

Abstract
The susceptibilities of gammaherpesviruses, including Epstein-Barr virus (EBV), Kaposi's sarcoma-associated herpesvirus (KSHV), and animal rhadinoviruses, to various nucleoside analogs was investigated in this work. Besides examining the antiviral activities and modes of action of antivirals currently marketed for the treatment of alpha- and/or betaherpesvirus infections (including acyclovir, ganciclovir, penciclovir, foscarnet, and brivudin), we also investigated the structure-activity relationship of various 5-substituted uridine and cytidine molecules. The antiviral efficacy of nucleoside derivatives bearing substitutions at the 5 position was decreased if the bromovinyl was replaced by chlorovinyl. 1-β-d-Arabinofuranosyl-(E)-5-(2-bromovinyl)uracil (BVaraU), a nucleoside with an arabinose configuration of the sugar ring, exhibited no inhibitory effect against rhadinoviruses but was active against EBV. On the other hand, the fluoroarabinose cytidine analog 2′-fluoro-5-iodo-aracytosine (FIAC) showed high selectivity indices against gammaherpesviruses that were comparable to those of brivudin. Additionally, we selected brivudin- and acyclovir-resistant rhadinoviruses in vitro and characterized them by phenotypic and genotypic (i.e., sequencing of the viral thymidine kinase, protein kinase, and DNA polymerase) analysis. Here, we reveal key amino acids in these enzymes that play an important role in substrate recognition. Our data on drug susceptibility profiles of the different animal gammaherpesvirus mutants highlighted cross-resistance patterns and indicated that pyrimidine nucleoside derivatives are phosphorylated by the viral thymidine kinase and purine nucleosides are preferentially activated by the gammaherpesvirus protein kinase.
INTRODUCTION
The gammaherpesvirus subfamily includes two major genera, the lymphocryptovirus and rhadinovirus, of which the human tumor viruses Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV), respectively, are the best-characterized members (1). A hallmark of these human herpesviruses is that they do not easily replicate in primary infection in cells in culture (2). In contrast, other members of the rhadinovirus genus, murine gammaherpesvirus 68 (MHV-68), herpesvirus saimiri (HVS), and rhesus rhadinovirus (RRV), are able to replicate to high titers in cell culture, and thus, they may serve as model systems for human gammaherpesviruses in experimental settings (3).
Overall, antiherpesvirus therapies are aimed at selectively inhibiting the lytic replication of the virus. At present, the antiviral agents used in EBV and KSHV viral infections are those that are approved for the treatment of other herpesvirus infections (4), in particular ganciclovir (GCV) for both EBV and KSHV and also acyclovir (ACV) in the case of EBV. Other structurally related antiherpetics that are currently marketed, such as penciclovir (PCV) and brivudin (BVDU), have also been evaluated in vitro against EBV and KSHV replication (5–8) but have not been used in the clinic. Differences in antiviral activities of GCV, ACV, PCV, and BVDU against EBV and KSHV in vitro have been well described. These nucleosides selectively inhibit EBV replication in vitro, whereas the antiviral activity of ACV and PCV against KSHV proved to be very weak (5–13). Acyclic or carbocyclic guanosine analogs have also been studied in the context of inhibiting EBV replication, and they include in particular H2G (omaciclovir), A-5021 [(1S′,2R′)-9-[[1′,2′-bis(hydroxymethyl)cycloprop-1-yl]methyl]guanine] and S2242 [2-amino-7-[1,3-dihydroxy-2-propoxymethyl)purine] (14–16).
To become biologically active, nucleoside analogs must first be phosphorylated to their active triphosphate forms to inhibit the viral DNA polymerase and/or to be incorporated into the viral DNA. The initial phosphorylation of nucleosides to their monophosphate form is carried out by viral kinases. ACV, GCV, PCV, and BVDU are converted to their monophosphate form by the viral thymidine kinase (TK) in alphaherpesvirus-infected cells. In contrast to ACV (which is not recognized by cellular nucleoside kinases), GCV is converted to its monophosphate form at low levels by cellular kinases. This may contribute to the difference in toxicity between the two drugs. Although the EBV and KSHV TKs are able to phosphorylate nucleoside analogs of thymidine, such as BVDU and azidodeoxythymidine, these enzymes appear to be inefficient at catalyzing the phosphorylation of ACV and GCV (17–19). This feature highlights the more restricted substrate specificity of the EBV and KSHV TKs compared to the TKs of herpes simplex virus (HSV) and varicella-zoster virus (VZV) (17–19). In regard to the phosphorylation of ACV and GCV by the gammaherpesvirus TK, different groups have reported conflicting findings. Some researchers have suggested that gammaherpesvirus TK is not able to phosphorylate ACV and GCV, while others have demonstrated that this enzyme does possess some activity toward these nucleosides (17, 18, 20–22). Nevertheless, there is a general agreement that ACV and GCV are phosphorylated to their monophosphate form by the viral protein kinase (PK) in gammaherpesvirus-infected cells (20, 21). The gammaherpesvirus protein kinase is the homolog of the human cytomegalovirus (HCMV) (a betaherpesvirus) protein kinase pUL97, which phosphorylates GCV and to a lesser extent ACV to their monophosphate forms in HCMV-infected cells (23).
The benzimidazole derivative maribavir (MBV) is a potent inhibitor of HCMV and EBV replication (24), but unlike the case with nucleoside analogs, its activity is not dependent on phosphorylation and this compound does not target the viral DNA polymerase. Instead, MBV directly inhibits pUL97 of HCMV, affecting viral assembly and nuclear egress (25). MBV also inhibits EBV replication, but the mechanism of action of MBV against the EBV lytic phase is more complex than that for HCMV. This may be linked to the indirect effects of MBV on the transcription of EBV genes through the interaction of EBV PK with multiple viral proteins (24, 26).
Drug-resistant HSV, VZV, and HCMV are found relatively frequently in the clinic, almost exclusively among severely immunocompromised patients receiving prolonged antiviral therapy, mainly ACV or GCV (27). Viral mutations conferring resistance to nucleoside analogs have been found in either the drug-activating genes, i.e., HSV TK, VZV TK, or HCMV UL97 gene, and/or in conserved regions of the viral DNA polymerase (27). In the majority of cases, the mechanism underlying resistance to ACV in HSV is TK deficiency (TK−) as a result of a deletion(s), insertion(s), or point mutation(s) in the encoding gene. Mutations in HSV TK occur preferentially at a limited number of hot spots within long homopolymer nucleotide stretches, rather than being randomly distributed throughout the gene (28).
Since GCV is the drug of choice for prophylactic and preemptive HCMV therapy, resistance mutations are detected most frequently (>90%) in the UL97 gene, consistently in a limited number of codons (27, 29). Additionally, there is a larger number of different mutations in the DNA polymerase, which tend to be selected only after extended periods of HCMV treatment with GCV (29). Interestingly, mutations in the UL97 gene associated with resistance to GCV or MBV do not overlap, since MBV-associated mutations occur upstream of known GCV resistance mutations (30).
In contrast, no drug-resistant EBV and KSHV strains have ever been isolated from patients, though monitoring the development of resistance might be useful, especially when immunocompromised hosts with EBV or KSHV diseases are treated with antiviral agents (31). In addition, no studies have reported the in vitro selection and characterization of drug-resistant EBV and KSHV strains, while this has been extensively investigated for other herpesviruses, such as HSV, VZV, and HCMV. Yet structure-function studies concerning the EBV and KSHV TKs have been described. This has been achieved by the engineering of different viral TK mutants by site-directed mutagenesis in order to characterize essential residues in the conserved ATP and substrate binding site of the EBV TK (32, 33) and investigate the contributions of the N- and C-terminal regions of KSHV TK (34).
