

Abstract
Along with thrombolytic therapy, which has a number of limitations, stroke outcome may be improved with neuroprotective therapies that disrupt ischemic cell death. Recent research has shown a neuroprotective role of ethanol administration during ischemic stroke, such as its ability to reduce infarct volume and neurologic deficit. In order to investigate this further, we assessed the hypothesis that ethanol’s neuroprotective effect is through reduction of apoptosis and the modulation of the important apoptotic PKC-δ and Akt signaling pathway. Ethanol (1.5 g/kg) was given by intraperitoneal injections to 54 Sprague-Dawley rats after 2 hours of middle cerebral artery (MCA) occlusion, followed by 3 or 24 hours of reperfusion. We measured apoptotic cell death, PKC-δ, and Akt mRNA and protein expressions in each of ischemic groups with or without ethanol treatment using ELISA, real-time PCR and Western blot analysis. Our results showed that cell death was significantly increased in rats following 2 hour MCA occlusion and 24 hour reperfusion. Subsequently, cell death was significantly reduced by an administration of ethanol. We further found that ethanol administration, prior to either 3 or 24 hours of reperfusion, significantly decreased the expression of PKC-δ while simultaneously increasing the expression Akt at both mRNA and protein levels at the two points. In conclusion, our study suggests that ethanol administration following ischemic stroke modulates the gene and protein profile in such a way that it increased expression of anti-apoptotic Akt and decreased the pro-apoptotic PKC-δ. This ultimately results in a decrease in neuronal apoptosis, thus conferring neuroprotection.
Keywords: Ischemia/reperfusion, neuroprotection, apoptosis, ethanol, PKC-δ, Akt/PKB
Ischemic stroke, the most prevalent type of stroke, causes a rapid disruption of blood flow and thus deprives the brain of vital nutrients due to a thrombosis or embolism all within minutes. This ischemic crisis initiates a cascade of intracellular events within affected cells, eventually leading to neuronal death and loss of functional brain tissue. Thrombolytics, the only FDA approved treatment, are limited because there is a short time window for administration and late treatment increases risks [1]. The main goal of thrombolytic therapy is to remove the occlusion; however, full treatment of the acute ischemic stroke will necessitate inhibiting the cellular dysfunction that is a result of vascular occlusion. Therefore a neuroprotective agent is needed that will confer effective protection and inhibit the deleterious ischemic cell-death cascade thus normalizing metabolic function.
The molecular basis of ischemia-induced apoptotic cell degradation, one of the major factors that contribute to the spread of neuronal cell loss, has been elucidated in previous studies [2]. Although advances have uncovered the cascade of molecular events underlying apoptosis, there is a scarcity of knowledge of how the apoptotic cascade can be modulated by various treatments. Amongst the limited treatments for ischemic stroke, ethanol remains to be one of the most promising due to its neuroprotective effect following stroke [3]. In our recent study we showed that ethanol can serve as a neuroprotective agent by reducing apoptotic cell death in rats subjected to middle cerebral artery (MCA) occlusion [4]. Another study showed that ethanol down regulated pro-apoptotic proteins [5]. Despite the many advances, however, the complete biochemical mechanism behind ethanol’s neuroprotective effect still needs to be explained.
Currently, there are two main pathways of apoptosis: the extrinsic (death receptor) pathway and the intrinsic (mitochondrial pathway). Although they differ in how they are initiative, both pathways converge upon a common-point: the cleavage, and thus activation, of caspase-3, known as the executioner caspase [6]. Unlike the extrinsic pathway, the intrinsic pathway is a non-receptor mediated pathway governed by the Bcl-2 family of proteins which causes crucial changes to the mitochondria, such as opening of the mitochondrial permeability transition (MPT) and releasing sequestered pro-apoptotic proteins into the cytosol, such as the deadly cytochrome c – an activator of caspase-3 [7]. Certain factors, however, can modulate this process and, in particular, affect the release of cytochrome c. A previous study has concluded that protein kinase C delta (PKC-δ), a key signaler of the apoptotic pathway, causes the release of cytochrome c [8]. On the other hand, studies have shown that Akt phosphorylates a group of molecules thereby blocking the release of mitochondrial cytochrome c [9]. Therefore, both PKC-δ and Akt serve as strategic regulators of apoptosis.
