

Using synthetic systems to mimic natural enzymes with high catalytic activity and distinct substrate selectivity has been a challenge for the last several decades. Hemin, the catalytic center for many protein families including cytochromes, peroxidases, myoglobins and hemoglobins, can catalyze a variety of oxidation reactions like peroxidase enzymes.[1] However, direct application of hemin as an oxidation catalyst is of significant challenge because of its molecular aggregation in aqueous solution to form catalytic inactive dimers and oxidative self-destruction in the oxidizing media, which causes passivation of its catalytic activity.[2] A potential solution to this problem is to synthetically modify the porphyrin structure to achieve a variety of iron porphyrin derivatives for improved catalytic activity or stability.[3] An alternative approach is to use high surface area materials such as zeolites, nanoparticles, silica or natural clay to support hemin to achieve improved stability or activity in epoxidation or other reactions in organic solutions.[4] For reactions in aqueous solutions, hydrogel-embedded hemin[5] or more elaborate hemin complex obtained by conjugating with specific antibodies[6] have shown activity significantly better than free molecules, which is, however, still orders of magnitude inferior to natural enzymes, not to mention the difficulties in the synthesis of such kinds of complex hemin conjugates. Therefore, the discovery and development of novel materials as supports to achieve biomimetic catalysts with enzyme-like activity is highly desired.
Graphene, a single layer of carbon arranged in a honeycomb structure, has attracted intense interest due to its fascinating electronic, thermal, and mechanical properties.[7] Graphene is typically prepared through mechanical cleavage or chemical methods.[8] In particular, chemical exfoliation of graphite oxide (GO) either by ultrasonic dispersion or rapid thermal expansion followed by chemical reduction provides a low-cost and scalable method to produce bulk quantities of graphene flakes for wide range of applications.[9] The resulting graphene usually has a rich variety of surface defects and functional groups such as carboxylic groups to enable it to disperse well in aqueous solution.[9a,b] With a two-dimensional sheet-like structure, graphene represents an interesting geometrical support for molecular catalysts with a large open surface area that is readily accessible to substrates/products with little diffusion barrier, which is distinct from conventional high surface area porous materials. Moreover, graphene also possesses a rich surface chemistry and has the potential to further promote the catalytic activity and stability of the supported molecular systems such as hemin and other porphyrin species through cation-π interactions or π-π stacking. Although the formation of porphyrin-graphene heterostructures[10] and their electrochemical applications[11] have been reported recently, many of these studies involve catalytically less active dimers[10d,11c] and the superior catalytic properties of hemin-graphene conjugates have not yet been adequately explored.
Here we report the synthesis of a hemin-graphene conjugate (Fig. 1) via π-π stacking interactions. Spectroscopic characterizations show that the hemin retains the catalytic active monomer form as in natural enzymes. The catalytic studies show that the hemin-graphene conjugates can function as effective catalysts in pyrogallol oxidation reaction with exceptionally high catalytic activity (kcat) and substrate binding affinity (KM) approaching that of natural enzymes. An iron porphyrin derivative, tetramethylpyridylporphyrin iron (FeTMPyP), was also immobilized on graphene with nearly enzyme-like activity, demonstrating the general applicability of graphene as a support for metalloprophyrin species.
Figure 1.

Schematic illustration of the formation of hemin-graphene conjugates through π-π stacking.
Graphene oxide is prepared through Hummer's method[12] and graphene is obtained by reducing graphene oxide with hydrazine.[9b] The graphene solution was directly used for the subsequent studies without further purification. Due to the insolubility of hemin in neutral aqueous solution, the conjugation experiments between hemin and graphene were carried out in methanol. The hemin-graphene conjugates were prepared by dispersing dry graphene in 1.5 mM of hemin in methanol followed by incubation for 120 minutes. The hemin-graphene conjugates were then separated from the reaction solution through a centrifugation process, and characterized using UV-Vis spectroscopy and atomic force microscopy (AFM).
