

Abstract
Primaquine (PQ) remains the sole available drug to prevent relapse of Plasmodium vivax malaria more than 60 years after licensure. While this drug was administered as a racemic mixture, prior studies suggested a pharmacodynamic advantage based on differential antirelapse activity and/or toxicities of its enantiomers. Oral primaquine enantiomers prepared using a novel, easily scalable method were given for 7 days to healthy rhesus macaques in a dose-rising fashion to evaluate their effects on the blood, liver, and kidneys. The enantiomers were then administered to Plasmodium cynomolgi-infected rhesus macaques at doses of 1.3 and 0.6 mg/kg of body weight/day in combination with chloroquine. The (−)-PQ enantiomer had higher clearance and apparent volume of distribution than did (+)-PQ and was more extensively converted to the carboxy metabolite. There is evidence for differential oxidative stress with a concentration-dependent rise in methemoglobin (MetHgb) with increasing doses of (+)-PQ greater than that seen for (−)-PQ. There was a marked, reversible hepatotoxicity in 2 of 3 animals dosed with (−)-PQ at 4.5 mg/kg. (−)-PQ in combination with chloroquine was successful in preventing P. cynomolgi disease relapse at doses of 0.6 and 1.3 mg/kg/day, while 1 of 2 animals receiving (+)-PQ at 0.6 mg/kg/day relapsed. While (−)-PQ was also associated with hepatotoxicity at higher doses as seen previously, this has not been identified as a clinical concern in humans during >60 years of use. Limited evidence for increased MetHgb generation with the (+) form in the rhesus macaque model suggests that it may be possible to improve the therapeutic window for hematologic toxicity in the clinic by separating primaquine into its enantiomers.
INTRODUCTION
The 8-aminoquinoline primaquine (PQ) remains the sole malaria antirelapse drug available after more than 60 years of clinical use. Red cell oxidative stress with manifestations ranging from methemoglobinemia (1) to frank life-threatening hemolysis (2) in patients with glucose-6-phosphate dehydrogenase (G6PD)-deficient red cells poses an important safety concern (3). The metabolic activation required for 8-aminoquinoline antimalarial activity is complex and has proven challenging to study due to the reactive, transient nature and low quantities of many of the potentially toxic and efficacious metabolites (4). Efforts to better understand the mechanisms of efficacy and toxicity have proven challenging due to differential metabolism, pharmacokinetics (PK), and pharmacodynamics (PD) and the resultant discordance between human and animal models. The chiral nature of the 8-aminoquinolines has led to successful attempts to optimize activity and reduce toxicity in animal models with enantiomer pairs from this drug class, as exemplified by analogs NPC1161A and NPC1161AB (5, 6). Primaquine is normally used as a racemic mixture of two opposite enantiomers [d (S/+) and l (R/−) forms] (Fig. 1) (7). The unmatched therapeutic utility of primaquine merits further exploration of the potential separation of antirelapse activity from toxicity using this approach. Given the lead time needed for developing new antimalarial agents, approaches to better utilization of PQ that can improve safety would be a signal advance (8).
FIG 1.

Mirror image structures of primaquine enantiomers. (A) (+)-Primaquine; (B) (−)-primaquine. Enantiomers were prepared at the National Center for Natural Products Research, University of Mississippi.
Primaquine enantiomers have previously been found to have differential antimalarial potency and toxicity and differential metabolism, though the precise mechanisms remain unclear. When racemic PQ was administered to laboratory rats, a majority of residual PQ excreted in the urine was the (+)-enantiomer (9). Recent studies in the mouse Plasmodium berghei sporozoite model of causal malaria prophylaxis revealed that the (+)-enantiomer had afforded complete protection at the standard oral dosing regimen of 25 mg/kg of body weight for 3 days, while for (−)-PQ, there was no effect at the same dose (10). Both antimalarial potency and systemic toxicity were greater for the d form [S-(+)] in a mouse malaria model (11). This is in contrast to what occurs in the nonhuman primate, as primaquine enantiomers in rhesus monkeys had equal antimalarial and antirelapse potency, but l-primaquine [R-(−)] was 3 to 5 times more hepatotoxic than d-primaquine [S-(+)] (11). Neither methemoglobin (MetHgb) formation nor drug levels were measured. Schmidt concluded that (+)-PQ might afford a better “therapeutic index” in humans than racemic PQ. However, given that the dose-limiting toxicity in humans is hemolysis in G6PD-deficient patients, it is more critical to understand the differential effects of the two enantiomers on oxidative red cell stress rather than hepatotoxicity, which has not been a clinical concern in humans (12).
Separating hemolytic toxicity from antimalarial activity remains an important clinically relevant objective for the 8-aminoquinoline class. Much of our current understanding is based on findings in rodent models of malaria and from in vitro metabolism studies that suggest that the stereochemistry of the PQ side chain is an important factor. To better understand the differential hepatic and erythrocytic toxicities of primaquine enantiomers in primates, we evaluated the concentration-effect relationships in healthy rhesus macaques on liver function tests, circulating red blood cell mass, reticulocyte count, and methemoglobin formation, escalating from the standard human-equivalent treatment dose (1.3 mg/kg/day) to a 3-fold-higher dose (4.5 mg/kg/day). Additionally, we compared the radical curative activity of the enantiomers in a rhesus macaque P. cynomolgi malaria challenge study at two dose levels (0.6 and 1.3 mg/kg/day), approximating low and normal human-equivalent clinical doses. This pharmacokinetic-pharmacodynamic characterization addresses the comparative “therapeutic index” of PQ enantiomers and contributes further to a pharmacological rationale for improving the safety of primaquine and potentially other structurally related 8-aminoquinoline antimalarials.
MATERIALS AND METHODS
Animals.