To understand the biochemical pathway associated with ethanol-induced neuroprotection, we first investigated ethanol’s effect on apoptotic cell death. This assured us of ethanol’s ability to counter apoptotic cell death. To further elucidate ethanol’s neuroprotective effect on the apoptotic pathway, we then focused on these two important proteins involved in apoptosis, protein kinase C delta (PKC-δ) and Akt, also known as Protein Kinase B (PKB).
MATERIALS AND METHODS
Subjects
All experimental procedures were approved by the Institutional Animal Investigation Committee of Wayne State University and were in accordance with the National Institutes of Health guidelines for care and use of laboratory animals. A total of 54 adult male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) were used. Rats were randomly divided into: (1) sham-operated group (n=6×2) in which no middle cerebral artery (MCA) occlusion; (2) stroke with reperfusion group for 3 hours (h) (n=6×2); (3) stroke with reperfusion for 24h (n=6×2) group. The stroke groups were randomly assigned to receive either saline (no treatment) or an intraperitoneal injection of ethanol (1.5 g/kg) immediately prior to reperfusion (n=6 per group). The apoptotic cell-death was determined at 24 h post-reperfusion and the PKC-δ and Akt mRNA and protein were analyzed at both 3 and 24 h post-reperfusion. Investigators were blinded during the selection and surgical portion of this experiment.
Transient MCA Occlusion
Middle cerebral artery occlusion (MCAO) was induced for 2 hours using the intraluminal filament model [10]. This was followed by reperfusion for 3 or 24 hours. A circulating heating pad and hearting lamp was used to maintain rectal temperature at 36.5–37.5 °C. The Animals were anesthetized with 1.5–2% enflurane in a mixture of 70% air and 30% O2. Blood pCO2 and pO2, mean arterial pressure (MAP), as well as rectal and brain temperature were monitored throughout the procedure. At 3 or 24 hours after reperfusion, brains were harvested for further analysis.
Cell death detection
A cell death ELISA kit (Roche Diagnostics, Indianapolis, IN, USA) was used to quantify the degree of apoptosis in each group by measuring the amount of cytoplasmic histone-associated DNA fragments generated by apoptosis using a photometric enzyme immunoassay as described previously by us [4, 11]. According to protocol, 10 mg of brain sample was transferred to a 0.1 M citric acid solution mixed with 0.5% Tween-20 and was then centrifuged for 15 min at 2,000 rpm. The supernatant was diluted by incubation buffer and was used for assay. Absorbance at the 405 nm wavelength was detected for cell death using a DTX-880 multimode detector (Beckman). We then presented the relative mean values for cell death.
mRNA Expression of PKC-δ and Akt
A real-time polymerase chain reaction (PCR) technique was used to determine gene expressions in each group after stroke as described previously by [4]. Total RNA was isolated from the cortex and striatum of the sham-operated, no-treatment, and treatment groups (n=6) by using STAT-60 Reagent (Tel-Test Co.) according to the manufacturer’s protocol. The samples were purified using TRIzol® (Invitrogen, Grand Island, NY). Random primers were used to create first-strand DNA synthesis using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Then, the cDNA was amplified using an ABI Prism 7900HT sequencing detection system for real-time PCR with Brilliant II SYBR® Green Master Mixes (Agilent Technologies, Santa Clara, CA). PKC-δ (forward 5′- GAG GCA CTC ACC ACA GAC -3′ and reverse primer 5′- AGG TCC AGC CAG AAC TCA -3′) and Akt specific primers (forward 5′-TCC TGC ACC TGG AGC TCT GTT A- 3′ and reverse primer 5′-CTC AGG GCA GCA GGA CAT GTA G -3′) were used. As an internal PCR control, the rat ribosomal protein L32 (rpL32) was used for each sample (forward: 5′-TGT CCT CTA AGA ACC GAA AAG CC-3′ and reverse: 5′-CGT TGG GAT TGG TGA CTCTGA-3′). There relative quantification of target gene expression was determined by using the comparative CT (threshold cycle) method with the arithmetic formula [12]. Subtracting the CT of the housekeeping gene from the CT of the target gene yielded the ΔCT in each group (control and experimental groups). By entering this value into the equation 2−ΔCT, we were able to calculate the exponential amplification of PCR. The mean amount of gene expression from the control group was arbitrarily assigned as 1 to serve as reference. The expression of the target gene from experimental groups, therefore, represents the fold-difference expression relative to the reference gene.
