

Abstract
Background
Ergot alkaloids are a group of highly bioactive molecules produced by a number of filamentous fungi. These compounds have been intensely studied for decades, mainly due to their deleterious effects in contaminated food and feeds, but also for their beneficial pharmaceutical and agricultural applications. Biosynthesis of ergot alkaloids goes via the common intermediate chanoclavine-I, and studies of the key enzymes, EasE and EasC, involved in chanoclavine-I formation, have relied on gene complementation in fungi, whereas further characterization has been hampered by difficulties of poor EasE protein expression. In order to facilitate the study of ergot alkaloids, and eventually move towards commercial production, the early steps of the biosynthetic pathway were reconstituted in the unicellular yeast Saccharomyces cerevisiae.
Results
The genomic sequence from an ergot alkaloid producer, Aspergillus japonicus, was used to predict the protein encoding sequences of the early ergot alkaloid pathway genes. These were cloned and expressed in yeast, resulting in de novo production of the common intermediate chanoclavine-I. This allowed further characterization of EasE and EasC, and we were able to demonstrate how the N-terminal ER targeting signal of EasE is crucial for activity in yeast. A putative, peroxisomal targeting signal found in EasC was shown to be nonessential. Overexpression of host genes pdi1 or ero1, associated with disulphide bond formation and the ER protein folding machinery, was shown to increase chanoclavine-I production in yeast. This was also the case when overexpressing host fad1, known to be involved in co-factor generation.
Conclusions
A thorough understanding of the enzymatic steps involved in ergot alkaloid formation is essential for commercial production and exploitation of this potent compound class. We show here that EasE and EasC are both necessary and sufficient for the production of chanoclavine-I in yeast, and we provide important new information about the involvement of ER and protein folding for proper functional expression of EasE. Moreover, by reconstructing the chanoclavine-I biosynthetic pathway in yeast we demonstrate the advantage and potential of this host, not only as a convenient model system, but also as an alternative cell factory for ergot alkaloid production.
Electronic supplementary material
The online version of this article (doi:10.1186/s12934-014-0095-2) contains supplementary material, which is available to authorized users.
Keywords: Ergot alkaloids, Chanoclavine-I, Pathway reconstruction, S. cerevisiae, Metabolic engineering
Background
Ergot alkaloids (EA) belong to a diverse group of natural compounds with a range of biological activities that have important applications in medicine and agriculture [1-3]. Some of these compounds have a notorious neurological effect on humans, possibly due to the structural similarity of these compounds to neurotransmitters like serotonin and dopamine [4]. EAs are produced by a variety of plant associated fungi, mainly of the genera Claviceps and Aspergillus. The production of ergot alkaloids has been linked to biosynthetic gene clusters found in several species, first in Claviceps purpurea [5] and later in Aspergillus fumigatus [6,7], Neotyphodium lolii [8], and others (see [3] for review).
The ergot alkaloids can be divided into three classes, clavines, ergoamides, and ergopeptines, depending on the substitutions found on the basic ergoline scaffold. The biosynthetic pathways leading to various ergot alkaloids have been partially elucidated, and the early steps leading to the common biosynthetic intermediate chanoclavine aldehyde are identical. After biosynthesis of chanoclavine aldehyde the biosynthetic intermediates diverge (Figure 1). Hence, in C. purpurea the pathway continues via agroclavine to ergotamine and in A. fumigatus via festuclavine to the fumigaclavines [3].
Figure 1.

Biosynthetic pathway to chanoclavine-I and chanoclavine aldehyde. Ergot alkaloids are synthesized from tryptophan and dimethylallylpyrophosphate (DMAPP, not shown), via dimethylallyltryptophan (DMAT), N-methyl-4-dimethylallyltryptophan (Me-DMAT), chanoclavine-I, and chanoclavine aldehyde. The aldehyde is considered to be the branch point in the biosynthetic pathways of different ergot alkaloids, typically via intermediates like agroclavine and festuclavine.
The first step of the common pathway is the electrophilic aromatic addition of dimethylallyl-pyrophosphate (DMAPP) to the 4 position of tryptophan to form dimethylallyl-tryptophan (DMAT). The reaction is catalysed by a prenyl transferase, DmaW (fgaPT2 in A. fumigatus) [6,9]. The second step, the methylation of DMAT to form N-methyl-4-dimethylallyltryptophan (Me-DMAT), is catalysed by the methyltransferase EasF (fgaMT in A. fumigatus) [10].