All 530 Indian-origin rhesus macaques (Macaca mulatta) greater than 6 months of age and successfully weaned in the Armed Forces Research Institute of Medical Science (AFRIMS; Bangkok, Thailand) primate colony were tested for G6PD deficiency during semiannual physical checks prior to the study, using a previously reported fluorescent-spot test method (13) with test kits from R&D Diagnostics, Papagos, Greece. All animals were normal for G6PD. A total of eight male non-malaria-naive rhesus monkeys, individually housed at the USAMC-AFRIMS Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International-accredited Veterinary Medicine facility, were used in the healthy-animal study, with median age and weight of 10.8 years and 9.3 kg, respectively. In the P. cynomolgi radical cure study, 10 malaria-naive Indian-origin rhesus, individually housed at the same facility, were used, with a median age of 4 years and weight of 5.4 kg. Animal weights were evenly distributed by treatment group. All care and use of animals were in compliance with the 2011 Institute of Laboratory Animal Resources, Guide for the Care and Use of Laboratory Animals. Animal use protocols were approved by the USAMC-AFRIMS Institutional Animal Care and Use Committee. Cages were cleaned daily and sanitized biweekly. The animals received commercially prepared monkey chow twice daily and supplemented mixed fresh vegetable or fruit treats four times per week with chlorinated water ad libitum via automatic watering valves. An enrichment program including toys and food enrichment items was routinely provided during the period of the study. The monkeys were clinically observed a minimum of 3 times daily by trained animal technicians and at least once daily by the veterinarian on duty. The monkeys were pole collar trained for 2 to 3 weeks prior to entering the study to become acclimated to handling and chair restraint, thus allowing frequent, safe venipuncture without anesthesia.
Drug and dosage.
The two enantiomers of primaquine [(+)-(S)-N4-(6-methoxyquinolin-8-yl)pentane-1,4-diamine diphosphate and (−)-(R)-N4-(6-methoxyquinolin-8-yl)pentane-1,4-diamine diphosphate] were prepared from racemic PQ phthalimide, which is an intermediate in the synthesis of PQ, readily prepared by treating commercially available PQ with phthalic anhydride. The enantiomers were resolved by fractional crystallization with (+)- and (−)-tartaric acid, as described in reference 10, with an optical purity of >99%. (+)-PQ and (−)-PQ (as the diphosphate salts) were completely dissolved in the dosing vehicle of 0.5% hydroxyethyl-cellulose and 0.1% Tween 80 (HECT). To assess toxicokinetics, the PQ enantiomers were administered to healthy rhesus monkeys in an escalating fashion at 1.3, 3.0, and 4.5 mg/kg/day (of the base) for 7 days with a 2-week wash-out period between doses and crossover of enantiomers between the 3.0- and 4.5-mg/kg/day dose groups. Group 1 consisted of a single animal given HECT vehicle control only (n = 1). Group 2 animals (n = 3) received 1.3 mg/kg/day (+)-PQ, followed by 3 mg/kg/day (+)-PQ and then 4.5 mg/kg (−)-PQ. Group 3 animals (n = 3) received 1.3 mg/kg (−)-PQ, 3 mg/kg (−)-PQ, and then 4.5 mg/kg (+)-PQ. Primaquine enantiomers were orally administered by nasogastric tube, followed by a 5- to 10-ml flush with distilled water. Intramuscular ketamine sedation at a dose of 10 mg/kg was used to facilitate all drug administration.
P. cynomolgi challenge to evaluate radical curative activity.
For the P. cynomolgi malaria radical cure study, 10 rhesus macaques were allocated to 5 groups of 2 animals each to receive 7 daily doses of (+)- or (−)-PQ base at 1.3 or 0.6 mg/kg/day in combination with the chloroquine base at 10 mg/kg/day, with two of the animals receiving chloroquine alone as negative controls. Animals were infected with P. cynomolgi and treated to prevent relapse using a previously described method (14, 15). Animals received 1 × 106 P. cynomolgi sporozoites intravenously on study day 0 and were treated when parasitemia reached ≥5,000 blood-stage parasites on peripheral blood film. Animals were monitored for relapse with clinical evaluation and periodic blood smears for at least 100 days after the last dose of treatment was administered.
Clinical observation and laboratory analysis.
The animals were observed a minimum of 3 times daily, and standard clinical assessments of health and well-being were made by a clinical veterinarian daily. Each day prior to treatment, 0.5 ml of blood in EDTA tubes was collected to monitor complete blood count (CBC). In addition, during the healthy-animal experiment, tests for reticulocyte count (Retic), hemoglobin (Hgb), methemoglobin (MetHgb), and serum creatinine as a measure of renal function and liver function tests to include alanine aminotransferase (ALT), aspartate aminotransferase (AST), and bilirubin levels were performed daily during the dosing period in lithium heparin tubes. Samples were stored at room temperature, quickly sent to the Department of Veterinary Medicine clinical lab after collection, and processed on arrival. Daily laboratory monitoring was continued past the dosing period only in the event of a significant deviation from baseline until resolution, defined as a return to within 10% of the baseline clinical laboratory value.
Pharmacokinetic-pharmacodynamic study.
In the healthy-animal study, pharmacokinetic blood sampling was done at 0, 0.5, 1, 2, 4, 8, 24, 72, 120, 168, and 216 h on the last day of the 3- and 4.5-mg/kg dose with 2 ml collected at each time point in heparinized sodium/lithium Vacutainer tubes. Samples were centrifuged at 2,500 rpm for 20 min, and then the supernatant (plasma) was transferred and kept at −80°C until analysis was performed. Plasma was separated into two tubes with 200 to 400 μl of plasma collected in each, and a third tube was used for erythrocytes.
LC-MS analysis.
The liquid chromatography-mass spectrometry (LC-MS) analysis methods of levels in plasma for each PQ enantiomer and their respective carboxyprimaquine (cPQ) metabolites in rhesus macaques were modified from our previous studies (16). Chromatography was performed on a Chiracel OD-H instrument (250 by 4.6-mm [inner diameter], 5-μm particle size, and precolumn of the same material, 910 by 4.0 mm [inner diameter]; Chiral Technologies Inc.). The mobile phase used for analysis was 20 mM ammonium formate, pH 3.56 (A), and 0.1% formic acid in ACN (B). The starting gradient had an initial composition of 81% A and 19% B for 10 min, increasing to 60% ACN at 2 min, and then was held for 16 min at a flow rate of 0.5 ml/min. Total analysis time was 30 min.
For mass spectrum analysis, the Waters ZQ LC-MS system was set in the positive electrospray ionization mode with single-ion recording. Instrument parameters included a cone voltage of 15 V, extractor voltage of 1 V, and a 2.8-kV capillary voltage. The source and desolvation temperatures were set at 120 and 350°C, respectively. The nitrogen generator was set at 100 lb/in2 to generate cone and desolvation gas flow of 30 and 500 liters/h, respectively. For the analyzer, both LM1 and HM1 resolution were set at 13 with ion energy set at 0.0, and the multiplier was set at 650 V. Enantiomers of PQ and cPQ yielded m/z ratios of 260 and 275, respectively. The enantiomers of (−)- and (+)-PQ eluted at 11.52 and 12.79 min, and those of (−)- and (+)-cPQ eluted at 24.91 and 25.58 min, respectively. MassLynx/Quanlynx chromatography software was used for quantification. The quantification ranges of (−/+)-PQ and (−/+)-cPQ were 10 to 1,000 and 25 to 2,500 ng/ml, respectively. Plasma extraction was done using a protein precipitation method with 100% recovery of 800 ng/ml of (−/+)-PQ and 200 ng/ml of (−/+)cPQ.