In this report, we evaluate the inhibitory activities of various nucleoside analogs against EBV, KSHV, MHV-68, HVS, and RRV. Since the currently established in vitro assays for EBV and KSHV do not allow efficient selection of drug-resistant viruses, cell culture systems with HVS and MHV-68 were used to select and characterize gammaherpesvirus mutants resistant to BVDU and ACV. Hence, we identified amino acids that are important for drug interactions within the gammaherpesvirus TK, PK, and DNA polymerase and defined the patterns of cross-resistance of the different in vitro-selected mutant viruses.
MATERIALS AND METHODS
Cell culture and viruses.
KSHV-infected BCBL-1 cells (NIH AIDS Research and Reference Reagent Program) and EBV-infected P3HR-1 cells (ATCC HTB-62) were cultured in RPMI 1640 medium (Life Technologies Europe BV, Ghent, Belgium). Murine fibroblasts (NIH 3T3 cells; ATCC CRL-1685), owl monkey kidney cells (OMK) (ATCC CRL-1556), and rhesus monkey fibroblasts (RF) (kindly provided by S. Wong, Oregon Health and Science University, Beaverton, OR, USA) were grown in Dulbecco's modified eagle's medium (DMEM). All media were supplemented with 10% heat-inactivated fetal calf serum (FCS), 2 mM l-glutamine, 1% nonessential amino acids, 1% sodium pyruvate, and 1% HEPES. Cultures were incubated at 37°C and 5% CO2. The following viral strains were used: MHV-68 (clone G2.4; provided by A. A. Nash, Edinburgh, United Kingdom), HVS (strain C-488; ATCC VR-1414), and RRV (strain 17577; kindly provided by S. Wong, Oregon Health and Science University, Beaverton, OR, USA) and were grown, respectively, in NIH 3T3, OMK, and RF cells.
Compounds.
The sources of the compounds that were used in this study are presented in Table 1.
TABLE 1.
Description and source of the compounds used in this study
| Group | Compound(s) (abbreviation) | Description | Source |
|---|---|---|---|
| Nucleoside analogs | Acyclovir (ACV) | 9-(2-Hydroxyethoxymethyl)guanine | GlaxoSmithKline, Stevenage, UK |
| Ganciclovir (GCV) | 9-(1,3-Dihydroxy-2-propoxymethyl)guanine | Roche, Basel, Switzerland | |
| Penciclovir (PCV) | 9-(4-Hydroxy-3-hydroxymethylbut-1-yl)guanine | Aventis, Frankfurt, Germany | |
| Brivudin (BVDU) | E-5-(2-bromovinyl)-2′-deoxyuridine | Searle, UK | |
| BVaraU | (E)-5-(2-bromovinyl)-1-β-d-arabino-pentafuranosyluracil | Soy sauce; Yamasa, Koshi, Japan | |
| BTDU | 5-(5-Bromo-2-thienyl)-2′-deoxyuridine | Rega Institute for Medical Research, Leuven, Belgium | |
| IDU | 5-Iodo-2′-deoxyuridine | Sigma Chemicals, St. Louis, MO | |
| EDU | 5-Ethyl-2′-deoxyuridine | Sigma Chemicals, St. Louis, MO | |
| (E)-CVDC | (E)-5-(2-chlorovinyl)-2′-deoxycytidine | A. Kumar, Department of Chemistry, Uttar Pradesh, India | |
| CVDU | 5-(2-Chlorovinyl)-2′-deoxyuridine | University of Birmingham, Birmingham, UK | |
| FIAC | 2′-Fluoro-5-iodo-aracytosine | Bristol-Myers Company, Research Parkway, Wallingford, CT | |
| Omaciclovir (H2G) | (R)-9-[4-hydroxy-2-(hydroxymethyl)butyl]guanine | Wellcome Research Laboratories, Kent, UK | |
| Vidarabine (AraA) | 9-β-d-Arabinofuranosyladenine | Sigma Chemicals, St. Louis, MO | |
| Pyrophosphate analog | Foscarnet (PFA) | Phosphonoformate sodium salt | Sigma Chemicals, St. Louis, MO |
| Acyclic nucleoside phosphonates | Cidofovir (HPMPC) | (S)-1-[3-hydroxy-2-(phosphonomethoxypropyl) cytosine] | Gilead Sciences, Foster City, CA |
| Adefovir (PMEA) | 9-[2- (Phosphonomethoxyethyl)adenine] | Gilead Sciences, Foster City, CA | |
| HPMPA | (S)-9-[3-hydroxy-2- (phosphonomethoxy)-propyl]adenine | Marcela Krecmerova, Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Prague, Czech Republic | |
| HPMPO-DAPy | (R)-[2,4-diamino-3-hydroxy-6-[2-(phosphonomethoxy)propoxy]]pyrimidine | M. Krecmerova | |
| HPMP-5-azaC | 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]-5-azacytosine | M. Krecmerova | |
| PMEADAP | 9-[2-(phosphonomethoxy)ethyl]-2,6-diaminopurine | M. Krecmerova | |
| PMEAO-DAPy | 2,4-Diamino-6-[2-(phosphonomethoxy)ethoxy]pyrimidine | M. Krecmerova |
Antiviral assays.
The drug susceptibility profiles of MHV-68, HVS, and RRV were evaluated by cytopathic effect (CPE)-reduction assays in their corresponding cell lines in 96-well plates as described previously (35). Briefly, after virus absorption for 2 h at 37°C, compounds diluted in fresh medium were added to the virus-infected cells. Viral CPE was recorded microscopically based on detectable alterations of the cell morphology as soon as it reached completion in the untreated, virus-infected cells. The scale used for scoring the CPE was as follows: (0, 0% CPE, 1, ∼20% CPE; 2, 20 to 40% CPE; 3, ∼40 to 60% CPE; 4, 60 to 80%; 5, 80 to 100% CPE. The antiviral activity was expressed as the EC50, the concentration required to reduce virus-induced CPE by 50% compared to the untreated controls, and was estimated from graphic plots. The antiviral assays for KSHV and EBV were performed in 48-well plates. Viral replication was induced in BCBL-1 and P3HR-1 cells by adding 20 ng/ml 12-O-tetradecanoylphorbol 13-acetate (TPA) (Sigma-Aldrich, Bornem, Belgium) to the growing cells in the presence or absence of the test compounds. After 5 days, the intracellular DNA was quantified by real-time quantitative PCR (qPCR). Forward and reverse primers, as well as TaqMan probe sequences, allowing the detection of the target ORF73 gene of KSHV and BNRF1 gene of EBV have been described elsewhere (35).
The cytostatic effects of the compounds were determined based on the inhibition of cell growth for NIH 3T3, OMK, RF, uninduced BCBL-1, and P3HR-1 cells, as previously described (35). The number of cells was determined using a Coulter counter, and the cytostatic concentration was calculated as the CC50, or the concentration of the compound required to reduce cell growth by 50% relative to the number of cells in the untreated controls.
Selection of drug-resistant viruses.
Drug-resistant HVS and MHV-68 were obtained by serial passages of the virus in the presence of increasing concentrations of BVDU or ACV, starting at a concentration equivalent to their EC50. OMK and NIH 3T3 cells were seeded in 25-cm2 flasks and infected with HVS (C488) or MHV-68 (G2.4) in the presence of the drug. When full CPE was reached, samples were frozen and virus was harvested and used to infect cells for the next passage. This process was repeated several times in increasing concentrations of the compound. After 12 (ACV) and 13 (BVDU) passages, several HVS resistant clones were isolated by limiting dilution. Drug-resistant MHV-68 clones were selected after 13 (ACV) and 27 (BVDU) passages. All viral clones were tested for their drug sensitivity by measuring the EC50s in CPE reduction assays. The TK, PK, and DNA polymerase of each drug-resistant clone was sequenced. The level of drug resistance (fold-resistance) was expressed as the ratio of the EC50 of the resistant virus to the EC50 of wild-type (WT) virus.
Sequence analysis.