While the biosynthesis of Me-DMAT is well understood, the mechanistic basis behind the conversion of Me-DMAT to chanoclavine-I is less clear. The conversion of Me-DMAT to chanoclavine-I was investigated by gene disruption and complementation studies in C. pupurea and A. fumigatus: Lorenz and co-workers [11] used a mutated C. purpurea strain P1 to show that deletion of the easE_Cp (ccsA) gene abolished production of chanoclavine-I and any downstream products. Instead an accumulation of Me-DMAT was seen, indicating a block in the pathway after this intermediate. Alkaloid biosynthesis could be restored by expressing a GFP fusion construct of the easE_Cp gene. Analogously, Goetz and co-workers [12] disrupted the easC_Af gene in A. fumigatus, and also observed accumulation of Me-DMAT and the absence of downstream products. Furthermore, a similar pattern was observed when easE_Af was disrupted, in concordance with the C. purpurea results (above). In both of the easE and easC deletion strains, the alkaloid pathway could be restored by re-introduction of the corresponding wild type allele. Most recently Ryan and co-workers [13] transferred part of the A. fumigatus EA cluster, comprising the four genes dmaW, easF, easC, and easE, into A. nidulans, a fungus known as a non-producer of any EA. This partial cluster conferred the ability to produce chanoclavine-I, further suggesting that EasE and EasC are sufficient for the conversion from Me-DMAT. However, the involvement of enzymes from cryptic EA clusters in A. nidulans cannot be excluded [14].
Interestingly, EasC_Af contains a C-terminal amino acid motif (SRL), which is a classic type 1 peroxisomal targeting signal (PTS1) [15]. Inspection of related published sequences reveals that similar PTS1 signals are found in many EasC homologues from Aspergillus spp., but not from Claviceps spp. EasE_Af, on the other hand, appears to have an N-terminal signal peptide for the secretory pathway, as previously suggested [12]. The sequence has the typical core of hydrophobic amino acids within the first 15–30 residues, and a signal peptide is predicted by the SignalP model [16]. However, the implication of these localisation signals remains unclear, particularly in light of the apparent co-operation between EasC and EasE. The EasC proteins have similarity to peroxisomal catalases [6,7,17]. EasC_Af (the easC_Af gene product) was purified after expression in E. coli, and in vitro catalase activity of EasC_Af was shown using H2O2 as substrate [12]. However, when the enzyme was incubated with Me-DMAT, no new product was detected. Extensive efforts were made to produce active EasE_Af from E. coli or S. cerevisiae [12]. However, in all cases, incubation of EasE with Me-DMAT, or of EasE with Me-DMAT and EasC, failed to produce any new product. Therefore, biochemical studies to understand the transformation of Me-DMAT to chanoclavine-I have not been possible.
As an alternative approach to gain information about the early EA pathway we undertook the reconstitution of the chanoclavine-I pathway in yeast (S. cerevisiae). Yeast is the work horse of eukaryotic gene expression and is easily amenable to genetic manipulation [18,19]. Heterologous genes and proteins are generally well expressed, and yeast has a long history of use for industrial production of a variety of products [20,21]. We report here the successful engineering of yeast for de novo production of chanoclavine-I, overcoming the previously reported difficulties of expressing EasE. Further, we provide new insight into the roles of EasC and EasE in the intriguing biochemical conversion from Me-DMAT into chanoclavine-I, the key step in the early ergot alkaloid pathway.
Results
Prediction of dmaW coding sequences
An ergot alkaloid gene cluster was recently identified in A. japonicus [22], one of several fungal species currently being investigated for their capacity to produce molecules of potential commercial importance [23-25]. A. japonicus was previously reported to produce cycloclavine [26], an EA of the clavine group, and the genome sequence of the cluster displays high homology to EA clusters of Claviceps spp. and Aspergillus spp. This cluster therefore provided an interesting alternative for studying the chanoclavine-I pathway. The putative coding sequences (CDS) of dmaW, easF, easC, and easE were predicted, using free online gene prediction software and by alignment to homologues in the GenBank database (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Analysis of the dmaW sequence in the A. japonicus genome revealed two different CDS predictions: one prediction was for a single open reading frame of 1602 bps, corresponding to a 534 amino acid protein, whereas another prediction, with the same translation start codon, was for two exons of 1287 bps and 150 bps separated by an 85 bps intron, corresponding to a 479 amino acid protein. Both of these predictions were slightly longer than similar DmaW enzymes found in the GenBank database, where the lengths of DmaW homologues were in the range of 435–465 amino acids. Moreover, a multiple protein alignment in GenBank, using either of the two A. japonicus DmaW predictions as query, showed a lack of homology beyond approx. 428 amino acids, i.e. beyond the predicted first exon. A somewhat similar situation was seen for the two entries of A. fumigatus DmaW (Additional file 1: Figure S1). Hence, we decided to test, not only the two predictions encoding 534 amino acids (dmaW_Aj1) and 479 amino acids (dmaW_Aj2), but also a shorter version encoding 429 amino acids (dmaW_Aj3), corresponding to the predicted first exon. For easF, representing the next step in the pathway, we tested the homologues easF_Af and easF_Aj, from A. fumigatus and A. japonicus, respectively.