UV-vis absorption spectra of the hemin methanol solution, a mixture of hemin and graphene in methanol, and separated hemin-graphene conjugates re-dispersed in methanol exhibit nearly the same absorption characteristics with a Soret band at 400 nm (Fig. 2a). The absorption band is consistent with that of monomeric hemin in methanol or dimethyl sulfoxide solution[13], suggesting that the adsorbed hemin species on graphene are monomeric — the same form that hemin takes in natural enzymes. This is also distinct from previous studies on graphene-hemin conjugates, in which hemin dimers are typically obtained[10d,11c]. A careful analysis of the spectra reveals that there is a spectral shift in the Q bands and charge transfer band upon the formation of the graphene-hemin conjugate. For example, the Q bands of hemin in the hemin-graphene conjugate blue-shift from 504 nm to 501 nm and 535 nm to 531 nm, respectively, compared to free hemin. The charge transfer band shows an even clearer blue-shift from 628 nm to 623 nm (Fig. 2b). The band shift suggests the formation of an axial ligation to the iron center of hemin[14] likely due to cation- π interaction between iron centers and graphene.
Figure 2.
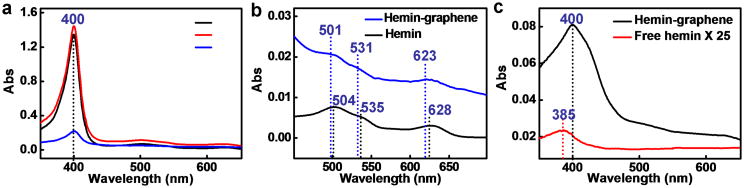
Characterization of hemin-graphene conjugates. (a) UV-vis spectroscopy of free hemin (black line), hemin/graphene mixture (red line) and separated hemin-graphene re-dispersed in methanol solution (blue line). All samples show a Soret band at 400 nm. (b) UV-vis spectroscopy of free hemin, and a hemin-graphene conjugates in methanol solution shows that the Q bands and charge transfer (CT) band exhibit a slight blue-shift upon formation of hemin-graphene conjugates. (c) UV-vis spectroscopy of free hemin and hemin-graphene conjugates in pH 7.4 Tris buffer, highlighting that hemin-graphene retains its monomeric form, while free hemin in water forms catalytic inactive dimers.
The separated hemin-graphene conjugates were then re-dispersed in pH 7.4 Tris buffer. The equivalent amount of free hemin was also dispersed in pH 7.4 Tris buffer to form a saturated solution. The UV-vis absorption spectra of hemin-graphene conjugates in pH 7.4 Tris buffer show the same absorption peaks as that in methanol, confirming the structural stability of the hemin-graphene conjugates in pH 7.4 Tris buffer. Quantitative analysis shows that the coverage of hemin on graphene in hemin-graphene conjugates is ∼0.3 monolayer (see methods). In contrast, the free hemin solution in pH 7.4 Tris buffer shows an extremely weak absorption at 385 nm, indicating the low solubility of hemin and formation of catalytically inactive dimers.[15]
The absorption of a monolayer hemin on graphene was also investigated by atomic force microscopy (AFM) studies. Specifically, graphene was first deposited onto silicon oxide substrate from aqueous solution. AFM images were then used to determine the thickness of selected graphene flakes (∼1.6 nm for 3-4 layer graphene as shown in Fig. 3a and ∼1.4 nm for 2-3 layer graphene as shown in Fig. 3b). The substrate was then immersed into the hemin methanol solution to allow for absorption of hemin on graphene. The absorption of hemin on a bare silicon oxide substrate is nearly negligible in this process (Supporting Fig. 1). After absorption, AFM studies show that the thickness of the exact same graphene flakes (∼2.0 nm in Fig. 3a and ∼1.8 nm in Fig. 3b, respectively) is increased by ∼0.4 nm, which can be attributed to the absorption of a monolayer of hemin molecules on graphene. Control studies conducted by immersing graphene into pure methanol shows no thickness change (Supporting Fig. 2), confirming the thickness increase observed above is indeed due to the absorption of hemin on graphene.
Figure 3.

AFM morphology for hemin-graphene conjugates. The scale bars are 50 nm. The grahene flakes exhibit a ∼0.4 nm increase in step height after immersing into a hemin solution, which can be attributed to the absorption of a monolayer hemin.