DNA of drug-resistant HVS or MHV-68 clones was extracted (Qiagen), and the viral TK, PK, and DNA polymerase were amplified by PCR using FastStart high-fidelity DNA polymerase (Roche Applied Science, Mannheim, Germany). PCR was performed, following the manufacturer's instructions. The PCR products were purified using the QIAquick purification kit (Qiagen), and gene amplicons were directly sequenced using the cycle sequencing kit BigDye Terminator kit, version 3.1, on an ABI 3730 sequencing system (Applied Biosystems) and a set of primers spanning the entire coding region of the genes. The sequencing results were computer assembled and compared with the sequence from the WT gene using the software program SeqScape version 2.7 (Applied Biosystems).
RESULTS
Activities and selectivities of nucleoside analogs.
The antiviral efficacies and selectivities of structurally related compounds against different gammaherpesviruses were determined, and the results are summarized in Table 2; their chemical structures are shown in Fig. 1. The guanosine analogs ACV, PCV, GCV, and H2G showed marked inhibitory activities against EBV replication (EC50 of approximately 4.5 μM for ACV, PCV, and GCV and 7.5 μM for H2G). In contrast, rhadinoviruses (i.e., KSHV, MHV-68, HVS, and RRV) were moderately sensitive to GCV (EC50s ranging from 11 μM to 82 μM) and weakly sensitive to ACV and PCV (EC50s > 100 μM), with the exception of the inhibitory activity of ACV against MHV-68 (EC50 of 9 μM). H2G did not inhibit KSHV, HVS, and RRV replication at the highest concentration evaluated, while a weak activity was measured against MHV-68 (EC50 of 59 μM).
TABLE 2.
Antiviral activities and selectivities of nucleoside analogs against human and animal gammaherpesviruses
| Compound | EC50 (μM)a |
CC50 (μM)b |
SIc |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EBV | KSHV | MHV-68 | HVS | RRV | P3HR-1 | BCBL-1 | NIH 3T3 | OMK | RF | EBV | KSHV | MHV-68 | HVS | RRV | |
| ACV | 4.0 ± 0.4 | 137 ± 44 | 9 ± 7 | 182 ± 80 | 809 ± 116 | 911 ± 89 | 533 ± 182 | 98 ± 62 | >900 | 560 ± 280 | 228 | 4 | 11 | >5 | 1 |
| PCV | 4.7 ± 1.6 | 201 ± 43 | 115 ± 36 | 367 ± 47 | 395 ± 0 | >700 | 138 ± 24 | 91 ± 43 | 308 ± 67 | 170 ± 103 | >149 | 1 | 1 | 1 | <1 |
| GCV | 4.3 ± 3.5 | 11 ± 9 | 18 ± 4 | 82 ± 35 | 31 ± 5.1 | >700 | 239 ± 86 | 137 ± 39 | >900 | >900 | >163 | 22 | 8 | >11 | >29 |
| H2G | 7.5 ± 3.2 | >50 | 59 ± 13 | >500 | >500 | >200 | >200 | >200 | >200 | >200 | >27 | 4 | >4 | <1 | <1 |
| BVDU | 2.4 ± 1.4 | 0.6 ± 0.3 | 0.09 ± 0.06 | 5.7 ± 4.5 | 0.3 ± 0.2 | 495 ± 60 | 138 ± 72 | 18 ± 6 | >900 | 411 ± 195 | 206 | 230 | 200 | >158 | 1370 |
| IDU | 10 ± 5 | >130 | 0.8 ± 0.6 | 73 ± 62 | >500 | ≤14 | 4 ± 2 | 7 ± 4 | 133 ± 4 | ≥367 | ≤1 | <1 | 9 | 2 | <1 |
| EDU | 74 ± 2 | >150 | 43 ± 31 | 484 ± 129 | ≥390 | 578 ± 65 | 215 ± 222 | 17 ± 9 | >700 | 602 ± 35 | 8 | <1 | <1 | >2 | <2 |
| BTDU | >130 | >130 | >500 | >500 | >500 | 262 ± 41 | >500 | 151 ± 8 | 257 ± 13 | 288 ± 59 | <2 | <4 | <1 | <1 | <1 |
| CVDU | 16 ± 1.4 | 5.5 0.7 | 1.4 ± 0.7 | 5.5 ± 2.1 | 14 ± 6 | >70 | >70 | >70 | >70 | >70 | >4 | >13 | >50 | >13 | >5 |
| (E)-CVDC | 12 ± 7 | 2.4 ± 1.4 | 1.0 ± 0.7 | 122 ± 56 | 8 ± 2 | 490 ± 83 | 184 ± 0 | 216 ± 7 | 278 ± 38 | 469 ± 108 | 41 | 77 | 216 | 2 | 59 |
| BVaraU | 11 ± 5 | >500 | 573 ± 0 | >500 | >500 | >500 | 340 ± 178 | 530 ± 140 | >250 | 670 ± 447 | >45 | <1 | 1 | <1 | <1 |
| AraA | 18 ± 17 | 99 ± 43 | 2.0 ± 1.2 | 29 ± 15 | 118 ± 16 | 38 ± 1 | 233 ± 4 | 21 ± 21 | 115 ± 24 | ≥233 | 2 | 2 | 1 | 3 | ≥1 |
| FIAC | 0.4 ± 0.3 | 1.1 ± 0.8 | 1.1 ± 0.5 | 2.4 ± 1.1 | <1.3 | 22 ± 8 | 49 ± 13 | 30 ± 30 | ≤49 | ≥383 | 55 | 45 | 27 | ≤20 | >294 |
EC50, 50% effective concentration: the drug concentration required to reduce 50% of CPE or viral DNA copies.
CC50, 50% cytostatic concentration: the drug concentration required to reduce 50% of cell growth.
SI, selectivity index: the ratio of CC50 to EC50. SI values that are ≥8 are indicated in bold.
FIG 1.

Chemical structures of BVDU and analogs, AraA, FIAC, and H2G.
All gammaherpesviruses tested were sensitive to the inhibitory effects of BVDU, with EC50s of 0.09 μM (MHV-68), 0.3 μM (RRV), 0.6 μM (KSHV), 2.4 μM (EBV). and 5.7 μM (HVS). Also, 5-(2-chlorovinyl)-2′-deoxyuridine (CVDU) and (E)-5-(2-chlorovinyl)-2′-deoxycytidine [(E)-CVDC] had substantial inhibitory effects against gammaherpesviruses, with the exception of (E)-CVDC against HVS (EC50 of 122 μM). BTDU, particularly active against VZV (36) and HSV (37), did not show any activity against gammaherpesviruses. 5-Iodo-2′-deoxyuridine (IDU) showed high inhibitory effects against MHV-68 replication, with an EC50 of 0.8 μM, and also possessed some activity against EBV (EC50 of 10 μM). In contrast, 5-ethyl-2′-deoxyuridine (EDU) showed weak antiviral activity against both MHV-68 and EBV (EC50 of 43 μM and 74 μM, respectively).
Unlike BVDU, its arabinofuranosyl counterpart {i.e., BVaraU [(E)-5-(2-bromovinyl)-1-β-d-arabino-pentafuranosyluracil]} showed only inhibitory effects against EBV replication (EC50 of 11 μM) but not against rhadinoviruses. The arabinofuranosyl adenosine analog AraA possessed low antiviral activity against EBV, KSHV, HVS and RRV, whereas MHV-68 replication was inhibited at an EC50 of 2 μM. The fluoroarabinose cytidine analog 2′-fluoro-5-iodo-aracytosine (FIAC) showed pronounced antiviral activity against all gammaherpesvirus tested, with EC50 of ≤2.4 μM.
Guanosine analogs had no cytostatic activity, but IDU and to a lesser extent FIAC and AraA showed the highest cytostatic effects on human lymphoblastoid cells (P3HR-1 and BCBL-1), murine fibroblasts (NIH 3T3), and simian epithelial cells (OMK). The molecules EDU and BVDU showed moderate cytostatic effects on NIH 3T3 cells, with CC50s of 17 μM and 18 μM, respectively. Weak or no toxicity was seen for the other drugs, since the CC50 values were >100 μM. All tested nucleoside analogs showed no noticeable cytostatic effects on RF cells.