Pharmacokinetic-pharmacodynamic analysis.
Noncompartmental analysis was used to derive pharmacokinetic parameters of PQ and cPQ using PK-Solutions 2.0 (Summit Research Services Co., USA). The PK parameters determined were the elimination half-life (t1/2), maximum concentration in plasma by inspection (Cmax), time to reach Cmax after dosing (Tmax), area under the concentration-time curve from time zero to infinity (AUC0-∞), and mean residence time (MRT). Because the fraction of dose absorbed cannot be estimated for extravascular models, apparent volume of distribution (V/F) and apparent clearance (CL/F) were substituted for V and CL. Pharmacodynamic (PD) endpoints were also monitored for the two doses, including percentages of decrease in hemoglobin (%Hgb), increase in reticulocytes (%Retic), decrease in methemoglobin (%MetHgb), and increase in alanine aminotransferase (%ALT) compared to the baseline values for each animal, with a two-tailed Student's t test comparison for difference in means between the dosing groups. The PK parameters for both PQ and cPQ were compared with PD variables of interest, including mean %Hgb, %Retic, %MetHgb, and %ALT. Data analysis and graphical representation were completed using SPSS, v.12.0, and GraphPad Prism, v5.0, respectively.
RESULTS
Clinical radical curative (antirelapse) activity.
Clinical antirelapse activity as defined by the absence of blood-stage P. cynomolgi 100 days after dosing in the rhesus monkey P. cynomolgi challenge model revealed slightly greater apparent curative potency of (−)-PQ, with 2/2 animals cured at both the 1.3- and 0.6-mg/kg/day dose levels, compared to that of (+)-PQ, with only 1 of 2 animals cured at the 0.6-mg/kg/day dose (Table 1).
TABLE 1.
Radical curative activity of primaquine enantiomers in 10 rhesus monkeys infected with P. cynomolgi malaria
| Compound | Daily dosea (mg base/kg) | Parasitemia at treatment (1/2)b | No. of days to parasite clearance | No. of days to relapse | Activity (no. of animals undergoing radical cure/no. treated) |
|---|---|---|---|---|---|
| (+)-Primaquine | 1.3 | 180,190 | 3 | 2/2 | |
| 55,808 | 2 | ||||
| 0.6 | 36,515 | 2 | 1/2 | ||
| 8,291 | 2 | 36 | |||
| (−)-Primaquine | 1.3 | 312,040 | 3 | 2/2 | |
| 12,077 | 2 | ||||
| 0.6 | 16,662 | 2 | 2/2 | ||
| 4,403 | 2 | ||||
| Control | CQ only | 4,635 | 2 | 11 | 0/2 |
| 8,544 | 3 | 11 |
Administered for 7 days in combination with 10 mg chloroquine base/kg of body weight.
1/2, animal 1/animal 2.
Clinical and laboratory adverse events. (i) Clinical adverse events.
In the healthy-animal study, monkeys displayed normal behavior without evidence of adverse events at clinically relevant doses of the study drug (1.3 mg/kg). At higher doses (3.0 and 4.5 mg/kg), monkeys showed a range of clinical symptoms, including depression, lethargy, reduced appetite, and anorexia. In the antirelapse study, at clinically used doses, only transient mildly reduced appetite was observed. All animals treated with (−)-PQ had normal appetites at a daily dose of 1.3 and 3.0 mg/kg for 7 consecutive days, while 1 animal at the 4.5-mg/kg dose among monkeys administered (+)-PQ showed reduced appetite and soft stool after the 4th dose. Culture result in this animal was positive for Campylobacter infection. After repeated doses of (−)-PQ at a higher dose of 4.5 mg/kg/day for 7 days, 2 of 3 monkeys had slight to moderate depression, loss of appetite, emesis, and diarrhea after the 4th and 5th doses, respectively. Both animals had severe but reversible hepatotoxicity with rises in ALT, AST, and bilirubin to >10 times the upper limit of normal (ULN) (Fig. 2). Dosing was halted, and aminotransferases and bilirubin declined rapidly over a period of 7 days, with liver function recovering fully without sequelae. No renal toxicity for any animal was observed based on serum creatinine measurements.
FIG 2.
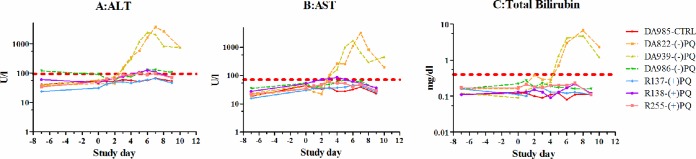
Hepatotoxicity in healthy animals following 7 days of dosing with (+)/(−)-PQ or vehicle control (CTRL) at 4.5 mg/kg/day. (A) Alanine aminotransferase (ALT); (B) aspartate aminotransferase (AST); (C) total bilirubin.
Hematologic adverse events.
One animal had eosinophilia with an erythematous maculopapular skin rash limited to the left axilla after receiving the first dose of 1.3 mg/kg (−)-PQ, which continued through the 7-day dosing period. Eosinophilia peaked at 24% of the differential leukocyte count on day 5 of dosing and returned to normal within 2 weeks. Eosinophilia recurred after the first dose of 3 mg/kg (−)-PQ, peaked at 20% on day 3, and persisted for more than 30 days. On the last day of observation (day 30 postdose), the eosinophil count rose dramatically to 31%, but no further observations were made after this time. The animal was replaced in the study by another animal and was not administered the next (highest) dose of 4.5 mg/kg (−)-PQ.
All animals had normal hematocrit and hemoglobin at baseline with little change over the course of PQ administration even at the highest dose, regardless of the enantiomer (data not shown). There was a modest rise in reticulocyte counts at the end of the 7-day dosing period at 4.5 mg/kg (Fig. 3A), but this appeared to be equivalent for the two enantiomers and increased the most following phlebotomy after the last dose. Animals receiving (+)-PQ had a greater and more consistent methemoglobin rise than those who received (−)-PQ, though one animal had a >5-fold MetHgb increase over baseline with both the (+)-enantiomer at 3.5 mg/kg and the (−)-enantiomer at 4.5 mg/kg (Fig. 3B).