Me-DMAT production in yeast
To analyse the biosynthesis of chanoclavine-I we first constructed a yeast strain designed to produce the chanoclavine-I precursor Me-DMAT. For this, plasmids were constructed with synthetic, yeast codon optimized genes corresponding to each of the three dmaW predictions described above, as well as the dmaW_Cp [GenBank:AJ011963] and dmaW_Af [GenBank:XM_751048] (Table 1). All genes were tested by co-expression with a codon optimized easF_Aj gene. The easF_Aj gene was cloned in pRS315-C/A, and each dmaW gene was cloned in the pRS316-C/A, allowing expression from the yeast Cup1 promoter. Combinations of easF_Aj and each of the dmaW genes were then introduced in yeast and expressed by inducing the Cup1 promoter. After 72 h of growth at 30°C, the culture supernatants were analysed by LC-MS, and the expected mass to charge ratio (m/z) of DMAT (m/z = 273.159 +/− 0.01) and Me-DMAT (m/z = 287.175 +/− 0.01) were extracted from the total ion chromatograms. The area under the corresponding peaks was calculated and compared, indicating production of both DMAT and Me-DMAT in the strains expressing dmaW_Aj2, dmaW_Aj3, and dmaW_Cp, but essentially no production with dmaW_Aj1 or dmaW_Af (Figure 2). We suspect that dmaW_Aj1 is likely to be an incorrect CDS prediction. We also noted that, compared to the homologue used in a previous study [6], dmaW_Af has a 24 bp deletion and speculate that the sequence of dmaW_Af may also be an incorrect prediction. (Additional file 1: Figure S1). Inspection of the total ion current chromatogram revealed the appearance of several additional minor peaks in strains accumulating the putative DMAT and Me-DMAT. These were not analysed further but we speculate that they could be the result of non-enzymatic conversions due to low pH in the culture medium in the late stationary phase (pH < 4 after 72 hours). Alternatively, they could be shunt products resulting from the activity of yeast metabolic enzymes. In any case, the apparent accumulation of DMAT suggested a relatively poor activity of the methyl transferase EasF_Aj. In a separate experiment we therefore tested the A. fumigatus homologue EasF_Af in comparison to EasF_Aj. Again, the combination of DmaW_Aj2 and EasF_Aj resulted in accumulation of DMAT (Figure 2,G) although the relative levels of DMAT and Me-DMAT showed some variation compared to the previous experiment (Figure 2,B). This could be due to some in vivo instability of the compounds, as indicated by the shunt product formation mentioned above, but since it was not the focus of this work it was not investigated further. More importantly, when combining DmaW_Aj2 with the A. fumigatus homologue EasF_Af it appeared that much more of the produced DMAT was converted to Me-DMAT (Figure 2,H). Hence, for further studies the combination of dmaW_Aj2 and easF_Af, which showed the highest production of Me-DMAT, was integrated into the yeast genome. The new strain was used to purify Me-DMAT, and the compound was analysed by 1NMR to confirm the compound identity (Additional file 1: Figure S2a).
Table 1.
Genes used in this study
| CDS name | Source | Amino acids |
|---|---|---|
| dmaW_Aj1 | WO2012/116935 | 534 |
| dmaW_Aj2 | WO2012/116935 | 479 |
| dmaW_Aj3 | WO2012/116935 | 428 |
| dmaW_Af | XM_751048 | 451 |
| dmaW_Cp | AJ011963 | 448 |
| easF_Aj | WO2012/116935 | 340 |
| easF_Af | XM_751050 | 339 |
| easC_Aj | WO2012/116935 | 510 |
| easC_Af | XM_751047 | 520 |
| easC_Aj -PTS1 | WO2012/116935 | 507 |
| easE_Aj | WO2012/116935 | 622 |
| easE_Af | XM_751049 | 628 |
| easE_Cp | AJ011965 | 483 |
| easE_Aj -N sig. | WO2012/116935 | 594 |
| pdi1/easE_Aj | D00842/WO2012/116935 | 615 |
| pdi1_Sc | D00842 | 522 |
| ero1_Sc | NM_001182493 | 563 |
| fad1_Sc | NM_001180104 | 306 |
Gene names are followed by a two-letter code to indicate species of origin: Aj: A. japonicus; Af: A. fumigatus; Cp: C. purpurea; Sc: S. cerevisiae. All genes were synthesized with yeast codon optimization, except for pdi1_Sc, ero1_Sc, and fad1_Sc, which were prepared by PCR on yeast genomic DNA. Synthesis and PCR primer design was based on the named sources to give proteins of the indicated length.