Protein Expression
Western Blot analysis was used to detect protein levels in the ischemic tissue as described previously [4]. Briefly, brain isolates from rats in the control group and one variable group (Stroke or Stroke + 1.5 g/kg) were loaded onto a single 10% sodium dodecyl sulfate-polyacrylamide gel for electrophoresis. Upon conclusion of electrophoresis, proteins were transferred to a polyvinylidene fluoride (PVDF) membrane (Bio-Rad). Membranes were incubated with a primary antibody (rabbit polyclonal anti- PKC-δ, 1:5000, Santa Cruz; rabbit polyclonal anti-Akt 1:1000, Cell Signaling) for 24 hours at 4 °C. Next, membranes were washed 3 times with PBS for 6 minutes each, and re-incubated with a secondary antibody (goat anti-rabbit IgG, Santa Cruz; goat anti-rabbit IgG, Santa Cruz) for one hour at room temperature. An ECL system was used to detect immunoreactive bands by luminescence. Western blot images for each antibody, including β-actin, were analyzed using an image analysis program (ImageJ 1.42, National Institutes of Health, USA), to quantify protein expression in terms of relative image density. The mean amount of protein expression from the control group was assigned a value of 1 to serve as reference. The expressions of the target genes were represented as fold-differences compared to the control.
Statistical Analysis
Statistical analysis was performed with SPSS for Windows, version 17.0 (SPSS, Inc.). The differences among multiple groups were assessed using both one-way and two-way analysis of variance (ANOVA) with a significance level at p<0.05. Post-hoc comparison between groups was also performed with the least significant difference (LSD) method. All data was expressed as mean +/− standard errors (SE).
RESULTS
Apoptotic Cell-death
Cell death was quantified by the amount of cytoplasmic histone-associated DNA fragments generated by apoptosis. Rats in the untreated stroke group showed, as expected, a significant increase in apoptosis when compared to the sham-operated animals [F (2, 15) =17.840, p=0.000] (Fig 1). Treatment of the stroke group with 1.5 g/kg of ethanol provided a significant decrease in cell death as compared to stroke without treatment [LSD: p = 0.001]. There was no significant difference in apoptosis between sham animals and treated stroke animals [LSD: p = 0.126].
Figure 1.

Ethanol significantly decreases apoptotic cell death. Apoptotic cell death by groups at 24h after reperfusion is represented as a relative value compared to a reference value of 1 based on the amount of cytoplasmic histone-associated DNA fragments formed by apoptosis. Stroke significantly increased cell death (*p<0.01) as compared to sham-operation. Ethanol treatment decreased apoptosis (#p<0.01) as compared to stroke without treatment.