To evaluate their catalytic activity, hemin-graphene conjugates were used as catalysts for the pyrogallol oxidation reaction (Fig. 4a)[5a]. The pyrogallol oxidation reaction, in which pyrogallol is oxidized into purpurogallin by hydrogen peroxide, is a commonly used standard assay to characterize the catalytic performance of various porphyrin derivatives.[5a] The catalytic reactions were carried out with a constant hemin-graphene catalyst concentration (5 μM hemin equivalent), variable pyrogallol concentrations of 0.1- 2 mM and a hydrogen peroxide concentration of 40 mM. The reaction progress was monitored at 420 nm by kinetic mode UV-vis spectroscopy. The reaction process follows the conventional enzymatic dynamic regulation of the Michiaelis-Menten equation (Fig. 4b). Based on the different oxidation rates with variable substrate concentrations, a Lineweaver–Burk plot can be obtained with a nearly perfect linear relationship (Fig. 4c), from which the important kinetic parameters such as kcat and KM can be derived (Table 1). The kcat gives a direct measure of the catalytic production of the product: i.e, it measures the maximum number of substrate molecules turned over per catalyst molecule per unit time under optimal conditions. It can also be viewed as the optimum turnover rate. KM is the Michaelis constant and is often associated with the affinity of the catalyst molecules with the substrate. KM is also a measure of the substrate concentration required for effective catalysis to occur. In this way, kcat/KM gives a measure of the catalytic efficiency. Either a large value of kcat (rapid turnover) or a small value of KM (high affinity for substrate) will make kcat/KM large enough to obtain improved catalyst efficiency.
Figure 4.

Pyrogallol oxidation reaction catalyzed by hemin-graphene conjugates. (a) Schematic illustration of the pyrogallol oxidation reaction, in which pyrogallol is oxidized to purpurogallin by hydrogen peroxide. (b) The initial pyrogallol oxidation profile catalyzed by hemin-graphene conjugates (5 μM hemin equivalent). The concentrations of pyrogallol range from 0.1 mM to 2 mM. (c) Lineweaver–Burk plot of the pyrogallol oxidation catalyzed by the hemin-graphene conjugates.
Table 1.
Kinetic parameters for the pyrogallol oxidation reaction catalyzed using different catalysts.
The derived kcat of the hemin-graphene catalyst shows a surprisingly high value of 246 min-1, which is more than one order of magnitude higher than a recently reported hemin-hydrogel catalyst (19 min-1) and about two orders of magnitude higher than free hemin (2.4 min-1) (Table 1). This Kcat value is nearly comparable to that of the natural enzyme horseradish peroxidase (HRP, ∼1750 min-1). The derived KM value (∼1.2 mM) is also comparable to that of the natural enzyme HRP (0.81 mM), indicating good affinity of substrate to the hemin-graphene conjugates. Together, these studies clearly demonstrate that the hemin-graphene catalyst exhibits excellent catalytic efficiency (kcat/KM ∼ 2×105), approaching that of the natural enzyme HRP (kcat/KM ∼ 2×106).
Other porphyrin derivatives such as FeTMPyP have also been explored as alternatives to hemin molecules with improved catalytic performance.[6] To demonstrate the general applicability of a graphene support to enhance the catalytic performance of porphyrin derivatives, we have synthesized FeTMPyP and immobilized them onto graphene using a similar method. Catalytic studies of the pyrogallol oxidation reaction with a FeTMPyP-graphene catalyst shows similar behavior to that of the hemin-graphene catalyst (Fig. 5a). The Lineweaver–Burk plot (Fig. 5b) gives a kcat value of 545 min-1, which is comparable to that of the complex antibody supported species (680 min-1). More notably, the KM of FeTMPyP-graphene (0.96 mM) is much lower than the antibody supported species (8.6 mM), and is similar to that of the natural enzyme HRP (0.81 mM), thus showing excellent binding affinity. The catalytic efficiency (kcat/KM) of FeTMPyP-graphene (5.7×105 M-1min-1) is also about one order of magnitude better than the antibody supported species (7.9×104 M-1min-1) and is nearly comparable to that of natural enzyme HRP (kcat/KM ∼ 2×106). Overall, the FeTMPyP-graphene catalyst shows a further improvement in catalytic performance, closely approaching the natural enzyme systems.
