dtsch. Arztebl. Int. 109:352–358. 10.3238/arztebl.2012.0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dieterich D, Rockstroh JK, Orkin C. 2014. Simeprevir (TMC435) plus PegIFN/ribavirin in HCV genotype-1/HIV-1 coinfection (study C212), abstr 24 Conf. Retroviruses Opportunistic Infect., Boston, MA. [Google Scholar]
- 16.Janssen Therapeutics. 2013. Simeprevir side effect. Janssen Therapeutics, Division of Janssen Products, LP, Titusville, NJ: http://www.olysio.com/shared/product/olysio/prescribing-information.pdf. [Google Scholar]
- 17.Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. 1999. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 6:937–943. 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- 18.Domingo E, Martínez-Salas E, Sobrino F, de la Torre JC, Portela A, Ortín J, López-Galindez C, Pérez-Breña P, Villanueva N, Nájera R. 1985. The quasispecies (extremely heterogeneous) nature of viral RNA genome populations: biological relevance—a review. Gene 40:1–8. 10.1016/0378-1119(85)90017-4. [DOI] [PubMed] [Google Scholar]
- 19.Herring BL, Tsui R, Peddada L, Busch M, Delwart EL. 2005. Wide range of quasispecies diversity during primary hepatitis C virus infection. J. Virol. 79:4340–4346. 10.1128/JVI.79.7.4340-4346.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lohmann V, Körner F, Koch J-O, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 21.Kato T, Date T, Miyamoto M, Zhao Z, Mizokami M, Wakita T. 2005. Nonhepatic cell lines HeLa and 293 support efficient replication of the hepatitis C virus genotype 2a subgenomic replicon. J. Virol. 79:592–596. 10.1128/JVI.79.1.592-596.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. 2003. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J. Virol. 77:3181–3190. 10.1128/JVI.77.5.3181-3190.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. 10.1053/j.gastro.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 24.Saeed M, Gondeau C, Hmwe S, Yokokawa H, Date T, Suzuki T, Kato T, Wakita T. 2013. Replication of hepatitis C virus genotype 3a in cultured cells. Gastroenterology 144:56–58. 10.1053/j.gastro.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 25.Saeed M, Scheel TKH, Gottwein JM, Marukian S, Dustin LB, Bukh J, Rice CM. 2012. Efficient replication of genotype 3a and 4a hepatitis C virus replicons in human hepatoma cells Antimicrob. Agents Chemother. 56:5365–5373. 10.1128/AAC.01256-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herlihy KJ, Graham JP, Kumpf R, Patick AK, Duggal R, Shi ST. 2008. Development of intergenotypic chimeric replicons to determine the broad-spectrum antiviral activities of hepatitis C virus polymerase inhibitors. Antimicrob. Agents Chemother. 52:3523–3531. 10.1128/AAC.00533-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemm JA, Liu M, Gentles RG, Ding M, Voss S, Pelosi LA, Wang YK, Rigat KL, Mosure KW, Bender JA, Knipe JO, Colonno RJ, Meanwell NA, Kadow JF, Santone KS, Roberts SB, Gao M. 2014. Preclinical characterization of BMS-791325, an allosteric inhibitor of hepatitis C virus NS5B polymerase. Antimicrob. Agents Chemother. 58:3485–3495. 10.1128/AAC.02495-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sims K, Lemm JA, Eley T, Liu M, Berglind A, Sherman D, Lawitz E, Vutikullird AB, Tebas P, Gao M, Pasquinelli C, Grasela DM. 2014. A randomized, placebo-controlled, single ascending dose study of BMS-791325, a hepatitis C virus NS5B polymerase inhibitor, in genotype 1 infection. Antimicrob. Agents Chemother. 58:3496–3503. 10.1128/AAC.02579-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Boyle DR, Nower PT, Lemm JA, Valera L, Sun JH, Rigat K, Colonno R, Gao M. 2005. Development of a cell-based high-throughput specificity screen using a hepatitis C virus-bovine viral diarrhea virus dual replicon assay. Antimicrob. Agents Chemother. 49:1346–1353. 10.1128/AAC.49.4.1346-1353.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR, II, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100. 10.1038/nature08960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang C, Sun JH, O'Boyle DR, II, Nower P, Valera L, Roberts S, Fridell RA, Gao M. 2013. Persistence of resistant variants in hepatitis C virus-infected patients treated with the NS5A replication complex inhibitor daclatasvir. Antimicrob. Agents Chemother. 57:2054–2065. 10.1128/AAC.02494-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang C, Valera L, Jia L, Kirk MJ, Gao M, Fridell RA. 2013. In vitro activity of daclatasvir on hepatitis C virus genotype 3 NS5A. Antimicrob. Agents Chemother. 57:611–613. 10.1128/AAC.01874-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun JH, O'Boyle DR, II, Zhang Y, Wang C, Nower P, Valera L, Roberts S, Nettles RE, Fridell RA, Gao M. 2012. Impact of a baseline polymorphism on the emergence of resistance to the hepatitis C virus nonstructural protein 5A replication complex inhibitor, BMS-790052. Hepatology 55:1692–1699. 10.1002/hep.25581. [DOI] [PubMed] [Google Scholar]