Pro-apoptotic PKC-δ mRNA and protein expression
Compared to sham-operated rats, levels of PKC-δ mRNA were significantly increased in ischemic rats at 3 hours after reperfusion, but with ethanol treatment there was a significant decrease from ischemic rats and no significant difference between control and ethanol-treated rats [F(2,18) = 17.046, p = 0.000. LSD: Control-Stroke p = 0.000, Control-Ethanol p = 0.118, Stroke-Ethanol p = 0.001] (Fig 2A). Similarly, at 3 hours after reperfusion, PKC-δ protein expression was significantly increased as compared to sham-operated rats, but not significantly increased in ischemic rats that had been treated with ethanol [F(2,15) = 79.347, p = 0.000. LSD: Control-Stroke p = 0.000, Control-Ethanol p = 0.573, Stroke-Ethanol p = 0.000] (Fig. 3A). The results at 24 hours post-reperfusion were similar for both mRNA [F(2,15) = 13.912, p = 0.000. LSD: Control-Stroke p = 0.000, Control-Ethanol p = 0.936, Stroke-Ethanol p = 0.000](Fig. 2A) and protein [F(2,15) = 22.666, p = 0.000. LSD: Control-Stroke p = 0.000, Control-Ethanol p = 0.584, Stroke-Ethanol p = 0.000] (Fig. 3A). Ischemia/reperfusion increased PKC-δ mRNA [F(1,22) = 19.065, p = 0.000] and protein [F(1, 20) = 59.143, p = 0.000] expression while ethanol significantly reduced them at 24 hours.
Figure 2.
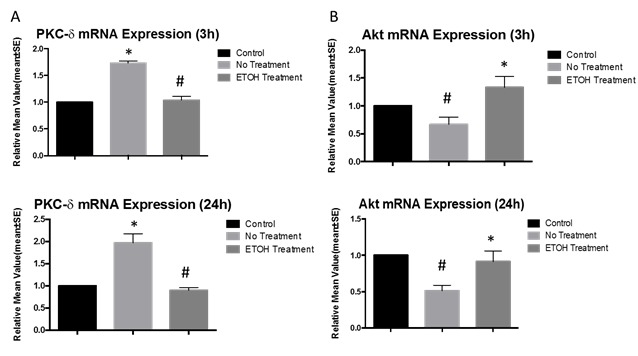
mRNA expressions of PKC-δ and Akt. As compared to the stroke without treatment group, ethanol suppresses PKC-δ mRNA expression at 3h (#p<0.01) and 24h (#p<0.01) (A). As compared to stroke without treatment, ethanol increases Akt mRNA expression at 3h (*p<0.01) and 24h (*p<0.01) (B). All values are set as a percentage of the sham-operation group (standardized at 1.0), and represented as means ± SE
Figure 3.

Protein expressions of PKC-δ and Akt. Pro-apoptotic protein expression of PKC-δ was significantly reduced with ethanol treatment as compared to stroke without treatment at 3h (#p<0.01) and 24h (#p<0.01) (A). Pro-survival protein expression of Akt was significantly increased with ethanol treatment as compared to stroke without treatment at 3h (*p<0.01) and 24h (*p<0.01) (B). All values are set as a percentage of the control (standardized at 1.0), and represented as means ± SE.
Pro-Survival Akt mRNA and protein expression
At 3 hours after reperfusion, animals in the stroke without treatment group had decreased levels of Akt mRNA (Fig. 3B) as compared to sham-operated rats, while ethanol treatment elevated Akt mRNA in ischemic rats. There was a significant difference in mRNA expression between the stroke and ethanol groups [F(2,15) = 5.889, p = 0.013. LSD: Control-Stroke p = 0.106, Control-Ethanol p = 0.106, Stroke-Ethanol p = 0.004]. Similarly, at 3 hours after reperfusion, phosphorylated Akt protein expression had significantly decreased as compared to sham-operated rats, while ethanol restored Akt protein levels [F(2,15) = 6.652, p = 0.009. LSD: Control-Stroke p = 0.003, Control-Ethanol p = 0.363, Stroke-Ethanol p = 0.021] (Fig. 3B). For mRNA (Fig. 3B), at 24 hours post-reperfusion, ethanol treatment has a significant effect on reduced expression by stroke [F(3,20) = 6.216, p = 0.004]. Acute ethanol administration before reperfusion significantly [F(1, 20) = 13.852, p = 0.004] increased the expression of Akt as compared to stroke without treatment. At 24 hours post-reperfusion, ischemia-injured animals demonstrated significantly decreased Akt protein levels as compared to sham-operated animals, while ethanol elevated Akt expression significantly [F(2,15) = 33.295, p = 0.000. LSD: Control-Stroke p = 0.001, Control-Ethanol p = 0.013, Stroke-Ethanol p = 0.000] (Fig. 3B).