Figure 5.

Pyrogallol oxidation catalyzed by FeTMPyP-graphene conjugates. (a) Initial pyrogallol oxidation profile catalyzed by FeTMPyP-graphene (5 μM FeTMPyP equivalent). The concentrations of pyrogallol range from 0.1 mM to 2 mM. (b) A Lineweaver–Burk plot of pyrogallol oxidation catalyzed by FeTMPyP-graphene.
It should be noted that hemin-graphene conjugates have been prepared and explored as catalysts recently.[11c] However, hemin-graphene conjugates obtained in this previous study has a dimeric form, with the catalytic activity comparable to that of free hemin (e.g. Vmax of 4.55×10-8 M s-1 for dimeric hemin-graphene vs. 4.69×10-8 M s-1 for free hemin in tetramethylbenzidine (TMB) oxidation reaction).[11c] We have also conducted pyrogallol oxidation reaction using free hemin or dimeric hemin-graphene conjugates prepared using the reported approach as the catalyst. Neither of the two catalysts showed a measurable catalytic activity in the pyrogallol assay. Overall, the catalytic activity of the dimeric hemin-graphene is comparable to that of free hemin, and does not show apparent performance enhancement, while that of our monomeric hemin-graphene conjugates is at least 100 times more active than free hemin or dimeric hemin on graphene reported recently. These studies clearly highlight the importance to retain the monomeric hemin graphene structure in the hemin-graphene conjugates.
Together, our studies demonstrate that graphene supported porphyrin derivatives show excellent catalytic characteristics that are more than two orders of magnitude better than free hemin, and more than one order of magnitude better than any other supported systems, which opens up the exciting possibility for other important oxidation reactions such as epoxidation and sulfoxidation. Our related studies have also shown that the hemin-graphene conjugate function as an effective catalyst to facilitate the L-arginine oxidation reaction (for nitric oxide generation) and toluene oxidation reaction. The fundamental reason for such a substantial enhancement of catalytic activity is a particularly interesting topic to investigate in the future both experimentally and theoretically. In general, several combined features of the graphene support may contribute to the performance enhancement. First, graphene supported hemin or FeTMPyP could prevent molecules from self dimerization to form catalytically inactive species. Second, graphene as a support can block one side of the porphyrin molecule which could prevent hydrogen peroxide attack from both sides, and thus lowering the possibility of oxidative destruction of the catalyst molecules themselves.[16] Third, compared to hydrogel support and other three-dimensional (3D) porous supports, graphene provides a two-dimensional (2D) support with a large open and accessible surface area; therefore, the diffusion of substrate to and product away from the catalytic centers are much easier, which could be beneficial to the reaction turn-over rate and the binding interactions. Previous studies also demonstrated metalloporphyrin immobilized on silica surfaces exhibit a higher catalytic activity than those trapped in a 3D silica matrix.[17] Fourth, graphene can function as a π donor to the iron centers of hemin through cation-π interactions. The cation-π interaction between the iron centers and graphene mimics the role of cysteine or histidine in enzymes, which has been proven as axial ligand to the hemin center from Glu mutation study.[18] Enzymatic studies[19] also showed that axial ligand can serve multiple functions to enhance the catalytic characteristics, such as enhancement of the rate of O-O cleavage, promoting heterolytic splitting rather than homolytic O-O splitting[20] and stabilization of the ferryl (FeO4+) moiety due to resonance and enhanced electrophilicity, which is crucial for catalytic activity.[21]
Supplementary Material
Acknowledgments
We acknowledge support by the NIH Director's New Innovator Award Program, part of the NIH Roadmap for Medical Research, through Grant 1DP2OD004342-01.
Contributor Information
Teng Xue, Department of Materials Science and Engineering, University of California, Los Angeles, CA 90095, U. S. A.
Shan Jiang, Department of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095, U. S. A.
Dr. Yongquan Qu, Department of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095, U. S. A
Qiao Su, Department of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095, U. S. A.
Rui Cheng, Department of Materials Science and Engineering, University of California, Los Angeles, CA 90095, U. S. A.
Sergey Dubin, Department of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095, U. S. A.