The selectivity indices (SIs) were determined for each of the compounds. GCV, BVDU, CVDU, (E)-CVDC, and FIAC showed high selectivity against the majority of the gammaherpesviruses. Furthermore, high SI values were calculated against EBV for the following compounds in decreasing order of SIs: ACV (SI of 228), PCV (SI of >149), BVaraU (SI of >45), and H2G (SI of >27). Moderate drug selectivity was also observed for EDU against EBV (SI of 8) and for ACV and IDU against MHV-68, with SIs of 11 and 9, respectively.
Isolation of HVS and MHV-68 strains resistant to BVDU and ACV.
HVS and MHV-68 were cultured for several passages in the presence of the antiviral agent BVDU or ACV. Of note, PCV was not included in these studies, since this compound only weakly inhibited HVS and MHV-68 replication (EC50 of 367 μM and 115 μM, respectively) (Table 2). Viral stocks of HVS and MHV-68 were prepared when the viruses were able to grow at a concentration of 200 to 400 μM of the compounds. When these viral stocks displayed resistance (i.e., increases in EC50s) to the drug of interest, viral clones were subsequently isolated by limiting dilution. These clones were used in all genotyping and phenotyping assays.
The viral TK, PK, and DNA polymerase genes were sequenced to map eventual drug-associated mutations, since the encoded proteins are known to be involved in the activation of these molecules or as the target of the activated drugs (Table 3).
TABLE 3.
Identification of mutations in drug-resistant HVS and MHV-68
| Virus | Compound (na) | No. of clones/total | Viral geneb | Nucleotide change(s)c | Amino acid change(s) |
|---|---|---|---|---|---|
| HVS | BVDU (13) | 2/6 | TK | C133T | R45stop |
| 2/6 | TK | C133T + G1090T | R45stop + E364stop | ||
| 1/6 | TK | C1193A | A398D | ||
| 1/6 | TK | C133T + C1193A | R45stop + A398D | ||
| ACV (12) | 1/3 | DNA polymerase | C1586A | S529Y | |
| PK | T insertion at nt 1016–1017 | Frameshift | |||
| 2/3 | DNA polymerase | C1891A | L631I | ||
| MHV-68 | BVDU (27) | 4/4 | TK | T1116G | C372W |
| ACV (13) | 3/3 | PK | GT deletion of nt 1175–1176 | Frameshift |
n, number of passages of the virus in cell culture in the presence of increasing concentrations of the compound.
The viral TK, PK, and DNA polymerase genes were sequenced for all clones. TK, thymidine kinase; PK, protein kinase.
nt, nucleotides.
After 13 passages of HVS under pressure of BVDU, four clonal populations bearing mutations in the TK gene were found (Table 3). In contrast, the sequences of the PK and DNA polymerase genes were identical to those for the WT viruses. Two HVS BVDU-resistant (BVDUr) clones harbored a mutation that resulted in the introduction of a stop codon at the N-terminal region of the viral TK (R45stop). In two other clones, this mutation was associated with another mutation, localized at the C-terminal region of the viral TK, that also led to the appearance of the stop codon E364stop. One HVS BVDUr clone had an amino acid substitution changing the alanine residue to an aspartic acid at codon 398 of the TK (A398D). The A398D change appeared also in combination with the R45stop change (Table 3).
Genotyping of HVS clones grown under pressure of ACV revealed the presence of mutations in the viral DNA polymerase and PK genes and not in TK gene. Two populations were identified. The first population harbored a substitution in the viral DNA polymerase, S529Y, together with a frameshift insertion in the PK, which was due to an insertion of one thymidine at nucleotides 1016 and 1017. The second population had a single amino acid substitution L631I in the viral DNA polymerase.
After 27 passages of MHV-68 in the presence of BVDU, the four isolated clones harbored one single amino acid substitution, C372W, in the viral TK. Both the PK and DNA polymerase genes had sequences identical to that of the WT.
Selection of ACVr MHV-68 mutants resulted in the identification of a frameshift deletion in the PK and not in the TK and DNA polymerase. It consisted of a deletion of two nucleotides (GT) at positions 1175 and 1176 in the C-terminal region of the viral PK gene.
Drug susceptibility profiles of BVDUr and ACVr gammaherpesvirus clones.
Phenotypic studies were performed for all drug-resistant HVS and MHV-68 clones listed in Table 3. Their sensitivities to the nucleoside analogs BVDU, ACV, and GCV, to the acyclic nucleoside phosphonates (HPMPC, HPMP-5-azaC, HPMPA, HPMPO-DAPy, PMEA, and PMEDAP), and to the pyrophosphate analog phosphonoformate sodium (PFA) were determined (Fig. 2 and 3). For each compound, the EC50 of the mutant virus was compared with the EC50 of WT virus, and the result is referred to as the fold -resistance at the tops of graphs.
FIG 2.

The susceptibilities of BVDUr and ACVr HVS clones to nucleoside analogs, acyclic nucleoside phosphonates, and PFA. The data are presented as dot plots of the EC50 (μM) for the drug-resistant clones versus the EC50 for the wild-type (WT) virus. The fold resistance (ratio of EC50 for mutant virus to EC50 for WT virus) was calculated and is shown at the top of each graph. Note that when a value is not given, the ratio was equal to 1. EC50s were determined by CPE reduction assays, and horizontal bars indicate the mean values for one representative clone tested in three independent experiments.
FIG 3.

Susceptibilities of BVDUr and ACVr MHV-68 clones to nucleoside analogs, acyclic nucleoside phosphonates, and PFA. The data are presented as dot plots of the EC50 (μM) for the drug-resistant clones versus the EC50 for the wild-type (WT) clones. If the fold resistance (ratio of EC50 for mutant virus to EC50 for WT virus) was >1, the value of the ratio of EC50 for the mutant to EC50 for the WT is indicated at the top of the graph. EC50s were determined by CPE reduction assays, and horizontal bars indicate the mean values for a representative clone tested in three independent experiments.
All HVS clones selected under pressure of BVDU harboring mutations in the viral TK gene were highly resistant to BVDU (fold resistance of ≥171) (Fig. 2). Their sensitivities to the other nucleoside analogs (i.e., ACV and GCV), to acyclic nucleoside phosphonates, and to PFA was not altered.
The HVS DNA polymerase L631I substitution (obtained under ACV pressure) conferred resistance not only to ACV (≥5-fold) but also to GCV and to HPMPC, HPMP-5-azaC, PMEA, PMEDAP, and PFA. The highest levels of cross-resistance were seen for HPMPC and HPMP-5-azaC (7- and 8-fold, respectively). The ACVr HVS strain that harbored mutations in the viral DNA polymerase and in the PK showed a similar drug resistance profile, a ≥5-fold resistance to ACV and GCV, and with resistance levels to HPMPC, HPMP-5-azaC, PMEA, PMEDAP, and PFA varying from 5- to 6-fold. Both HVS strains were still sensitive to the inhibitory effects of BVDU, HPMPA, and HPMPO-DAPy.
As shown in Fig. 3, the BVDUr MHV-68 clones bearing the C372W substitution had a mean EC50 for BVDU increased by 19-fold compared to that of the WT strain, while their sensitivities to the other evaluated drugs did not change. The MHV-68 ACVr clones that harbored a frameshift deletion in the viral PK were resistant to ACV (13-fold increase in EC50) and GCV (5-fold increase in EC50) and remained sensitive to BVDU. Similarly to the BVDUr clones, these mutants were inhibited by different acyclic nucleoside phosphonates and PFA.
Cross-resistance of drug-resistant TK and PK mutants to a selection of nucleoside derivatives.