Figure 2.
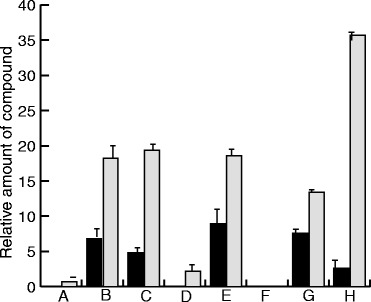
Activity of different DmaW and EasF enzymes. Relative production of DMAT (black bars) and Me-DMAT (grey bars) were analysed in strains co-expressing EasF_Aj in combination with DmaW_Aj1 (A), DmaW_Aj2 (B), DmaW_Aj3 (C), DmaW_Af (D), or DmaW_Cp (E). Expression of DmaW_Aj1 and DmaW_Af resulted in only small amounts of compounds compared to the other three DmaW homologues. Similarly, DmaW_Aj2 was co-expressed with the two homologues EasF_Aj (G) or EasF_Af (H) and relative DMAT and Me-DMAT amounts were measured. The EasF_Af resulted in more than double the amount of Me-DMAT compared to EasF_Aj. The control strain (F) carried an empty plasmid with no DmaW expression. The vertical axis shows arbitrary units based on area under the HPLC peak.
Chanoclavine-I production in yeast
Having established the production of Me-DMAT in yeast, we next addressed the conversion of this compound into chanoclavine-I. Preliminary results in our laboratory had shown poor expression of a C-terminal GFP-fusion construct of EasE_Aj, which is consistent with the reported difficulties of purifying the corresponding enzyme, EasE_Af, from A. fumigatus [12]. Therefore, in this study, we tested several easE homologues from A. japonicus, A. fumigatus, as well as from C. purpurea. To investigate the hypothesis that EasC and EasE are both required for this conversion [12,13] we expressed easC_Aj, in combination with each of the three different easE homologues, in the background of the Me-DMAT producing yeast (see above). Each easE homologue was cloned into pRS313-G/C, and easC_Aj into pRS315-P/A. When combining easC_Aj and easE_Aj we saw the appearance of a new compound with a retention time of 4.3 min, which had the expected m/z of chanoclavine-I (m/z = 257.165 +/− 0.01). The compound co-eluted with a compound of identical mass found in an A. japonicus mycelium extract (Figure 3). The compound at 4.3 min was purified, and the identity was confirmed by 1H NMR to be chanoclavine-I (Additional file 1: Figure S2b). Chanoclavine-I was not detected in strains expressing easC_Aj in combination with either easE_Af or easE_Cp, indicating that the A. fumigatus and C. purpurea homologues used in this study may not produce functional enzymes in yeast. In strains expressing only an easE or an easC homologue no chanoclavine-I was detected, supporting the hypothesis that both EasE and EasC are required for its biosynthesis. The combination of easC_Aj and easE_Aj was integrated into the genome of the Me-DMAT producing strain (see above), and the resulting strain was used to purify chanoclavine-I, which was again characterized by 1H NMR.
Figure 3.
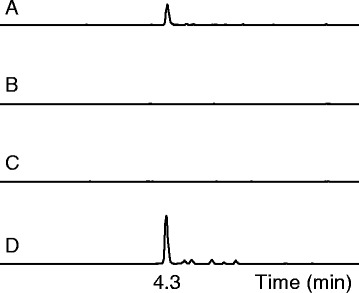
Production of chanoclavine-I enabled by EasE from A. japonicus . Yeast strains co-expressing DmaW_Aj2, EasF_Af, and EasC_Aj in combination with one of three different homologues, EasE_Aj (A), EasE_Af (B), or EasE_Cp (C), were analysed by UPLC-TOF for chanoclavine-I production. Only the strain co-expressing EasE_Aj produced chanoclavine-I, whereas no chanoclavine-I was detected in strains expressing either EasE_Af or EasE_Cp. Retention time and m/z of (A) corresponded to a chanoclavine-I reference extracted from A. japonicus mycelium (D).