Chin-Yi Chiu, Department of Materials Science and Engineering, University of California, Los Angeles, CA 90095, U. S. A.
Prof. Richard Kaner, Department of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095, U. S. A.; California Nanosystems Institute, University of California, Los Angeles, CA 90095, U. S. A
Prof. Yu Huang, Email: yhuang@seas.ucla.edu, Department of Materials Science and Engineering, University of California, Los Angeles, CA 90095, U. S. A.; California Nanosystems Institute, University of California, Los Angeles, CA 90095, U. S. A.
Prof. Xiangfeng Duan, Email: xduan@chem.ucla.edu, Department of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095, U. S. A.; California Nanosystems Institute, University of California, Los Angeles, CA 90095, U. S. A.
References
- 1.Genfa Z, Dasgupta PK. Analytical Chemistry. 1992;64:517–522. doi: 10.1021/ac00029a013. [DOI] [PubMed] [Google Scholar]
- 2.Bruice TC. Accounts of Chemical Research. 1991;24:243–249. [Google Scholar]
- 3.a) Mansuy D. Coordination Chemistry Reviews. 1993;125:129–141. [Google Scholar]; b) Shema-Mizrachi M, Pavan GM, Levin E, Danani A, Lemcoff NG. Journal of the American Chemical Society. 2011;133:14359–14367. doi: 10.1021/ja203690k. [DOI] [PubMed] [Google Scholar]
- 4.Bedioui F. Coordination Chemistry Reviews. 1995;144:39–68. [Google Scholar]
- 5.a) Wang QG, Yang ZM, Zhang XQ, Xiao XD, Chang CK, Xu B. Angew Chem-Int Edit. 2007;46:4285–4289. doi: 10.1002/anie.200700404. [DOI] [PubMed] [Google Scholar]; b) Wang Q, Yang Z, Ma M, Chang CK, Xu B. Chemistry-a European Journal. 2008;14:5073–5078. doi: 10.1002/chem.200702010. [DOI] [PubMed] [Google Scholar]
- 6.Yamaguchi H, Tsubouchi K, Kawaguchi K, Horita E, Harada A. Chemistry-a European Journal. 2004;10:6179–6186. doi: 10.1002/chem.200305692. [DOI] [PubMed] [Google Scholar]
- 7.a) Geim AK, Novoselov KS. Nature Materials. 2007;6:183–191. doi: 10.1038/nmat1849. [DOI] [PubMed] [Google Scholar]; b) Schwierz F. Nature Nanotechnology. 2010;5:487–496. doi: 10.1038/nnano.2010.89. [DOI] [PubMed] [Google Scholar]; c) Bai J, Cheng R, Xiu F, Liao L, Wang M, Shailos A, Wang KL, Huang Y, Duan X. Nature Nanotechnology. 2010;5:655–659. doi: 10.1038/nnano.2010.154. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liao L, Lin YC, Bao M, Cheng R, Bai J, Liu Y, Qu Y, Wang KL, Huang Y, Duan X. Nature. 2010;467:305–308. doi: 10.1038/nature09405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Park S, Ruoff RS. Nature Nanotechnology. 2009;4:217–224. doi: 10.1038/nnano.2009.58. [DOI] [PubMed] [Google Scholar]; b) Allen MJ, Tung VC, Kaner RB. Chemical Reviews. 2010;110:132–145. doi: 10.1021/cr900070d. [DOI] [PubMed] [Google Scholar]; c) Jiao L, Zhang L, Wang X, Diankov G, Dai H. Nature. 2009;458:877–880. doi: 10.1038/nature07919. [DOI] [PubMed] [Google Scholar]