DISCUSSION
In the present study, we demonstrated that ethanol treatment following ischemic stroke affected the apoptotic protein profile of PKC-δ and Akt in the brain. Following 2 hours of MCA occlusion, the administration of 1.5 g/kg ethanol led to a significant increase in the anti-apoptotic Akt mRNA and protein expression in the ischemic brain. This increase was achieved when ethanol was administered at 3 and 24 hours after reperfusion. Moreover, ethanol decreased the levels of PKC-δ mRNA and protein expression significantly when administered following ischemic stroke at 3 and 24 hours after reperfusion. This was in line with our results of a significant decrease in cellular apoptosis and cell death seen in stroke at 24 hours when ethanol was given at the onset of reperfusion [13].
Although multiple proteins are involved in apoptosis, Protein Kinase C- δ (PKC-δ) and Akt /PKB are strategic signalers of the pathway. As a class of proteins, protein kinases serve as tactical regulators of signal transduction pathways in cells by phosphorylating, and thus functionally modulating, other target proteins. Studies have shown that PKC-δ has been involved in many diseases, including reperfusion injury [14]. Furthermore, both PKC-δ and Akt are kinases that control major cellular functions and thus any disruption to their functions can lead to major downstream effects. PKC-δ is a pro-apoptotic protein while Akt is an anti-apoptotic kinase that has been the focus of major previous studies [8, 9, 14–18]. Previous research demonstrated that PKC-δ is up-regulated in ischemic stroke [8,14,19]. This is because in ischemic stroke there is a deprivation of oxygen and ATP in brain tissues. This causes the Na+/K+ pump to dysfunction, which causes K+ accumulation in the exracellular space leading to a depolarized space. Consequently, this depolarized state causes Ca+ release through voltage-dependent Ca+ channels. Increases Ca+ releases glutamate which in turn causes the activation of PKC-δ. In addition, the activation of PKC-δ is aided by the accumulation of reactive oxygen species (ROS)[14]. Previous studies have theorized the mechanisms by which PKC-δ causes apoptosis in ischemic stroke. For example, a study showed that PKC-δ increases apoptosis by facilitation the release of cytochrome c from the mitochondria via the Bad pathway [8]. Other studies theorize that PKC phosphorylation alters gene expression so that protein synthesis is decreased. Nevertheless, PKC’s activation of the apoptotic pathway has been established; however, to our knowledge, no previous studies have shown a successful and efficient treatment to reverse PKC-δ’s effect on apoptosis. Thus we showed that ethanol down-regulates PKC-δ.
Furthermore, ethanol’s simultaneous effect on Akt protein is also studied in this paper. Akt, also known as PKB, is a protein kinase that is involved in multiple pathways such as in the cell cycle and insulin signaling pathways. In detail, studies have elucidated that Akt carries out the downstream effects controlled by a class of kinases known as phosphoinositide 3-kinases (PI 3-kinases). P1 3-kinases have been shown to be stimulated by, for example, growth factors. Recent data suggests that, in all, Akt modulates three important pathways of the cell: the cell cycle, apoptosis, and insulin signaling [20]. Relevant to metabolism, studies have demonstrated that the Akt gene is highly expressed in insulin-responsive tissues, such as adipose tissue, and serves as a downstream target for PI 3-kinase in the insulin signaling pathway. This is corroborated with another study that showed that the Akt gene is crucial for normal glucose homeostasis, and even for expression for the main glucose transporter in muscle tissue, Glut 4 [21]. Akt also has been shown to regulate the conversation of nutrients to macromolecules. Interestingly, 6-phosphofructo 2-kinase is phosphorylated and activated by Akt – a key glycolytic enzyme (Lawlor and Alessi 2001). Therefore, Akt has a very large role in cellular metabolism and will indirectly feed in substrates to the metabolic enzyme, PDH (pyruvate dehydrogenase). More importantly to our study, Akt has an anti-apoptotic function especially after stroke. It prevents cell death through its effect on the Bad pathway and inhibiting the release of cytochrome c from the mitochondria. In detail, Akt phosphorylates BAD and subsequently prevents it from binding to Bcl-X L, preventing apoptosis. Akt has been shown to also modulate apoptosis in other ways, such as directly inhibiting caspase proteases, inhibition of apoptosis signal-regulating kinase 1 (Ask-1), or through the promotion of survival factors. Additionally, Akt’s involvement in insulin signaling could be another method by which Akt prevents apoptosis of neuronal cells following stroke because it promotes normal metabolic process and recovery [20]. Although the exact mechanisms of Akt’s inhibition of apoptosis is complex and needs to elucidated, our results show that Akt levels increase post-ischemia while ethanol administration increases levels significantly.