Peroxisomal targeting signal is not needed in yeast
As noted by Goetz and co-workers [12] EasC_Af has a classical PTS1 peroxisomal targeting sequence, the tri-peptide SRL, at its carboxy-terminal end similar to PTS1 signals commonly found in yeast [15]. One such putative signal, ARL, is also present in the A. japonicus EasC_Aj sequence, as well as in other homologues within the Aspergillus genus. However, in the Claviceps genus no obvious PTS1 signal is found and the C. purpurea EasC enzyme instead has a C-terminal IVE tri-peptide, which has no resemblance to the PTS1 consensus sequence. Assuming the EA enzymes from these fungi have similar function, and therefore localization, this is somewhat puzzling and we therefore wondered if the PTS1 of Aspergilli is important for function. Hence, we prepared an EasC-Aj version in which the ARL triplet was deleted. A set of strains were prepared expressing the integrated dmaW_Aj2 and easF_Af, together with easE_Aj (in pRS313-G/C) and either the new easC-Aj -PTS1 version (in pRS315-P/A) or the original full length easC_Aj (in pRS315-P/A). To our surprise we saw no major difference in the ability to produce Me-DMAT and chanoclavine-I. In fact, the version without the putative PTS1 sequence resulted in slightly higher concentrations of Me-DMAT and chanoclavine-I (Figure 4).
Figure 4.
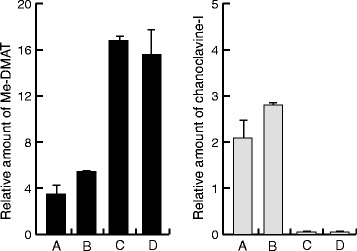
Roles of EasE signal peptide, and EasC tripeptide ARL. Relative production of Me-DMAT (left panel) and chanoclavine-I (right panel) was analysed, of a Me-DMAT producing strain, co-expressing EasC and EasE proteins with different localization signals. Wild type EasE_Aj was co-expressed in combination with either wt EasC_Aj (A) or EasC_Aj -PTS1which has no PTS1 (B). Removal of the PTS1 tripeptide ARL resulted in a slight increase of Me-DMAT and chanoclavine-I. However, when an N-terminally truncated EasE_Aj –N sig. was co-expressed with either wt EasC_Aj (C) or EasC_Aj -PTS1 (D), production of chanoclavine-I was essentially abolished. The loss of function, due to the N-terminal truncation of EasE_Aj, resulted in increased accumulation of the precursor Me-DMAT (C and D compared to A and B). The vertical axes show arbitrary units based on areas under the HPLC peaks.
N-terminal signal is crucial for EasE function
A common feature observed for EasE enzymes is a high number of hydrophobic amino acids at the N-terminal end, which is typically associated with signal peptides for the secretory pathway. Analysis of the amino acid sequence of EasE_Aj, using the online SignalP server [16], confirmed this notion by predicting a signal peptide. A putative cleavage site in EasE_Aj was predicted after pos. 29, i.e. between A and V. We used this information to prepare a truncated version of the enzyme, which lacked the N-terminal sequence, and expressed the corresponding gene from the pRS313-G/C plasmid, in the Me-DMAT producing strain, together with easC_Aj (in pRS315-P/A). The N-truncation seriously impaired the functional expression of EasE_Aj, and essentially no chanoclavine-I was detected using this enzyme. Instead, accumulation of the precursor compound Me-DMAT was observed (Figure 4).
To further evaluate the function of the putative signal peptide we prepared a version of EasE_Aj, in which we replaced the N-terminal 31 amino acids (predicted signal peptide including the cleavage site) with the known signal peptide from the native yeast Pdi1 protein. DNA sequences were fused to encode the N-terminal 24 amino acids from Pdi1, which includes the cleavage site, followed by the EasE_Aj peptide from pos. 32 (Additional file 1: Table S2). The fusion protein was tested as described for the truncated version. When expression was compared to the original EasE_Aj approximately half the chanoclavine-I production level was observed, indicating that the Pdi1 signal peptide provided functionality to the enzyme (Figure 5). This was taken as an indication that for proper function the EasE needs to enter the secretory pathway. It is not clear why the yields were lower when the Pdi1 sequence was used but possibly the cleavage site of the EasE_Aj could have been wrongly predicted and proper cleavage thus affected. Translocation across the ER membrane is a complex matter [27], but presumably a lack of cleavage would cause proteins to getting stuck in the membrane, and/or to being rapidly degraded, but this was not investigated. Testing alternative fusions of EasE_Aj to the Pdi1 signal, or fusions to other known signal peptides, might also shed more light on the function and requirements of the N-terminal signal sequence of EasE_Aj.
Figure 5.