FIG 3.

Individual hematologic profiles. (A) Reticulocytes as percentages of total red blood cell counts following 7 days of oral dosing at 4.5 mg/kg/day in 6 healthy animals dosed with (+)-PQ or (−)-PQ and 1 vehicle control (CTRL). (B) Percent methemoglobin levels at 1.3, 3.0, and 4.5 mg/kg/day. Note that enantiomers were switched to opposite groups between 3.0 and 4.5 mg/kg/day.
Pharmacokinetic-pharmacodynamic analysis.
The concentration-time profiles of PQ enantiomers and their carboxy metabolites on day 7 of dosing with 4.5 mg/kg/day are shown in Fig. 4. Concentrations in plasma reached the maximum level in 3 to 5 h and then rapidly and monoexponentially declined below the limit of detection within 72 h for both enantiomers of PQ and cPQ at all dose levels. The carboxy metabolite quickly appeared in plasma at levels 2-fold higher than the parent (+)-enantiomer, but for the (−)-PQ, the carboxy metabolite was 10-fold higher than the parent. cPQ metabolite formation increased substantially between doses of 3.0 and 4.5 mg/kg for the (−)-enantiomer, while levels of cPQ changed little for the (+)-enantiomer between 3.0 and 4.5 mg/kg.
FIG 4.
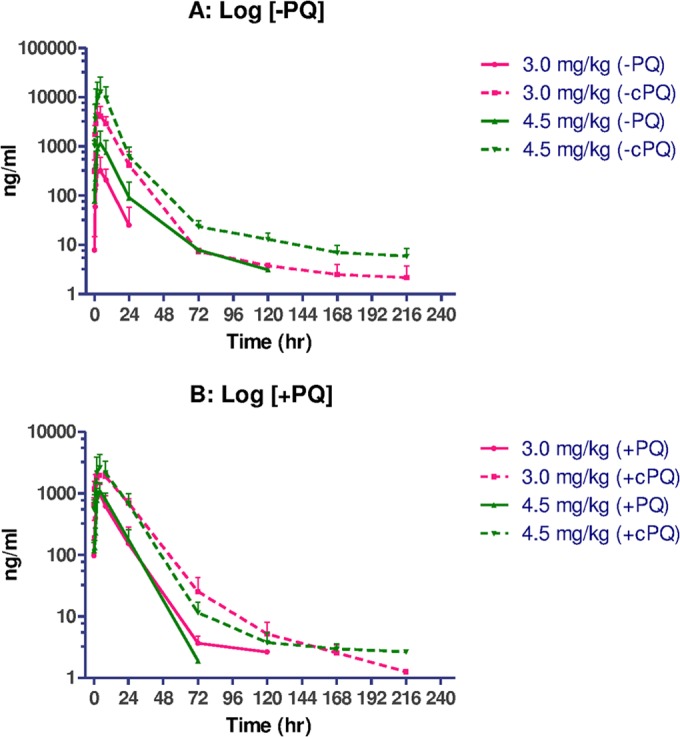
Semi-log plots of means (± standard deviations [SD]) of concentration in plasma-time profiles of (−)-PQ/cPQ (A) and (+)-PQ/cPQ (B) enantiomers following 7 days of oral administration at 3.0 and 4.5 mg/kg/day.
There was no enantiomeric interconversion of primaquine in vivo as measured by LC-MS using a chiral separation column (not shown). The noncompartmental PK parameters at day 7 following dosing with 3.0 and 4.5 mg/kg/day are shown in Table 2. Overall exposure and pharmacokinetic parameters for the parent compounds of the two enantiomers were comparable, and for both enantiomers, carboxyprimaquine was formed in much higher concentrations, particularly for (−)-PQ. Notably, V was higher for the (−) form of the parent compound, suggesting greater tissue distribution than in the (+) form. There was lower clearance of (+)-PQ with a longer half-life than (−)-PQ. After receiving 3 mg/kg PQ, Cmax and AUC of (+)-cPQ were approximately 2- and 4-fold greater than for (+)-PQ, respectively, while (−)-cPQ had Cmax and AUCs 20- to 30-fold greater than (−)-PQ. Mean Cmax values of (+)-PQ following 3 and 4.5 mg/kg/day for 7 days were similar. However, administration of 4.5 mg/kg of (−)-PQ showed a significant increase in mean Cmax and AUC of both (−)-PQ and (−)-cPQ compared to those at 3.0 mg/kg.
TABLE 2.
Noncompartmental PK parameters of (−)-PQ/cPQ and (+)-PQ/cPQ enantiomers following the 7th (last) dose of oral administrationa
| Parameter | (−)-Primaquine |
(+)-Primaquine |
||||||
|---|---|---|---|---|---|---|---|---|
| 3 mg/kg |
4.5 mg/kg |
3 mg/kg |
4.5 mg/kg |
|||||
| (−)PQ | (−)cPQ | (−)PQ | (−)cPQ | (+)PQ | (+)cPQ | (+)PQ | (+)cPQ | |
| T1/2 (h) | 4.9 (2.8) | 8.9 (1.7) | 6.5 (3.3) | 10.7 (1.8) | 7.9 (1.9) | 9.7 (1.7) | 6.5 (0.3) | 8.1 (0.2) |
| Cmax (μg/liter) | 357 (315) | 4,667 (2,605) | 1,232 (901) | 13,403 (11,931) | 1,049 (479) | 2,006 (137) | 1,098 (372) | 2,642 (1,662) |
| Tmax (h) | 4.7 (3.1) | 4.0 (3.5) | 3.3 (1.2) | 5.3 (2.3) | 3.3 (1.2) | 5.3 (2.3) | 4.00 (0) | 4.00 (0) |
| AUC0-∞ (μg/h/liter) | 4,128 (290) | 65,146 (14,185) | 15,585 (12,409) | 176,431 (136,466) | 15,616 (10,130) | 52,646 (7,835) | 17,030 (5,000) | 56,093 (26,904) |
| MRT (h) | 8.5 (4.3) | 10.5 (5.0) | 8.9 (2.5) | 9.2 (1.4) | 11.0 (2.0) | 15.0 (0.9) | 11.1 (2.6) | 13.8 (2.1) |
| V (liter/kg) | 8.1 (6.2) | 4.6 (4.4) | 2.7 (1.3) | 2.6 (0.8) | ||||
| CL (liter/h/kg) | 1.3 (1.3) | 0.6 (0.6) | 0.2 (0.1) | 0.28 (0.1) | ||||
Values are means (SD).