It has been shown previously that PKC-δ down-regulates the Akt signaling pathway [22]. When ethanol was administered, a significant up-regulation of Akt was obtained. On the other hand, as we have demonstrated, ethanol down regulates PKC-δ. Thus, utilizing the negative regulation of PKC-δ on Akt found in previous studies [19], it is likely that ethanol increases Akt not only by acting on it directly, but also indirectly down-regulating its inhibitor PKC-δ.
Despite our vast knowledge of the hallmarks of brain injury and ischemic stroke, no significant protective treatments are applied clinically today. Ischemic injury produces a necrotic ischemic core and a penumbra that has a more apoptotic phenotype. Current reperfusion strategies, such as thrombolytic therapy, primarily aim at reducing the volume and injury severity of both the core and penumbra. These therapies have various limitations and side effects that render them ineffective in limiting brain injury after stroke. This necessitates further research to develop new clinically-promising treatments that are effective and safe, and can prevent brain damage from ischemic injury with or without reperfusion. Therefore neuroprotective therapies, as opposed to reperfusion therapies, primarily focus on salvaging injured neurons found in the penumbra. Ethanol has been proven to have neuroprotective effects on brain cells after stoke by creating a pro-survival environment in the cell. The pathway that leads to neural cell loss, however, is very complex and ethanol’s actions on its players are not well understood. Ethanol’s effects on two regulators of the apoptotic pathway are revealed in this paper. Its effects on other regulators and proteins of the pathway are the subject of future investigation in our lab.
References
- [1].Hacke W, Donnan G, Fieschi C, Kaste M, von Kummer R, Broderick JP, et al. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet. 2004;363:768–774. doi: 10.1016/S0140-6736(04)15692-4. [DOI] [PubMed] [Google Scholar]
- [2].Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke; a journal of cerebral circulation. 2009;40:e331–339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- [3].Wang F, Wang Y, Geng X, Asmaro K, Peng C, Sullivan JM, et al. Neuroprotective effect of acute ethanol administration in a rat with transient cerebral ischemia. Stroke; a journal of cerebral circulation. 2012;43:205–210. doi: 10.1161/STROKEAHA.111.629576. [DOI] [PubMed] [Google Scholar]
- [4].Fu P, Peng C, Ding JY, Asmaro K, Sullivan JM, Guthikonda M, et al. Acute administration of ethanol reduces apoptosis following ischemic stroke in rats. Neuroscience research. 2013;76:93–97. doi: 10.1016/j.neures.2013.02.011. [DOI] [PubMed] [Google Scholar]
- [5].Yuan Y, Peng C, Li K, Hussain M, Sikharam C, Guthikonda M, et al. Ethanol reduces expression of apoptotic proteins after hypoxia/reoxygenation in a brain slice model. Neurological research. 2012;34:373–378. doi: 10.1179/1743132812Y.0000000030. [DOI] [PubMed] [Google Scholar]
- [6].Elmore S. Apoptosis: a review of programmed cell death. Toxicologic pathology. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, Vandenabeele P. Toxic proteins released from mitochondria in cell death. Oncogene. 2004;23:2861–2874. doi: 10.1038/sj.onc.1207523. [DOI] [PubMed] [Google Scholar]