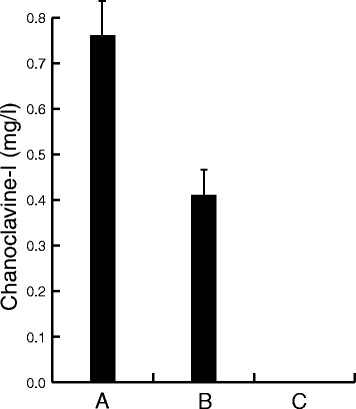
Signal peptide from Pdi1 partially restores EasE function. Concentration (mg/l) of chanoclavine-I, was measured in the growth medium, of a strain expressing DmaW_Aj2, EasF_Af, and EasC_Aj after co-expression of different versions of EasE. Expression of the wt EasE_Aj (A) resulted in production of 0.75 mg/l chanoclavine-I, whereas expression of a modified version (B), comprising the N-terminal signal peptide from Pdi1, resulted in production of approximately half of this amount. Complete deletion of the N-terminal sequence (C) abolished the function of EasE_Aj, and essentially no chanoclavine-I was detected.
Pdi1 or Ero1 overexpression improves the activity of EasE in yeast
We speculated that the importance of the N-terminal sequence is linked to a requirement for disulphide bond formation as part of the protein maturation process, which would happen during ER associated translation. EasE_Aj has a total of 9 cysteines which could potentially form disulphide bridges, and the majority of these cysteines are highly conserved among EasE enzymes (Additional file 1: Figure S3). For a preliminary evaluation of the importance of these cysteines we mutated the first seven of these individually, replacing them with alanine. All of these mutations resulted in complete loss of function (data not shown). The native yeast enzymes Pdi1 and Ero1 are known to have a function in the formation of disulphide bonds in ER (see e.g. [28] for review). Hence, a pair of chanoclavine-I producing strains were prepared in which we over-expressed one of the two genes, either pdi1 or ero1, to test whether this would have any impact on the production of chanoclavine-I. Over-expression of pdi1 (in pRS313-G/C) resulted in an approx. 50% increase in chanoclavine-I production, whereas ero1 over-expression (in pRS313-C/A) caused an almost 3 fold increase (Figure 6), supporting a possible involvement of disulphide bond formation in proper EasE folding. However, all chanoclavine-I producing strains also accumulated substantial amounts of Me-DMAT, indicating continued limitations in the conversion of Me-DMAT to chanoclavine-I.
Figure 6.
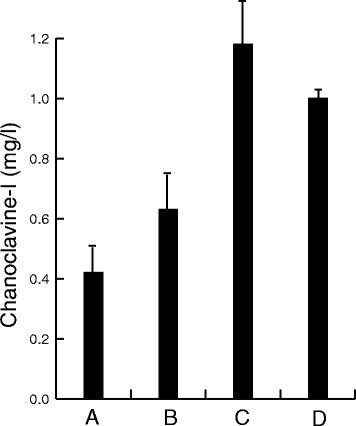
Effects of host engineering on chanoclavine-I production. Concentration (mg/l) of chanoclavine-I, was measured in the growth medium, of a chanoclavine-I producing control strain (A) and after over-expression of Pdi1 (B), Ero1(C), or Fad1 (D). Over-expression of these native yeast genes all resulted in an increased production of chanoclavine-I, relative to the control. All strains expressed the integrated, heterologous pathway to chanoclavine-I.
Fad1 overexpression improves the activity of EasE in yeast
EasE has previously been described as a flavin adenine dinucleotide (FAD) dependent reductase or dehydrogenase, and in support of this the structural similarity to the class of berberine bridge enzymes (BBE) has previously been pointed out [11]. In plants the BBE enzyme is involved in alkaloid biosynthesis, and it has been associated with transport from ER to the vacuole [29]. BBE was shown to bi-covalently bind a flavin co-factor [30] and the binding site, involving a histidine and a cysteine, seems to be conserved in EasE_Aj and other EasE enzymes (Additional file 1: Figure S4). With ample evidence that EasE is FAD dependent, we speculated whether an increased supply of this co-factor would directly improve the activity of EasE. An attempt to increase the supply via the growth medium showed no effect on chanoclavine-I production (data not shown), so instead we cloned the native yeast fad1 gene, which encodes the enzyme responsible for synthesis of FAD from flavin mono nucleotide (FMN). Over-expression of this gene, in a strain harbouring the chanoclavine-I pathway, led to a 2.5 fold increase in chanoclavine-I production (Figure 6) which we interpreted as an effect of improved co-factor supply.