We further evaluated the sensitivities of three drug-resistant viral strains, i.e., an HVS TK mutant (R45stop, BVDUr), an MHV-68 TK mutant (C372W, BVDUr), and an MHV-68 PK mutant (GT deletion at nucleotides 1175 to 1176, ACVr), to a selection of nucleoside analogs, which are presented in Table 1 [i.e., IDU, EDU, (E)-CVDC, FIAC, H2G, and AraA]. Additionally, we included in this study an MHV-68 TK mutant, previously selected in our laboratory, which harbored an insertion of one cytosine at nucleotides 661 to 665 in the N-terminal region of the TK, resulting in a frameshift (MHV-68 TK C insertion at nucleotides 661 to 665). The latter was selected under pressure of the thiothymidine derivative KAH-39-149 (38). While an MHV-68 PK mutant was included in this study, we were unable to include an HVS PK mutant since none has been selected yet.
Figure 4 depicts the fold resistance profiles of these four HVS and MHV-68 mutants toward six nucleoside derivatives. The three HVS and MHV-68 TK mutants showed resistance to the pyrimidine analogs IDU, EDU, and (E)-CVDC, with levels of resistance ranging from >4 to ≥52. A 2- to 4-fold resistance to FIAC was seen for mutants encoding a truncated TK (R45stop and C insertion at nucleotides 661 to 665), while the presence of the C372W substitution in MHV-68 TK did not confer cross-resistance to FIAC. Also, a C insertion at nucleotides 661 to 665 in MHV-68 TK conferred higher levels of resistance against IDU and EDU than the C372W substitution.
FIG 4.
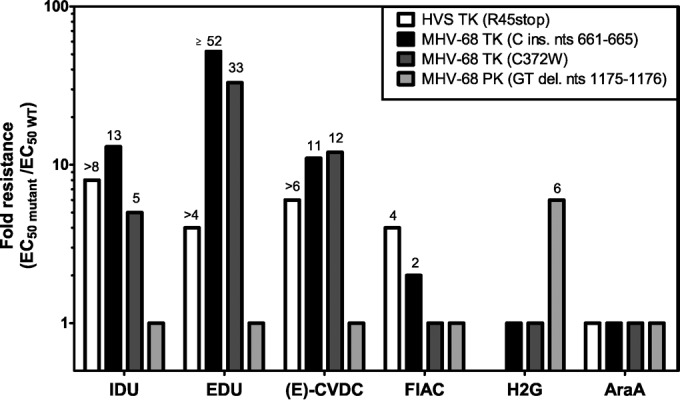
Levels of drug resistance of HVS and MHV-68 clones bearing a mutation in the viral thymidine kinase (TK) or protein kinase (PK). The data are presented as the fold changes in EC50s (EC50 for mutant virus to EC50 of wild-type virus). The fold resistance is given in log scale to facilitate comparison of drug resistance levels, which is given by a ratio ≥ 2.
While the purine derivative H2G remained active against TK MHV-68 mutants, it showed decreased inhibitory effects against the MHV-68 PK mutant, since a 6-fold increase in mean EC50over that of the WT virus was observed. H2G was not evaluated against the HVS TK mutant virus, since the drug does not possess antiviral activity against WT HVS. The four mutant gammaherpesviruses did not show altered sensitivity to AraA.
DISCUSSION
The selectivity of nucleoside analogs against herpesviruses is determined by the ability and efficiency of viral kinases (i.e., TK and/or PK) to catalyze the first phosphorylation step in drug activation compared to that of cellular kinases (4, 23, 39). Whereas HSV-1 and VZV TKs are able to convert ACV, GCV, PCV, and BVDU to their monophosphate forms, gammaherpesvirus TK substrate specificity is narrow, and it preferentially phosphorylates pyrimidine derivatives, such as BVDU. Similar to the case with HCMV, EBV and KSHV PKs are the viral enzymes that phosphorylate the guanosine analogs ACV and GCV (17, 18, 20, 21).
This study highlighted the difference in antiviral activities of various nucleoside analogs against EBV on the one hand and discrepancies in the spectra of activity among rhadinoviruses on the other hand. While ACV, PCV, GCV, and H2G were active against EBV, the sensitivities of rhadinoviruses (KSHV, MHV-68, HVS, and RRV) to these guanosine analogs drugs were markedly reduced, in particular for ACV (except for MHV-68), PCV, and H2G. These results suggested a low affinity of guanosine analogs for KSHV, HVS, and RRV PKs, in contrast to the case with EBV PK and to a lesser extent MHV-68 PK. Yet this hypothesis needs to be further investigated in enzymatic studies using purified gammaherpesvirus PKs. Notably, ACV showed high activity against MHV-68 (EC50 of 9 μM), indicating that MHV-68 PK could be functionally more related to EBV PK than to KSHV, HVS, or RRV PKs, although sequence identity of the PKs is similar (approximately 23%) between gammaherpesviruses.
The antiviral tests showed the high activity of H2G against EBV and to a lesser extent against MHV-68 replication, whereas no activity was observed against KSHV, HVS, and RRV. These data further support the assumption made above, that MHV-68 PK functionally more closely resembles EBV PK, and H2G may be a substrate of MHV-68 PK. Indeed, results obtained from the phenotypic assays with the MHV-68 PK mutant (GT deletion at nucleotides 1175 to 1176) demonstrated that this mutation conferred cross-resistance to H2G and to ACV and GCV, whereas such a drug-resistant phenotype was not observed with the MHV-68 TK-deficient (C insertion at nucleotides 661 to 665) or TK C372W mutant. This strongly supports that MHV-68 PK is partially responsible for the antiviral activity of H2G in vitro. However, Lowe and colleagues observed that the EBV TK was able to phosphorylate H2G to the monophosphate form in enzymatic studies, yet this enzyme was only partially purified (40). In fact, we may speculate that EBV TK is able to recognize and phosphorylate H2G but that the viral PK is the predominant kinase that activates the drug in vivo, which would be analogous to what is seen with GCV (20, 21).
H2G triphosphate has been shown to possess a longer intracellular half-life than its structural analog ACV (40), and similar to the case with ACV, H2G has an orally bioavailable prodrug, i.e., valomaciclovir (41). Valomaciclovir has been evaluated in clinical trials for the treatment of zoster (phase IIb) (41, 42) and for the treatment of acute infectious mononucleosis (phase I/II), which have been successfully completed (clinicaltrials.gov identifier NCT00575185). In the oral compartment of 11 individuals with infectious mononucleosis treated with valomaciclovir (4 g/day for 21 days), the mean EBV load was significantly reduced versus that with a placebo (43).
Unlike the other purine nucleoside derivatives tested in this report, AraA is known to be phosphorylated to its active triphosphate form by cellular kinases and is not dependent for its activation on viral kinases (44). Hence, no alterations in antiviral activity of AraA were seen for the MHV-68 PK mutant or for the HVS and MHV-68 TK mutant strains in this study.
The cross-resistance study performed using HVS and MHV-68 TK mutants revealed that uridine derivatives (BVDU, IDU, and EDU), as well as cytosine derivatives [(E)-CDVC and FIAC], depend on gammaherpesvirus TK for their activation. However, these compounds might have different affinities for the viral TKs, since the inhibitory activities differed among the various gammaherpesviruses tested (Table 2). These variations reflect the low-level amino acid sequence homology (approximately 20% to 30%) between the TKs of gammaherpesviruses. Diversity in antiviral activities of TK-dependent drugs against EBV and KSHV might also be a consequence of a poor TK activity of KSHV as opposed to that of EBV. The low KSHV TK activity was previously described based on the low incorporation of thymidine into DNA (18). It remains to be elucidated whether MHV-68, HVS, and RRV have similarly low TK activities.
Previously, the antiviral activities of deoxyuridine analogs against EBV and KSHV were shown to be dependent on the substitution on the 5′ position of the uracil, showing that the presence of a halogen group, such as chloro, bromo, or iodo, in 5-substituted deoxyuridines increased affinity for the viral TK compared to results with the methyl counterpart (18, 45). Here, we observed that the inhibitory activities of 5-substitued deoxyuridines against gammaherpesviruses are greatest when the halogen atom is substituted to bromovinyl, such as in BVDU. A substitution with iodo was inhibitory only to EBV and MHV-68. Moreover, replacement of the uracil by a cytosine [CVDU to (E)-CVDC] did not alter the antiviral potency against gammaherpesviruses, with the exception of HVS.