- 34.Wang C, Jia L, Huang H, Qiu D, Valera L, Huang X, Sun JH, Nower PT, O'Boyle DR, II, Gao M, Fridell RA. 2012. In vitro activity of BMS-790052 on hepatitis C virus genotype 4 NS5A. Antimicrob. Agents Chemother. 56:1588–1590. 10.1128/AAC.06169-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fridell RA, Wang C, Sun JH, O'Boyle DR, II, Nower P, Valera L, Qiu D, Roberts S, Huang X, Kienzle B, Bifano M, Nettles RE, Gao M. 2011. Genotypic and phenotypic analysis of variants resistant to hepatitis C virus nonstructural protein 5A replication complex inhibitor BMS-790052 in humans: in vitro and in vivo correlations. Hepatology 54:1924–1935. 10.1002/hep.24594. [DOI] [PubMed] [Google Scholar]
- 36.McPhee F, Sheaffer AK, Friborg J, Hernandez D, Falk P, Zhai G, Levine S, Chaniewski S, Yu F, Barry D, Chen C, Lee MS, Mosure K, Sun LQ, Sinz M, Meanwell NA, Colonno RJ, Knipe J, Scola P. 2012. Preclinical profile and characterization of the hepatitis C virus NS3 protease inhibitor asunaprevir (BMS-650032). Antimicrob. Agents Chemother. 56:5387–5396. 10.1128/AAC.01186-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gardiner D, Lalezari J, Lawitz E, DiMicco M, Ghalib R, Reddy KR, Chang KM, Sulkowski M, Marro SO, Anderson J, He B, Kansra V, McPhee F, Wind-Rotolo M, Grasela D, Selby M, Korman AJ, Lowy I. 2013. Randomized study of asunaprevir plus peginterferon alfa and ribavirin for previously untreated genotype 1 chronic hepatitis C. PLoS One 8:e63818. 10.1371/journal.pone.0063818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lemm JA, Liu M, Rose RE, Fridell R, O'Boyle DR, Colonno R, Gao M. 2005. Replication-competent chimeric hepatitis C virus subgenomic replicons. Intervirology 48:183–191. 10.1159/000081747. [DOI] [PubMed] [Google Scholar]
- 39.Pelosi LA, Voss S, Liu M, Gao M, Lemm JA. 2012. Effect on hepatitis C virus replication of combinations of direct-acting antivirals, including NS5A inhibitor daclatasvir. Antimicrob. Agents Chemother. 56:5230–5239. 10.1128/AAC.01209-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rydberg EH, Cellucci A, Bartholomew L, Mattu M, Barbato G, Ludmerer SW, Graham DJ, Altamura S, Paonessa G, De Francesco R, Migliaccio G, Carfi A. 2009. Structural basis for resistance of the genotype 2b hepatitis C virus NS5B polymerase to site A non-nucleoside inhibitors. J. Mol. Biol. 390:1048–1059. 10.1016/j.jmb.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 41.Tomei L, Altamura S, Bartholomew L, Biroccio A, Ceccacci A, Pacini L, Narjes F, Gennari N, Bisbocci M, Incitti I, Orsatti L, Harper S, Stansfield I, Rowley M, De Francesco R, Migliaccio G. 2003. Mechanism of action and antiviral activity of benzimidazole-based allosteric inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 77:13225–13231. 10.1128/JVI.77.24.13225-13231.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Marco S, Volpari C, Tomei L, Altamura S, Harper S, Narjes F, Koch U, Rowley M, De Francesco R, Migliaccio G, Carfí A. 2005. Interdomain communication in hepatitis C virus polymerase abolished by small molecule inhibitors bound to a novel allosteric site. J. Biol. Chem. 280:29765–29770. 10.1074/jbc.M505423200. [DOI] [PubMed] [Google Scholar]
- 43.Rigat K, Wang Y, Hudyma TW, Ding M, Zheng X, Gentles RG, Beno BR, Gao M, Roberts SB. 2010. Ligand-induced changes in hepatitis C virus NS5B polymerase structure. Antiviral Res. 88:197–206. 10.1016/j.antiviral.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 44.Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, Layden TJ, Perelson AS. 1998. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 282:103–107. 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 45.Everson GT, Sims KD, Rodriguez-Torres M, Hézode C, Lawitz E, Bourlière M, Loustaud-Ratti V, Rustgi V, Schwartz H, Tatum H, Marcellin P, Pol S, Thuluvath PJ, Eley T, Wang X, Huang SP, McPhee F, Wind-Rotolo M, Chung E, Pasquinelli C, Grasela DM, Gardiner DF. 2014. Efficacy of an interferon- and ribavirin-free regimen of daclatasvir, asunaprevir, and BMS-791325 in treatment-naive patients with HCV genotype 1 infection. Gastroenterology 146:420–429. 10.1053/j.gastro.2013.10.057. [DOI] [PubMed] [Google Scholar]