Discussion
Heterologous expression in yeast of the four enzymes DmaW_Aj2, EasF_Af, EasC_Aj, and EasE_Aj led to production of chanoclavine-I, demonstrating that yeast is a suitable host for producing ergot alkaloids. This work also confirms, in accordance with previous published studies, that EasC and EasE are solely responsible for the conversion of Me-DMAT to chanoclavine-I without the involvement of other EA enzymes. Secondary metabolism in S. cerevisiae is quite limited and it seems unlikely that any yeast enzyme would be involved in the highly specialized metabolic process of ergot alkaloid biosynthesis. The production of chanoclavine-I depended on the expression of EasE from A. japonicus, whereas EasE from A. fumigatus or C. purpurea were not active when expressed in yeast. We speculate that the predicted coding regions of easE_Af and easE_Cp, along with dmaW_Aj3, could be incorrect (see below, and Additional file 1: Figure S3 and S5). Certainly, the prediction of intron and exon sequences in filamentous fungi is still a challenge.
Our results support the notion that EasE and EasC co-operate to produce chanoclavine-I, but it is still not clear how or where this process occurs. Knockout experiments of easC and easE in Aspergillus fumigatus [12], along with the data presented here, indicate that the catalytic activities of two classes of enzymes, a catalase encoded by easC and a FAD-dependent oxidoreductase encoded by easE, are both required to convert Me-DMAT to chanoclavine-I. Deletion mutants of easC and easE accumulate Me-DMAT, which suggests, though does not prove, that these two enzymes act in a coordinated fashion. It is also possible that the ergot alkaloid pathway requires disproportionation of H2O2, and that the function of EasC is to remove H2O2 derived from the oxidative activities of EasE. More extensive biochemical experiments with active EasE enzyme would be able to shed further light on these questions.
As shown here the putative PTS1 signal of EasC_Aj was not essential for function, and it is possible that even with the PTS1 signal the natural cellular distribution involves multiple locations, as seen for some native yeast proteins [31,32]. The N-terminal signal of the EasE, on the other hand, was clearly crucial for function, and the fact that a native yeast signal peptide from Pdi1 partially restored function indicates that EasE contains a genuine ER targeting signal. This would suggest that folding and disulphide bond formation in the ER is needed for proper maturation of EasE and that it passes through the secretory pathway before, possibly, joining up with EasC. Cellular localization studies might help elucidate this puzzle, just as further progress toward obtaining in vitro activity of EasC and EasE should allow us to explore the interdependence of these two enzymes on one another.
The case for oxidative folding of EasE in ER is supported by the observed increase in chanoclavine-I production after over-expression of either of the two key enzymes in the yeast disulphide bridge formation machinery. Ero1 is a sulfhydryl oxidase responsible for generating disulphide bonds that are passed on to Pdi1, which in turn oxidizes the cysteines of newly translated proteins [28,33,34]. The EasE family contains several highly conserved cysteines that would be available for oxidation, although one cysteine is likely to be involved in binding the FAD co-factor.
Interestingly, a multiple sequence alignment of the EasE_Aj and its closest homologues showed some unexpected dissimilarity regarding EasE_Af and EasE_Cp (Additional file 1: Figure S5). The first approx. 130 amino acids of EasE_Af showed no similarity to the consensus sequence, whereas EasE_Cp seemed to be completely lacking the N-terminal domain. An earlier prediction of EasE_Cp [11] suggested a 483 amino acid peptide derived from two exons. A newer GenBank entry [GenBank:JN186799], however, predicts a third exon at the 5-end and encodes a 595 amino acid protein, including a signal peptide as predicted by the SignalP model [16]. The study [11] reported complementation of an easE_Cp gene knock-out after integration of a PCR fragment comprising the same easE gene including its native promoter region. We speculate that this PCR fragment may by chance have included the first exon, allowing correct splicing and, hence, full complementation. Complementation by re-integration into the easE locus was excluded, since integration at the niaD locus was confirmed by Southern hybridization. A similar approach was used for studying the easE gene in A. fumigatus [12]. The native easE gene was first disrupted, which abolished function, and the WT sequence was then re-integrated along with 1445 bps 5’-flanking sequence. The inclusion of upstream sequence would allow the fungus to splice the gene correctly, and any divergence between the mature mRNA and the predicted CDS of easE_Af would not have been detected. We analysed the genomic region of A. fumigatus chromosome II using an online intron prediction model, which suggested an 1809 bps easE_Af coding sequence, encoding a protein of 602 amino acids including a putative N-terminal signal peptide. This newly predicted protein shows very high sequence similarity to other EasE proteins in the multiple alignment (Additional file 1: Figure S3). We synthesized a yeast codon optimized version of this predicted easE_Af, and tested its ability to support chanoclavine-I formation. In a strain co-expressing dmaW_Cp, easF_Af, and easC_Aj, the expression of either easE_Af or easE_Aj resulted in similar levels of chanoclavine-I production (data not shown). This would indicate that, at least in this case, the EasC and EasE enzymes from different fungi are capable of working together. However, further studies are needed to confirm the results, and to investigate the nature of co-operation between EasC and EasE.