BVDU was the most active uridine analog against all tested gammaherpesviruses in vitro; however, this drug failed to exert its inhibitory effect in a mouse model of gammaherpesvirus infection (14, 35). The lack of efficacy of BVDU in vivo may result from the degradation of the drug by phosphorylases, converting it into BVU, which is inactive as an antiviral agent (46). On the contrary, (E)-CVDC and CVDU are resistant to degradation by these enzymes (36) and thus are expected to have a longer intracellular half-life, which could lead to potentially higher efficacy when evaluated in vivo against gammaherpesvirus infections.
Modification of the sugar ring, e.g., ribose nucleoside configuration of BVDU to an arabinose nucleoside conformation, as in BVaraU, is associated with a loss of inhibitory activity against rhadinoviruses but not against EBV. Introduction of fluorine on the sugar ring gives the arabinose nucleoside analog FIAC, with potency comparable to that seen for 5′-halogenated nucleoside analogs (e.g., BVDU) against all gammaherpesviruses. The selectivity of FIAC is probably partially dependent on virus TK, since low levels of resistance to FIAC were seen with TK− HVS and MHV-68 mutants. Yet the commercial development of this compound, once considered for the treatment of HCMV and hepatitis B virus infections, is no longer pursued due to toxicity (47).
Six conserved regions have been identified among herpesvirus TKs, including an ATP-binding site and a nucleoside-binding site. In HVS mutants selected in vitro under pressure of BVDU, we identified four different mutations in the viral TK, and they were all substitutions resulting in an amino acid change or the insertion of a stop codon, leading to the production of a truncated TK. A double amount of passages was needed to select the BVDUr MHV-68 TK mutant bearing the C372W substitution. However, the C372W change in MHV-68 TK conferred 10-fold less resistance to BVDU compared to the BVDUr HVS TK mutants. Yet both A398D and C372W are located in the active site of viral TK.
ACVr MHV-68 acquired mutations in viral PK, whereas ACVr HVS showed mutations predominantly in viral DNA polymerase (although one clone had an additional mutation in viral PK). As mentioned above, this might suggest that ACV is a better substrate for MHV-68 PK than for HVS PK, explaining the high antiviral activity of ACV against MHV-68. The data obtained with the MHV-68 PK mutant that showed resistance to ACV (approximately 13-fold) and GCV (5-fold) is in line with the results reported by Meng et al., who engineered EBV PK mutants containing stop codon insertions using a bacterial artificial chromosome and demonstrated that PK was required for the activation of ACV and GCV (21). It should be mentioned that no drug-associated mutations were identified in viral TK, PK, and/or DNA polymerase under pressure from GCV after 21 passages in cell culture for HVS and 45 passages for MHV-68.
The ACVr HVS double mutant acquired an insertion of thymidine in the substrate binding site of the viral PK, at a position homologous to those of mutations mapped in HCMV pUL97 conferring GCV resistance (27, 31). The HVS mutant was resistant to ACV and GCV; however, since it acquired another mutation in the viral DNA polymerase, we could not assess the impact of the PK mutation alone on the activation of ACV and GCV. Notably, whereas the acquisition of GCVr in the HCMV strain consisted of an in-frame deletion in pUL97 (31), the nucleotide insertion or deletion identified in the HVS or MHV-68 mutants led to a frameshift and the production of a truncated PK.
Understanding the impact of each mutation at the molecular level is hampered by the lack of a three-dimensional structure of gammaherpesvirus TK and of the PK and DNA polymerases. Nevertheless, the crystal structure of HSV-1 TK (PDB code 1KI8) and DNA polymerases (PDB code 2GV9) can be used as putative models, as depicted in Fig. 6. The substitutions here described (i.e., A398D and C372W) are located in the active site of the viral TK and are expected to impair the activity of the enzyme. Other amino acid changes (i.e., P326T, Q401R, E358D, and T364P) previously identified following selection with the novel pyrimidine nucleoside derivative HDVD (35) are also located in the active site of the viral TK. The E358D substitution is a minor change that accounted for resistance to HDVD but not to BVDU. The model of HSV-1 TK shows that the homologous glutamic acid at position 83 in HSV-1 is pointed toward the bromovinyl group of BVDU, and the E83K substitution in HSV-1 TK is known to confer drug resistance. This may explain why the substitution of glutamic acid into aspartic acid (which has a shorter side chain and a smaller volume [117 Å versus 140.1 Å] than glutamic acid) conferred resistance to HDVD and not to BVDU.
FIG 6.

Visualization of the mutations identified in HVS and MHV-68, at the homologous positions of HSV-1 TK and DNA polymerase three-dimensional (3D) structures. (A) BVDU (blue) is bound to the phosphate acceptor site of the HSV-1 TK (PDB code 1KI8). The p-loop (magenta) motif present in all the ATP-binding proteins is important for the interactions with the β and γ phosphates of the phosphate donor. The positions of the mutations identified in HVS are shown in cyan, and those identified in MHV-68 are in red. Amino acid changes at positions C372 (MHV-68) and A398 (HVS) described in this study were found under selection with BVDU. The other amino acid changes (shown in red at position P326 in HVS and positions Q401, E358, and T364 in MHV-68) were identified after selection with a novel pyrimidine nucleoside derivative (i.e., HDVD) possessing high levels of selectivity and potency against gammaherpesviruses. All the identified mutations are present in the first shell of residues surrounding the substrate (here BVDU) in the active site. (B) 3D structure of the HSV-1 DNA polymerase, showing the different domains: pre-NH2 domain (gray), NH2-terminal domain (red), 3′-5′-exonuclease domain (green), polymerase palm subdomain (blue), finger subdomain (black), and thumb subdomain (orange). Amino acid changes at the homologous position (indicated in blue) are present in the NH2-terminal domain and the finger subdomain. All the pictures were generated using PyMol graphic system (open source; v0.99rc9).
The amino acid substitutions identified in the two ACVr HVS clones, i.e., S529Y and L631I, are located in the conserved δ region C and region VI of the viral DNA polymerase, respectively (Fig. 5 and 6). The substitution L631I may induce a local structure change in the finger domain of conserved region VI, which may directly affect the interaction with incoming deoxynucleoside triphosphates (dNTPs) or alter the interaction with other residues involved in the formation of the polymerase active site (48, 49). Although of the change from leucine to isoleucine may be a minor change, it still affected the inhibitory activities of nucleoside analogs (ACV and GCV), acyclic nucleoside phosphonates (HPMPC, HPMP-5-azaC, PMEA, and PMEDAP), and PFA. In line with this, the L782I substitution, also located in the conserved region VI of the HSV-1 DNA polymerase, was previously characterized by producing a recombinant HSV-1 mutant and was shown to confer resistance to ACV (6-fold), HPMPC (2-fold), PMEA (4-fold), and PFA (2-fold) (50).
FIG 5.

Sequence alignment of sites in HVS and MHV-68 TK and DNA polymerase in which drug resistance mutations occurred. The TK and DNA polymerase sequences of the human herpesviruses (HSV-1, HSV-2, VZV, HCMV, EBV, and KSHV) belonging to three subfamilies (α, β, and γ) were aligned. In addition, three additional gammaherpesviruses, HVS, MHV-68, and RRV, were included. The two substitutions in the DNA polymerase gene occurred in the conserved regions δ region C and VI.
The S529Y substitution in HVS DNA polymerase is located in δ region C, and mutations in this region of DNA polymerases of herpesviruses are known to affect the DNA polymerase activity and/or the 3′-to-5′ exonuclease activity (50). Nevertheless, the exact impact of this mutation on the antiviral activity of ACV and GCV could not be evaluated because this mutant also harbored a mutation in the viral PK. Yet, since a substitution in the viral PK is not expected to affect the activity of acyclic nucleoside phosphonates and PFA, changes in susceptibility to these drugs may be attributed to the effect of the S529Y amino acid change in the viral DNA polymerase.