Comparison of the PK and PD variables of interest revealed few statistically significant differences between mean changes in hematologic values over baseline based on drug exposure (Table 3). There were nonstatistically significant trends in mean % methemoglobin increase from baseline, with 237% for (+)-PQ versus 94% for (−)-PQ at 3.0 mg/kg, but wide standard deviations. Individual responses were more revealing, with very large increases for one animal with the highest MetHgb values at both 3.0 and 4.5 mg/kg, despite animal dosing groups being crossed over to a different enantiomer. There was a very large increase in ALT over baseline for two animals in the (−) group at 4.5 mg/kg, but this was not statistically significant compared to the (+)-enantiomer, likely due to a very large difference between animals resulting in a wide standard deviation.
TABLE 3.
Mean percent changes in pharmacodynamic variables of interest from baseline after 7 days of dosing at escalating doses
| Dose and change measured | Mean % change (SD) |
|
|---|---|---|
| (−)-PQ | (+)-PQ | |
| 1.3 mg/kg/day | ||
| Hgb decrease | 1.01 (1.75) | 5.24 (2.50) |
| Rectic increase | 1.56 (2.69) | 28.5 (21.1) |
| MetHgb increase | 29.2 (11.8) | 178 (122) |
| ALT increase | 36.6 (12.7) | 28.3 (24.7) |
| 3.0 mg/kg/day | ||
| Hgb decrease | 2.49 (3.00) | 6.97 (3.00) |
| Rectic increase | 19.0 (20.0) | 59 (20.5) |
| MetHgb increase | 94.1 (43.2) | 237 (82.0) |
| ALT increase | 69.2 (44.5) | 54.2 (19.0) |
| 4.5 mg/kg/day | ||
| Hgb decrease | 6.47 (4.11) | 5.34 (3.30) |
| Rectic increase | 104 (38.0) | 126 (87.5) |
| MetHgb increase | 157 (194) | 149 (28.2) |
| ALT increase | 5,289 (4921) | 158 (44.4) |
Cmax and AUC of the parent compound were correlated with an increase in MetHgb formation at both 3.0 mg/kg (Spearman's ρ = 0.786, P = 0.048 for Cmax; and ρ = 0.786, P = 0.048 for AUC0-∞) and 4.5 mg/kg (ρ = 0.857, P = 0.024 for Cmax; and ρ = 0.893, P = 0.012 for AUC0-∞). However, this association was seen only when (+)- and (−)-enantiomers were combined and not when they were used separately. There were no other significant associations between PK parameters and PD variables to include methemoglobin, reticulocytosis, ALT, or hemoglobin and concentrations of either parent or carboxy metabolites, nor were there discernible differences in PK-PD relationships between the (+)- and (−)-enantiomers.
DISCUSSION
Basic efficacy findings in primates—contrast.
This study sheds additional light on the issue of stereoisomerism and its potential impact on the toxicity and antimalarial activity of PQ in different species and provides further impetus for the evaluation of these parameters in humans.
Schmidt et al. reported that the two PQ enantiomers had equivalent radical curative potency in primates (11). They tested 3 dose levels in groups of 3 animals for each enantiomer at 0.375, 0.5, and 0.75 mg/kg/day, with racemate given to one animal at each dose. This relatively narrow dose range managed to delineate important information: while the low dose was ineffective, the high dose was 100% curative, with the 0.5-mg/kg dose comprising the approximate 50% effective dose (ED50). In our case, while both animals were cured at 1.3 and 0.6 mg/kg (standard and ED50 doses, respectively) with l-/(−)-PQ, one of the two animals treated with d-/(+) PQ was not at the 0.6-mg/kg (ED50) dose. Although our findings largely support Schmidt's, our results suggest but do not confirm possible greater potency with the (−) form than with the (+) form.
Sample sizes were small in our experiment, and the results cannot be considered definitive, though it is important to note that the potential potency advantage observed here was despite higher systemic clearance and greater conversion of the (−)-PQ to the inert carboxy metabolite. Early studies in rats established the differential metabolism and disposition of (+)- and (−)-PQ, with greater conversion of the (−)-enantiomer to the carboxy metabolite both in isolated perfused liver (17) and in liver microsomes, while (+)-PQ was the predominant form excreted in urine (9). Our studies in mice were consistent with this (10), and administration of racemic PQ resulted in the preferential metabolism of (−)-PQ to the carboxy metabolite (16). If cPQ is inert as an antimalarial and not toxic, as assumed, it would be expected that (−)-PQ would show reduced potency compared to the (+) form, but we found the opposite in our present experiment in primates. Based on these findings, carboxyprimaquine formation appears to predominate with the (−) form in both primates and rodents. However, there is discordance between the primate and rodent models in terms of efficacy, with (+)-primaquine showing 3-fold-greater potency in suppressing mouse P. berghei blood-stage parasitemia and 5 to 7-fold-greater potency in the mouse P. berghei causal prophylaxis model than (−)-PQ (10). This is in contrast to similar activity of the two forms against primate P. cynomolgi malaria seen here and elsewhere.
While the enantiomers were not compared directly with racemic primaquine in the present experiments, racemic primaquine is used as the standard therapy to halt further relapses in the experimental rhesus P. cynomolgi model. Based on AFRIMS historical data, daily doses of 1.3 and 1.78 mg/kg/day have been 100% curative, preventing further relapse (14). Doses of 0.6 mg/kg/day have given variable results and appear to approximate ED50, while doses of 0.3 mg/kg for 7 days have been ineffective. Thus, it appears that the enantiomers in our hands had approximately the same radical curative efficacy against P. cynomolgi as racemic primaquine.
Hepatotoxicity/toxicity.
Overall, both primaquine enantiomers were clinically well tolerated by macaques at 1.3 and 3.0 mg/kg administered orally for 7days, approximating the 1× and 2× human-equivalent doses, respectively. However, one animal taking the (−) form needed to be excluded due to a rash and eosinophilia at 1.3 mg/kg/day, which resolved during the wash-out period and recurred on rechallenge with 3.0 mg/kg/day, suggesting an allergic reaction. At 4.5 mg/kg/day, (−)-PQ caused hepatotoxicity with a striking increase in AST, ALT, and bilirubin levels in 2 of 3 monkeys. This was not observed with the (+)-enantiomer. In contrast to the mouse model in which (+)-PQ had 4 times more acute toxicity (lethality) in mice than (−)-PQ, Schmidt et al. likewise found a 5-fold-greater therapeutic index of d-/(+)-PQ over the l-/(−) form and a 2-fold advantage over racemic PQ in nonhuman primates, based primarily on hepatotoxicity (11).