- [8].Dave KR, Bhattacharya SK, Saul I, DeFazio RA, Dezfulian C, Lin HW, et al. Activation of protein kinase C delta following cerebral ischemia leads to release of cytochrome C from the mitochondria via bad pathway. PloS one. 2011;6:e22057. doi: 10.1371/journal.pone.0022057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhao H, Sapolsky RM, Steinberg GK. Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Molecular neurobiology. 2006;34:249–270. doi: 10.1385/MN:34:3:249. [DOI] [PubMed] [Google Scholar]
- [10].Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke; a journal of cerebral circulation. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- [11].Geng X, Parmar S, Li X, Peng C, Ji X, Chakraborty T, et al. Reduced apoptosis by combining normobaric oxygenation with ethanol in transient ischemic stroke. Brain Res. 2013;1531:17–24. doi: 10.1016/j.brainres.2013.07.051. [DOI] [PubMed] [Google Scholar]
- [12].Hayes K, Sprague S, Guo M, Davis W, Friedman A, Kumar A, et al. Forced, not voluntary, exercise effectively induces neuroprotection in stroke. Acta neuropathologica. 2008;115:289–296. doi: 10.1007/s00401-008-0340-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Geng X, Parmar S, Li X, Peng C, Ji X, Chakraborty T, et al. Reduced apoptosis by combining normobaric oxygenation with ethanol in transient ischemic stroke. Brain Research. 2013;1531:17–24. doi: 10.1016/j.brainres.2013.07.051. [DOI] [PubMed] [Google Scholar]
- [14].Chou WH, Messing RO. Protein kinase C isozymes in stroke. Trends in cardiovascular medicine. 2005;15:47–51. doi: 10.1016/j.tcm.2005.01.003. [DOI] [PubMed] [Google Scholar]
- [15].Gliki G, Wheeler-Jones C, Zachary I. Vascular endothelial growth factor induces protein kinase C (PKC)-dependent Akt/PKB activation and phosphatidylinositol 3′-kinase-mediates PKC delta phosphorylation: role of PKC in angiogenesis. Cell biology international. 2002;26:751–759. doi: 10.1016/s1065-6995(02)90926-1. [DOI] [PubMed] [Google Scholar]
- [16].Taylor JM, Crack PJ. Impact of oxidative stress on neuronal survival. Clinical and experimental pharmacology & physiology. 2004;31:397–406. doi: 10.1111/j.1440-1681.2004.04017.x. [DOI] [PubMed] [Google Scholar]
- [17].Zhao H. Ischemic postconditioning as a novel avenue to protect against brain injury after stroke. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2009;29:873–885. doi: 10.1038/jcbfm.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhong M, Lu Z, Foster DA. Downregulating PKC delta provides a PI3K/Akt-independent survival signal that overcomes apoptotic signals generated by c-Src overexpression. Oncogene. 2002;21:1071–1078. doi: 10.1038/sj.onc.1205165. [DOI] [PubMed] [Google Scholar]
- [19].Gao X, Zhang H, Takahashi T, Hsieh J, Liao J, Steinberg GK, et al. The Akt signaling pathway contributes to postconditioning's protection against stroke; the protection is associated with the MAPK and PKC pathways. Journal of neurochemistry. 2008;105:943–955. doi: 10.1111/j.1471-4159.2008.05218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? Journal of cell science. 2001;114:2903–2910. doi: 10.1242/jcs.114.16.2903. [DOI] [PubMed] [Google Scholar]
- [21].Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, et al. Insulin Resistance and a Diabetes Mellitus-Like Syndrome in Mice Lacking the Protein Kinase Akt2 (PKBβ) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- [22].Wen HC, Huang WC, Ali A, Woodgett JR, Lin WW. Negative regulation of phosphatidylinositol 3-kinase and Akt signalling pathway by PKC. Cellular signalling. 2003;15:37–45. doi: 10.1016/s0898-6568(02)00047-5. [DOI] [PubMed] [Google Scholar]