It is interesting to note that three point mutations found in the TK and DNA polymerase of HVS occurred at conserved amino acid positions in EBV and/or KSHV (Fig. 5). The alanine residue at position 398 in the TK of HVS is conserved at a homologous position in EBV. In the DNA polymerase, the serine at position 529 is conserved in both human gammaherpesviruses, while leucine 631 is also found in KSHV. Reintroduction of the A398D change in TK and/or of S529Y and L631I in the viral DNA polymerase in the genome of EBV and/or KSHV would be an interesting approach in order to examine their effects on drug susceptibilities toward EBV or KSHV.
Our findings highlighted the differences existing in substrate recognition (pyrimidine versus purine analogs) among the viral kinases (TK versus PK) of gammaherpesviruses. Therefore, combinations of pyrimidine- and purine-based nucleoside analogs should be investigated, since any synergistic antiviral effects against gammaherpesviruses could potentiate the impact of antiviral treatment. Additionally, this would decrease the likelihood of developing antiviral resistance in the clinic if drug-associated mutations were to occur in EBV or KSHV. At present, ACVr and/or GCVr EBV mutants have not yet been described; nevertheless, Walling et al. provided evidence for an ACVr EBV phenotype in a few immunocompromised patients with HIV-associated oral hairy leukoplakia (51). Therefore, EBV PK and DNA polymerase should be examined when ACVr and GCVr are suspected.
Overall, we investigated for the first time the role of conserved and nonconserved amino acids in TK, PK, and DNA polymerase of MHV-68 and HVS through the selection of drug-resistant viruses, regarding substrate recognition and virus drug sensitivity.
ACKNOWLEDGMENT
This work was supported by a grant from the Geconcertreerde Onderzoeksacties (no. 10/014).
Footnotes
Published ahead of print 29 September 2014
REFERENCES
- 1.Ambinder R, Cesarmen E. 2007. Clinical and pathological aspectes of EBV and KSHV infection, p 885–903 In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 2.Ackermann M. 2006. Pathogenesis of gammaherpesvirus infections. Vet. Microbiol. 113:211–222. 10.1016/j.vetmic.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 3.Blossom D. 2007. EBV and KSHV-related herpesviruses in non-human primates, p 1093–1114 In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 4.Andrei G, De Clercq E, Snoeck R. 2009. Viral DNA polymerase inhibitors. In Cameron CE, Matthias G, Kevin R. (ed), Viral genome replication. Springer, New York, NY. [Google Scholar]
- 5.Friedrichs C, Neyts J, Gaspar G, De Clercq E, Wutzler P. 2004. Evaluation of antiviral activity against human herpesvirus 8 (HHV-8) and Epstein-Barr virus (EBV) by a quantitative real-time PCR assay. Antiviral Res. 62:121–123. 10.1016/j.antiviral.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 6.Medveczky MM, Horvath E, Lund T, Medveczky PG. 1997. In vitro antiviral drug sensitivity of the Kaposi's sarcoma-associated herpesvirus. AIDS 11:1327–1332. 10.1097/00002030-199711000-00006. [DOI] [PubMed] [Google Scholar]
- 7.Meerbach A, Holy A, Wutzler P, De Clercq E, Neyts J. 1998. Inhibitory effects of novel nucleoside and nucleotide analogues on Epstein-Barr virus replication. Antivir. Chem. Chemother. 9:275–282. [DOI] [PubMed] [Google Scholar]
- 8.Neyts J, De Clercq E. 1997. Antiviral drug susceptibility of human herpesvirus 8. Antimicrob. Agents Chemother. 41:2754–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bacon TH, Boyd MR. 1995. Activity of penciclovir against Epstein-Barr virus. Antimicrob. Agents Chemother. 39:1599–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin JC, De Clercq E, Pagano JS. 1987. Novel acyclic adenosine analogs inhibit Epstein-Barr virus replication. Antimicrob. Agents Chemother. 31:1431–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin JC, Machida H. 1988. Comparison of two bromovinyl nucleoside analogs, 1-beta-D-arabinofuranosyl-E-5-(2-bromovinyl)uracil and E-5-(2-bromovinyl)-2′-deoxyuridine, with acyclovir in inhibition of Epstein-Barr virus replication. Antimicrob. Agents Chemother. 32:1068–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meerbach A, Klocking R, Meier C, Lomp A, Helbig B, Wutzler P. 2000. Inhibitory effect of cyclosaligenyl-nucleoside monophosphates (cycloSal-NMP) of acyclic nucleoside analogues on HSV-1 and EBV. Antiviral Res. 45:69–77. 10.1016/S0166-3542(99)00076-5. [DOI] [PubMed] [Google Scholar]
- 13.Sergerie Y, Boivin G. 2003. Evaluation of susceptibility of human herpesvirus 8 to antiviral drugs by quantitative real-time PCR. J. Clin. Microbiol. 41:3897–3900. 10.1128/JCM.41.8.3897-3900.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neyts J, De Clercq E. 1998. In vitro and in vivo inhibition of murine gamma herpesvirus 68 replication by selected antiviral agents. Antimicrob. Agents Chemother. 42:170–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Clercq E, Naesens L, De Bolle L, Schols D, Zhang Y, Neyts J. 2001. Antiviral agents active against human herpesviruses HHV-6, HHV-7 and HHV-8. Rev. Med. Virol. 11:381–395. 10.1002/rmv.336. [DOI] [PubMed] [Google Scholar]
- 16.Neyts J, Naesens L, Ying C, De Bolle L, De Clercq E. 2001. Anti-herpesvirus activity of (1′S,2′R)-9-[[1′,2′-bis(hydroxymethyl)-cycloprop-1′-yl]methyl] × guanine (A-5021) in vitro and in vivo. Antiviral Res. 49:115–120. 10.1016/S0166-3542(00)00144-3. [DOI] [PubMed] [Google Scholar]
- 17.Gustafson EA, Chillemi AC, Sage DR, Fingeroth JD. 1998. The Epstein-Barr virus thymidine kinase does not phosphorylate ganciclovir or acyclovir and demonstrates a narrow substrate specificity compared to the herpes simplex virus type 1 thymidine kinase. Antimicrob. Agents Chemother. 42:2923–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gustafson EA, Schinazi RF, Fingeroth JD. 2000. Human herpesvirus 8 open reading frame 21 is a thymidine and thymidylate kinase of narrow substrate specificity that efficiently phosphorylates zidovudine but not ganciclovir. J. Virol. 74:684–692. 10.1128/JVI.74.2.684-692.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lock MJ, Thorley N, Teo J, Emery VC. 2002. Azidodeoxythymidine and didehydrodeoxythymidine as inhibitors and substrates of the human herpesvirus 8 thymidine kinase. J. Antimicrob. Chemother. 49:359–366. 10.1093/jac/49.2.359. [DOI] [PubMed] [Google Scholar]