The present study confirms Schmidt's findings regarding the hepatotoxicity of (−)-PQ in nonhuman primates and extends them with some important additional observations. They acknowledged, and subsequent clinical literature makes plain, that the greater hepatotoxicity of l-/(−)-PQ is not likely to be clinically significant. A review published in 1981 concluded that despite known issues with hepatotoxicity in animal models, these findings have not been borne out in humans (12). More recently, clinical trials of daily primaquine for malaria prophylaxis in 43 volunteers in Indonesia for 1 year and 122 Colombian soldiers for 4 months did not reveal any evidence for even mild subclinical hepatotoxicity (18, 19). We argue here that after conducting a similar experiment, our results do not necessarily support the same conclusion favoring d-/(+)-primaquine.
Hematologic toxicity.
The limiting adverse reaction with PQ utilization is hematological toxicity in patients with G6PD deficiency (3, 4, 8). Though Schmidt et al. recognized the hematological issues as important, hematological parameters were described only qualitatively as “low grade methemoglobinemia and anemia” (11). In our experiment, the (−) form appeared to be a less potent elicitor of methemoglobinemia, indicative of lower oxidative stress in erythrocytes than that elicited by the (+) form, even with increasing doses. At the lower doses (1.3 and 3.0 mg/kg), mild to moderate increases in MetHgb were seen with (+)-PQ but not with (−)-PQ. At the highest dose (4.5 mg/kg), when the two enantiomer dosing groups were “crossed over,” (+)-PQ elicited an even higher MetHgb increase in 3 of 3 animals than did the 3-mg/kg dose, while 2 of 3 showed no appreciable MetHgb in the (−) group. Overall, these results suggest that in primates, the (−) form may have an advantage compared to (+)-PQ, given that (−)-PQ appeared to generate less MetHgb yet still maintained equivalent antimalarial potency. Given that the therapeutic index in humans is primarily dependent on hemolytic rather than hepatic toxicity, this has potentially important implications for improving the therapeutic index of primaquine. Both the prior conclusions and ours here remain suggestive rather than definitive, given the small sample sizes.
The significance of the rise in MetHgb should be weighed carefully. It has been suggested that though MetHgb is an important early marker of toxicity for the 8-AQ class, signaling oxidative stress in erythrocytes, methemoglobinemia alone does not directly correlate to red cell hemolysis for many agents (20) and little association has been found following PQ administration (3). Our understanding of the mechanisms of 8-AQ-induced red cell oxidative stress causing methemoglobinemia and hemolysis remains incomplete, and these are likely separate mechanisms (6). A recent in vitro study revealed that while there was evidence of red cell oxidative stress in the presence of primaquine alone, hepatic metabolism was required to generate significant dose-dependent methemoglobinemia and this effect was similar in both normal and G6PD-deficient erythrocytes (21). It is well established that the hematological toxicity of PQ in individuals with normal G6PD is minimal, despite a commonly observed rise in MetHgb (22–24).
The specific nature and mechanisms of action of metabolites generated by primaquine, including those forming methemoglobin, are complex and poorly understood. 5,6-Dihydroxy-8-(4-amino-1-methylybutylamino)quinoline(6-desmethyl-5-hydroxyprimaquine) was identified as a methemoglobin-forming metabolite of primaquine in a mouse liver microsomal system and also depleted glutathione levels in glucose-6-phosphate dehydrogenase-deficient erythrocytes (25). This metabolite is a powerful hemoglobin oxidant compared to primaquine, but similar activity was not found in carboxyprimaquine, the major in vivo metabolite (26). The 5-hydroxy and 6-desmethyl-5-hydroxy metabolites were found to exert greater oxidative stress than the parent compounds (27). Though not directly measured, it is likely that the correlations between parent drug levels and methemoglobin formation observed in our experiment reflect metabolite formation rather than direct activity of the parent compound.
Pharmacokinetics/explanations.
With regard to pharmacokinetics, concentrations in plasma reached maximum levels in about 3 to 5 h for both enantiomers of PQ and cPQ at the doses measured (3.0 and 4.5 mg/kg), consistent with prior reports in this model. There were few if any clear plasma PK-PD correlations between variables of interest. (−)-PQ was found to have 3- to 4-fold-higher V, suggesting greater tissue distribution, which may serve as an explanation for the selective hepatotoxicity of the (−)-enantiomer reported here and in previous studies (11, 28), given the far more abundant formation of (−)-cPQ. Despite substantial elevation in ALT in two animals at 4.5 mg/kg, there was no correlation in the percentage of increase in ALT with plasma PK parameters. While possibly due to the small sample size, the lack of concentration dependence, despite apparent dose dependency, suggests a first-pass effect creating hepatotoxic metabolites. The lack of an observed rise in ALT in the (+)-enantiomer group confirms the most striking toxicokinetic advantage identified previously in this model (11).
Our results agree with prior findings indicating that a substantial fraction of PQ is metabolized to carboxyprimaquine. Carboxyprimaquine formation results from PQ side chain biotransformation to PQ-aldehyde by an amine oxidase, likely monoamine oxidase A. The aldehyde is subsequently converted to the carboxylic acid by an aldehyde dehydrogenase and likely undergoes further transformation, based on the lack of excretion observed in human urine (25). cPQ formation primarily occurs via a non-cytochrome P450 (non-CYP450)-mediated mechanism (29), with a far smaller conversion by hepatic cytochrome CYP450s (30). On the other hand, CYP450s appear to also metabolize PQ to hydroxylated metabolites, which have been shown to have hemolytic potential (31). Differential formation of the carboxy and hydroxylated metabolites in liver could conceivably explain the differential hepatotoxicity seen in our studies. In the perfused rat liver, (−)-PQ showed higher clearance and increased formation of cPQ compared to (+)-PQ, attributed to either increased binding site affinity or decreased binding to intrahepatic albumin, resulting in higher hepatic uptake of (−)-PQ (17). Consistent with this, the increased V for (−)-PQ seen here could reflect greater hepatic uptake and explain not only the modestly increased antirelapse activity against liver-stage parasites but also a greater propensity for liver toxicity at high doses. Similar to what is seen in the primate model, peak cPQ serum levels are up to 10-fold higher than the parent drug 3 to 12 h postdose in humans (32). Thus, higher serum levels of (−)-cPQ in nonhuman primates may also reflect increased intrahepatic levels of the parent drug and lead to the hepatotoxicity seen here. Further study of the intrahepatic metabolism of primaquine may help to better understand the hepatotoxic effects and mitigate this potential risk if (−)-PQ is selected for clinical use, thereby helping to separate antirelapse activity from toxicity.