- 9.a) Stankovich S, Dikin DA, Piner RD, Kohlhaas KA, Kleinhammes A, Jia Y, Wu Y, Nguyen ST, Ruoff RS. Carbon. 2007;45:1558–1565. [Google Scholar]; b) Li D, Mueller MB, Gilje S, Kaner RB, Wallace GG. Nature Nanotechnology. 2008;3:101–105. doi: 10.1038/nnano.2007.451. [DOI] [PubMed] [Google Scholar]; c) Dai BY, Fu L, Liao L, Liu N, Yan K, Chen YS, Liu ZF. Nano Res. 2011;4:434–439. [Google Scholar]; d) Tung VC, Huang JH, Tevis I, Kim F, Kim J, Chu CW, Stupp SI, Huang J. Journal of the American Chemical Society. 2011;133:4940–4947. doi: 10.1021/ja1103734. [DOI] [PubMed] [Google Scholar]; e) Liang Y, Li Y, Wang H, Zhou J, Wang J, Regier T, Dai H. Nature Materials. 2011;10:780–786. doi: 10.1038/nmat3087. [DOI] [PubMed] [Google Scholar]
- 10.a) Xu Y, Zhao L, Bai H, Hong W, Li C, Shi G. Journal of the American Chemical Society. 2009;131:13490–13497. doi: 10.1021/ja905032g. [DOI] [PubMed] [Google Scholar]; b) Geng J, Jung HT. Journal of Physical Chemistry C. 2010;114:8227–8234. [Google Scholar]; c) Ghosh A, Rao KV, George SJ, Rao CNR. Chemistry-a European Journal. 2010;16:2700–2704. doi: 10.1002/chem.200902828. [DOI] [PubMed] [Google Scholar]; d) Guo Y, Deng L, Li J, Guo S, Wang E, Dong S. Acs Nano. 2011;5:1282–1290. doi: 10.1021/nn1029586. [DOI] [PubMed] [Google Scholar]
- 11.a) Chen J, Zhao L, Bai H, Shi G. Journal of Electroanalytical Chemistry. 2011;657:34–38. [Google Scholar]; b) Zhang S, Tang S, Lei J, Dong H, Ju H. Journal of Electroanalytical Chemistry. 2011;656:285–288. [Google Scholar]; c) Guo CX, Lei Y, Li CM. Electroanalysis. 2011;23:885–893. [Google Scholar]
- 12.Hummers WS, Offeman RE. Journal of the American Chemical Society. 1958;80:1339–1339. [Google Scholar]
- 13.Ryabova ES, Dikiy A, Hesslein AE, Bjerrum MJ, Ciurli S, Nordlander E. Journal of Biological Inorganic Chemistry. 2004;9:385–395. doi: 10.1007/s00775-004-0532-5. [DOI] [PubMed] [Google Scholar]
- 14.Droghetti E, Sumithran S, Sono M, Antalik M, Fedurco M, Dawson JH, Smulevich G. Archives of Biochemistry and Biophysics. 2009;489:68–75. doi: 10.1016/j.abb.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silver J, Lukas B. Inorganica Chimica Acta-Bioinorganic Chemistry. 1983;78:219–224. [Google Scholar]
- 16.Ullrich V. Archives of Biochemistry and Biophysics. 2003;409:45–51. doi: 10.1016/s0003-9861(02)00410-1. [DOI] [PubMed] [Google Scholar]
- 17.Moreira MSM, Martins PR, Curi RB, Nascimento OR, Iamamoto Y. Journal of Molecular Catalysis a-Chemical. 2005;233:73–81. [Google Scholar]
- 18.Smulevich G, Neri F, Willemsen O, Choudhury K, Marzocchi MP, Poulos TL. Biochemistry. 1995;34:13485–13490. doi: 10.1021/bi00041a028. [DOI] [PubMed] [Google Scholar]
- 19.a) Feiters MC, Rowan AE, Nolte RJM. Chemical Society Reviews. 2000;29:375–384. [Google Scholar]; b) Szacilowski K, Chmura A, Stasicka Z. Coordination Chemistry Reviews. 2005;249:2408–2436. [Google Scholar]
- 20.Harris D, Loew G, Waskell L. Journal of the American Chemical Society. 1998;120:4308–4318. [Google Scholar]
- 21.a) Walker FA. Journal of Inorganic Biochemistry. 2005;99:216–236. doi: 10.1016/j.jinorgbio.2004.10.009. [DOI] [PubMed] [Google Scholar]; b) Weichsel A, Maes EM, Andersen JF, Valenzuela JG, Shokhireva TK, Walker FA, Montfort WR. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:594–599. doi: 10.1073/pnas.0406549102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.