- 20.Cannon JS, Hamzeh F, Moore S, Nicholas J, Ambinder RF. 1999. Human herpesvirus 8-encoded thymidine kinase and phosphotransferase homologues confer sensitivity to ganciclovir. J. Virol. 73:4786–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng Q, Hagemeier SR, Fingeroth JD, Gershburg E, Pagano JS, Kenney SC. 2010. The Epstein-Barr virus (EBV)-encoded protein kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J. Virol. 84:4534–4542. 10.1128/JVI.02487-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Littler E, Arrand JR. 1988. Characterization of the Epstein-Barr virus-encoded thymidine kinase expressed in heterologous eucaryotic and procaryotic systems. J. Virol. 62:3892–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gershburg E, Pagano JS. 2008. Conserved herpesvirus protein kinases. Biochim. Biophys. Acta 1784:203–212. 10.1016/j.bbapap.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitehurst CB, Sanders MK, Law M, Wang FZ, Xiong J, Dittmer DP, Pagano JS. 2013. Maribavir inhibits Epstein-Barr virus transcription through the EBV protein kinase. J. Virol. 87:5311–5315. 10.1128/JVI.03505-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gershburg E, Hong K, Pagano JS. 2004. Effects of maribavir and selected indolocarbazoles on Epstein-Barr virus protein kinase BGLF4 and on viral lytic replication. Antimicrob. Agents Chemother. 48:1900–1903. 10.1128/AAC.48.5.1900-1903.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang FZ, Roy D, Gershburg E, Whitehurst CB, Dittmer DP, Pagano JS. 2009. Maribavir inhibits Epstein-Barr virus transcription in addition to viral DNA replication. J. Virol. 83:12108–12117. 10.1128/JVI.01575-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilbert C, Bestman-Smith J, Boivin G. 2002. Resistance of herpesviruses to antiviral drugs: clinical impacts and molecular mechanisms. Drug Resist. Updat. 5:88–114. 10.1016/S1368-7646(02)00021-3. [DOI] [PubMed] [Google Scholar]
- 28.Sasadeusz JJ, Tufaro F, Safrin S, Schubert K, Hubinette MM, Cheung PK, Sacks SL. 1997. Homopolymer mutational hot spots mediate herpes simplex virus resistance to acyclovir. J. Virol. 71:3872–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lurain NS, Chou S. 2010. Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 23:689–712. 10.1128/CMR.00009-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou S. 2008. Cytomegalovirus UL97 mutations in the era of ganciclovir and maribavir. Rev. Med. Virol. 18:233–246. 10.1002/rmv.574. [DOI] [PubMed] [Google Scholar]
- 31.Romain CA, BHJ, Vezina HE, Holman CJ. 2010. A method for evaluating antiviral drug susceptibility of Epstein-Barr virus. Virus Adapt. Treat. 2010:1–7. 10.2147/VAAT.S8575. [DOI] [Google Scholar]
- 32.Wu CC, Chen MC, Chang YR, Hsu TY, Chen JY. 2004. Identification and characterization of the conserved nucleoside-binding sites in the Epstein-Barr virus thymidine kinase. Biochem. J. 379:795–803. 10.1042/BJ20031832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu CC, Hsu TY, Chen JY. 2005. Characterization of three essential residues in the conserved ATP-binding region of Epstein-Barr virus thymidine kinase. Biochemistry 44:4785–4793. 10.1021/bi0484872. [DOI] [PubMed] [Google Scholar]
- 34.Gill MB, Murphy JE, Fingeroth JD. 2005. Functional divergence of Kaposi's sarcoma-associated herpesvirus and related gamma-2 herpesvirus thymidine kinases: novel cytoplasmic phosphoproteins that alter cellular morphology and disrupt adhesion. J. Virol. 79:14647–14659. 10.1128/JVI.79.23.14647-14659.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coen N, Singh U, Vuyyuru V, Van den Oord JJ, Balzarini J, Duraffour S, Snoeck R, Cheng YC, Chu CK, Andrei G. 2013. Activity and mechanism of action of HDVD, a novel pyrimidine nucleoside derivative with high levels of selectivity and potency against gammaherpesviruses. J. Virol. 87:3839–3851. 10.1128/JVI.03338-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andrei G, Snoeck R, Reymen D, Liesnard C, Goubau P, Desmyter J, De Clercq E. 1995. Comparative activity of selected antiviral compounds against clinical isolates of varicella-zoster virus. Eur. J. Clin. Microbiol. Infect. Dis. 14:318–329. [DOI] [PubMed] [Google Scholar]
- 37.Andrei G, Snoeck R, Goubau P, Desmyter J, De Clercq E. 1992. Comparative activity of various compounds against clinical strains of herpes simplex virus. Eur. J. Clin. Microbiol. Infect. Dis. 11:143–151. [DOI] [PubMed] [Google Scholar]
- 38.Coen N, Duraffour S, Haraguchi K, Balzarini J, van den Oord JJ, Snoeck R, Andrei G. 2014. Antiherpesvirus activities of two novel 4′-thiothymidine derivatives, KAY-2-41 and KAH-39-149, are dependent on viral and cellular thymidine kinases. Antimicrob. Agents Chemother. 58:4328–4340. 10.1128/AAC.02825-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pepper SD, Stewart JP, Arrand JR, Mackett M. 1996. Murine gammaherpesvirus-68 encodes homologues of thymidine kinase and glycoprotein H: sequence, expression, and characterization of pyrimidine kinase activity. Virology 219:475–479. [DOI] [PubMed] [Google Scholar]
- 40.Lowe DM, Alderton WK, Ellis MR, Parmar V, Miller WH, Roberts GB, Fyfe JA, Gaillard R, Ertl P, Snowden W. 1995. Mode of action of (R)-9-[4-hydroxy-2-(hydroxymethyl)butyl]guanine against herpesviruses. Antimicrob. Agents Chemother. 39:1802–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li F, Maag H, Alfredson T. 2008. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J. Pharm. Sci. 97:1109–1134. 10.1002/jps.21047. [DOI] [PubMed] [Google Scholar]
- 42.Andrei G, Snoeck R. 2013. Advances in the treatment of varicella-zoster virus infections. Adv. Pharmacol. 67:107–168. 10.1016/B978-0-12-405880-4.00004-4. [DOI] [PubMed] [Google Scholar]
- 43.Balfour HH, Vezina HE, Hogquist RC, Brundage RC, Anderson BJ, Odumade OA. 2009. Randomized, placebo-controlled, double blind trial of valomaciclovir (VALM) for infectious mononucleosis (IM), abstr. V1256a, p 221 Abstr. 49th Intersci. Conf. Antimicrob. Agents Chemother.-47th Annu. Meet. American Society for Microbiology and Infectious Diseases Society of America, San Francisco, CA. [Google Scholar]
- 44.Suzuki M, Okuda T, Shiraki K. 2006. Synergistic antiviral activity of acyclovir and vidarabine against herpes simplex virus types 1 and 2 and varicella-zoster virus. Antiviral Res. 72:157–161. 10.1016/j.antiviral.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 45.Tung PP, Summers WC. 1994. Substrate specificity of Epstein-Barr virus thymidine kinase. Antimicrob. Agents Chemother. 38:2175–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Clercq E. 2005. (E)-5-(2-bromovinyl)-2′-deoxyuridine (BVDU). Med. Res. Rev. 25:1–20. 10.1002/med.20011. [DOI] [PubMed] [Google Scholar]
- 47.Straus SE. 2012. Unanticipated risks in clinical development, p 77–95 In Ognibene FP, Gallin JI. (ed), Principles and practice of clinical research. Academic Press, Waltham, MA. [Google Scholar]
- 48.Hwang YT, Zuccola HJ, Lu Q, Hwang CB. 2004. A point mutation within conserved region VI of herpes simplex virus type 1 DNA polymerase confers altered drug sensitivity and enhances replication fidelity. J. Virol. 78:650–657. 10.1128/JVI.78.2.650-657.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tian W, Hwang YT, Hwang CB. 2008. The enhanced DNA replication fidelity of a mutant herpes simplex virus type 1 DNA polymerase is mediated by an improved nucleotide selectivity and reduced mismatch extension ability. J. Virol. 82:8937–8941. 10.1128/JVI.00911-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bestman-Smith J, Boivin G. 2003. Drug resistance patterns of recombinant herpes simplex virus DNA polymerase mutants generated with a set of overlapping cosmids and plasmids. J. Virol. 77:7820–7829. 10.1128/JVI.77.14.7820-7829.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walling DM, Flaitz CM, Nichols CM. 2003. Epstein-Barr virus replication in oral hairy leukoplakia: response, persistence, and resistance to treatment with valacyclovir. J. Infect. Dis. 188:883–890. 10.1086/378072. [DOI] [PubMed] [Google Scholar]