Conclusions.
There does not in our view appear to be an advantage suggested for one compound over the other when they are compared head to head in the rhesus macaque P. cynomolgi model, though reduced methemoglobin formation and a potential potency advantage with the l-/(−) form were observed here. Given the caveats of apparent model discordance, further animal studies may not be informative. Primaquine has been used in the clinic for more than 60 years, and detailed clinical trials in humans would seem the next logical step to answer the remaining questions of hemolytic toxicity and radical curative efficacy in detail. Such studies have already been carried out with the racemate, and with tafenoquine, a primaquine analogue currently in advanced clinical development.
Given the similarities in activity and the potential model discordance between P. cynomolgi and P. vivax, further animal testing may not be informative in this regard. The most efficient development strategy in our view would be advancing both enantiomers to human testing to tease out potential toxicokinetic advantages and estimate the therapeutic index in great detail in healthy human subjects with mild to moderate G6PD deficiency. This would be done by dose escalation up to current clinical doses and include at least an initial comparison with racemic primaquine. Because there is no reliable method of establishing definitive P. vivax antirelapse activity, healthy-human studies should be followed by clinical trials in patients with uncomplicated P. vivax infections to attempt to discern whether a clinical advantage exists. The two enantiomers should be compared unless a significant dose-limiting toxicity beyond that of racemic primaquine is discovered for either form in healthy human volunteers. Primaquine enantiomers should be prepared for clinical trials using the simplified, scalable synthesis method.
ACKNOWLEDGMENTS
The opinions or assertions contained here are our views and are not to be construed as official or as reflecting true views of the Department of the Army or the Department of Defense.
Footnotes
Published ahead of print 29 September 2014
REFERENCES
- 1.Brewer GJ, Tarlov AR, Kellermeyer RW, Alving AS. 1962. The hemolytic effect of primaquine. XV. Role of methemoglobin. J. Lab. Clin. Med. 59:905–917. [PubMed] [Google Scholar]
- 2.Myint HY, Berman J, Walker L, Pybus B, Melendez V, Baird JK, Ohrt C. 2011. Improving the therapeutic index of 8-aminoquinolines by the use of drug combinations: review of the literature and proposal for future investigations. Am. J. Trop. Med. Hyg. 85:1010–1014. 10.4269/ajtmh.2011.11-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beutler E. 1959. The hemolytic effect of primaquine and related compounds: a review. Blood 14:103–139. [PubMed] [Google Scholar]
- 4.Vale N, Moreira R, Gomes P. 2009. Primaquine revisited six decades after its discovery. Eur. J. Med. Chem. 44:937–953. 10.1016/j.ejmech.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Nanayakkara NP, Ager AL, Jr, Bartlett MS, Yardley V, Croft SL, Khan IA, McChesney JD, Walker LA. 2008. Antiparasitic activities and toxicities of individual enantiomers of the 8-aminoquinoline 8-[(4-amino-1-methylbutyl)amino]-6-methoxy-4-methyl-5-[3,4-dichlorophenoxy]quinoline succinate. Antimicrob. Agents Chemother. 52:2130–2137. 10.1128/AAC.00645-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tekwani BL, Walker LA. 2006. 8-Aminoquinolines: future role as antiprotozoal drugs. Curr. Opin. Infect. Dis. 19:623–631. 10.1097/QCO.0b013e328010b848. [DOI] [PubMed] [Google Scholar]
- 7.Zhang C, Zhu C, Lin X, Gao F, Wei Y. 2002. Enantiomeric separation of primaquine, an anti-malarial drug, by cyclodextrin-modified micellar electrokinetic capillary chromatography. Anal. Sci. 18:595–597. 10.2116/analsci.18.595. [DOI] [PubMed] [Google Scholar]
- 8.John GK, Douglas NM, von Seidlein L, Nosten F, Baird JK, White NJ, Price RN. 2012. Primaquine radical cure of Plasmodium vivax: a critical review of the literature. Malar. J. 11:280. 10.1186/1475-2875-11-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker JK, McChesney JD. 1988. Differential metabolism of the enantiomers of primaquine. J. Pharm. Sci. 77:380–382. 10.1002/jps.2600770503. [DOI] [PubMed] [Google Scholar]
- 10.Nanayakkara NPD, Tekwani BL, Herath HMTB, Sahu R, Gettayamacin M, Tungtaeng A, van Gessel Y, Baresel P, Wickham K, Bartlett MS, Fronczek FR, Melendez V, Ohrt C, Reichard GA, McChesney JD, Rochford R, Walker LA. 2014. Primaquine enantiomers: scalable preparation and differential pharmacologic and toxicologic profiles. Antimicrob. Agents Chemother. 58:4737–4744. 10.1128/AAC.02674-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt LH, Alexander S, Allen L, Rasco J. 1977. Comparison of the curative antimalarial activities and toxicities of primaquine and its d and l isomers. Antimicrob. Agents Chemother. 12:51–60. 10.1128/AAC.12.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clyde DF. 1981. Clinical problems associated with the use of primaquine as a tissue schizontocidal and gametocytocidal drug. Bull. World Health Organ. 59:391–395. [PMC free article] [PubMed] [Google Scholar]
- 13.Solem E, Pirzer C, Siege M, Kollmann F, Romero-Saravia O, Bartsch-Trefs O, Kornhuber B. 1985. Mass screening for glucose-6-phosphate dehydrogenase deficiency: improved fluorescent spot test. Clin. Chim. Acta 152:135–142. 10.1016/0009-8981(85)90184-6. [DOI] [PubMed] [Google Scholar]
- 14.Deye GA, Gettayacamin M, Hansukjariya P, Im-erbsin R, Sattabongkot J, Rothstein Y, Macareo L, Fracisco S, Bennett K, Magill AJ, Ohrt C. 2012. Use of a rhesus Plasmodium cynomolgi model to screen for anti-hypnozoite activity of pharmaceutical substances. Am. J. Trop. Med. Hyg. 86:931–935. 10.4269/ajtmh.2012.11-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dow GS, Gettayacamin M, Hansukjariya P, Imerbsin R, Komcharoen S, Sattabongkot J, Kyle D, Milhous W, Cozens S, Kenworthy D, Miller A, Veazey J, Ohrt C. 2011. Radical curative efficacy of tafenoquine combination regimens in Plasmodium cynomolgi-infected Rhesus monkeys (Macaca mulatta). Malar. J. 10:212. 10.1186/1475-2875-10-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Avula B, Khan SI, Tekwani BL, Nanayakkara NP, McChesney JD, Walker LA, Khan IA. 2011. Analysis of primaquine and its metabolite carboxyprimaquine in biological samples: enantiomeric separation, method validation and quantification. Biomed. Chromatogr. 25:1010–1017. 10.1002/bmc.1557. [DOI] [PubMed] [Google Scholar]
- 17.Nicholl DD, Edwards G, Ward SA, Orme ML, Breckenridge AM. 1987. The disposition of primaquine in the isolated perfused rat liver. Stereoselective formation of the carboxylic acid metabolite. Biochem. Pharmacol. 36:3365–3369. [DOI] [PubMed] [Google Scholar]
- 18.Fryauff DJ, Baird JK, Basri H, Sumawinata I, Purnomo Richie TL, Ohrt CK, Mouzin E, Church CJ, Richards AL, Wignall S, Mouzin E, Ohrt C, Subianto B, Sandjaja B, Hoffman SL. 1995. Randomised placebo-controlled trial of primaquine for prophylaxis of falciparum and vivax malaria. Lancet 346:1190–1193. 10.1016/S0140-6736(95)92898-7. [DOI] [PubMed] [Google Scholar]
- 19.Soto J, Toledo J, Rodriquez M, Sanchez J, Herrera R, Padilla J, Berman J. 1998. Primaquine prophylaxis against malaria in nonimmune Colombian soldiers: efficacy and toxicity. A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 129:241–244. [DOI] [PubMed] [Google Scholar]
- 20.Clark BB, Morrisey RW, Blair D. 1951. Relation of methemoglobin to hemolysis. Blood 6:532–543. [PubMed] [Google Scholar]
- 21.Ganesan S, Chaurasiya ND, Sahu R, Walker LA, Tekwani BL. 2012. Understanding the mechanisms for metabolism-linked hemolytic toxicity of primaquine against glucose 6-phosphate dehydrogenase deficient human erythrocytes: evaluation of eryptotic pathway. Toxicology 294:54–60. 10.1016/j.tox.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 22.Carmona-Fonseca J, Alvarez G, Maestre A. 2009. Methemoglobinemia and adverse events in Plasmodium vivax malaria patients associated with high doses of primaquine treatment. Am. J. Trop. Med. Hyg. 80:188–193. [PubMed] [Google Scholar]
- 23.Baird JK, Lacy MD, Basri H, Barcus MJ, Maguire JD, Bangs MJ, Gramzinski R, Sismadi P, Krisin Ling J, Wiady I, Kusumaningsih M, Jones TR, Fryauff DJ, Hoffman SL. 2001. Randomized, parallel placebo-controlled trial of primaquine for malaria prophylaxis in Papua, Indonesia. Clin. Infect. Dis. 33:1990–1997. 10.1086/324085. [DOI] [PubMed] [Google Scholar]
- 24.Kantor GS. 1992. Primaquine-induced methemoglobinemia during treatment of Pneumocystis carinii pneumonia. N. Engl. J. Med. 327:1461. 10.1056/NEJM199211123272016. [DOI] [PubMed] [Google Scholar]
- 25.Mihaly GW, Ward SA, Edwards G, Orme ML, Breckenridge AM. 1984. Pharmacokinetics of primaquine in man: identification of the carboxylic acid derivative as a major plasma metabolite. Br. J. Clin. Pharmacol. 17:441–446. 10.1111/j.1365-2125.1984.tb02369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kristensen S, Nord K, Orsteen AL, Tonnesen HH. 1998. Photoreactivity of biologically active compounds, XIV: influence of oxygen on light induced reactions of primaquine. Pharmazie 53:98–103. [PubMed] [Google Scholar]
- 27.Agarwal S, Gupta UR, Daniel CS, Gupta RC, Anand N, Agarwal SS. 1991. Susceptibility of glucose-6-phosphate dehydrogenase deficient red cells to primaquine, primaquine enantiomers, and its two putative metabolites. II. Effect on red blood cell membrane, lipid peroxidation, MC-540 staining, and scanning electron microscopic studies. Biochem. Pharmacol. 41:17–21. [DOI] [PubMed] [Google Scholar]
- 28.Brocks DR, Mehvar R. 2003. Stereoselectivity in the pharmacodynamics and pharmacokinetics of the chiral antimalarial drugs. Clin. Pharmacokinet. 42:1359–1382. 10.2165/00003088-200342150-00004. [DOI] [PubMed] [Google Scholar]
- 29.Frischer H, Mellovitz RL, Ahmad T, Nora MV. 1991. The conversion of primaquine into primaquine-aldehyde, primaquine-alcohol, and carboxyprimaquine, a major plasma metabolite. J. Lab. Clin. Med. 117:468–476. [PubMed] [Google Scholar]
- 30.Constantino L, Paixao P, Moreira R, Portela MJ, Do Rosario VE, Iley J. 1999. Metabolism of primaquine by liver homogenate fractions. Evidence for monoamine oxidase and cytochrome P450 involvement in the oxidative deamination of primaquine to carboxyprimaquine. Exp. Toxicol. Pathol. 51:299–303. [DOI] [PubMed] [Google Scholar]
- 31.Bowman ZS, Oatis JE, Jr, Whelan JL, Jollow DJ, McMillan DC. 2004. Primaquine-induced hemolytic anemia: susceptibility of normal versus glutathione-depleted rat erythrocytes to 5-hydroxyprimaquine. J. Pharmacol. Exp. Ther. 309:79–85. 10.1124/jpet.103.062984. [DOI] [PubMed] [Google Scholar]
- 32.Bhatia SC, Saraph YS, Revankar SN, Doshi KJ, Bharucha ED, Desai ND, Vaidya AB, Subrahmanyam D, Gupta KC, Satoskar RS. 1986. Pharmacokinetics of primaquine in patients with P. vivax malaria. Eur. J. Clin. Pharmacol. 31:205–210. 10.1007/BF00606660. [DOI] [PubMed] [Google Scholar]