Conclusion
We show here that EasC and EasE are responsible for the conversion of Me-DMAT to chanoclavine-I in yeast. Although the activity of these enzymes appears to be linked, the cellular localization of the enzymatic reaction is unclear. The N-terminal peptide sequence of EasE was identified as a signal for the secretory pathway, and a role for ER-associated folding was supported by a beneficial effect of overexpressing host enzymes involved in protein disulphide bond formation.
Surprisingly, a putative C-terminal PTS1 in EasC appeared to be non-essential, at least in a yeast host background, and the cellular localization of EasC cannot be predicted. Further studies will be needed to address questions regarding cellular localization of the enzymes, and much work is still pending towards a full understanding of the biochemical reactions involved. Nonetheless, the knowledge we have obtained in this study should enable further elucidation of these reactions.
Importantly, we also demonstrate that the biosynthetic pathway for chanoclavine-I, the central biosynthetic precursor for all ergot alkaloids, can be transferred to the industrially important host, S. cerevisae. This discovery will greatly facilitate further genetic and metabolic engineering of the ergot alkaloid pathway, which will potentially have a broad range of pharmaceutical and agrochemical applications. Our results therefore provide an entry point on the road to commercial scale production of known or novel ergot alkaloids with important industrial applications.
Methods
DNA and protein sequence analysis
Computer-aided sequence analysis was done using Vector NTI 9.1.0 software (Invitrogen Corp. 2004) and the free online software FGENESH (http://linux1.softberry.com/berry.phtml), GENSCAN (http://genes.mit.edu/GENSCAN.html), and the NCBI server (http://www.ncbi.nlm.nih.gov). Signal peptides were predicted using the SignalP 4.0 tool [16].
Preparation and cloning of genes in yeast expression vectors
Synthetic genes, codon optimized for expression in yeast, were manufactured by DNA2.0 Inc., Menlo Park, CA, USA or GeneArt AG, Regensburg, Germany. All sequences were derived from A. japonicus, A. fumigatus, or C. purpurea. The genes encode the amino acid sequences, plus a translation stop codon, of DmaW_Af [GenBank: XP_756141], DmaW_Cp [GenBank:CAB39314], EasF_Af [GenBank:XP756143], EasC_Af [GenBank:XP756140], EasE_Af [GenBank:XP756142], and EasE_Cp [GenBank:CAB39328]. The sequences dmaW_Aj1, easF_Aj, easC_Aj, and easE_Aj were predicted, based on the genomic DNA sequence of the cycloclavine gene cluster of A. japonicus [22]. All genes were synthesized with the DNA sequence AAGCTTAAA, containing a HindIII restriction recognition site, at the 5’-end and with a SacII recognition site at the 3’-end (CCGCGG), and these sites were used for cloning. All PCR primers used for sub-cloning contained these sequences. Standard PCR conditions were used according to manufacturer’s recommendations (BioRad iProof High Fidelity DNA polymerase, Cat. #172-5302).
Gene dmaW_Aj3 was prepared by PCR using dmaW_Aj1 as template, and dmaW_Aj2 was prepared by sequential extension PCR, using dmaW_Aj3 as template, thus adding the second exon in two steps (Additional file 1: Table S1). The easC_Aj version without C-terminal PTS1 signal was prepared by PCR amplification of the coding sequence (CDS) without the 9 bps before the stop codon. The EasE_Aj N-terminal truncation (EasE_Aj -N sig.) was done by PCR, amplifying the corresponding CDS without the first 87 bps. The forward PCR primer inserted an alternative ATG translation start site. The fusion of an N-terminal signal peptide from Pdi1 to the N-truncated EasE_Aj was done by overlapping extension PCR, fusing the two amplicons to encode Pdi1-EasE_Aj (Additional file 1: Table S2). The CDS of the native yeast genes pdi1 [GenBank:D00842], ero1 [GenBank:NM_001182493], and fad1 [GenBank:NM_001180104] were amplified from genomic DNA by PCR.
For expression, all genes were cloned into expression vectors based on pRS313, pRS315, and pRS316 [35]. These vectors had been provided with a new multi-cloning site (MCS) linker, inserted between the two PvuII sites. The basic design of the MCS was SrfI-AscI-BglII-HindIII-SfiI(a)-SfiI(b)-SacII-SphI-AscI-SrfI [36]. The linker allowed cloning of promoter sequences into BglII and HindIII, and terminators into SacII and SphI restriction sites to create yeast expression cassettes. Promoters and terminators were amplified from yeast genomic DNA by PCR (Additional file 1: Table S3) for preparing three expression cassettes containing 1) a GPD1 promoter and a CYC1 terminator (G/C), 2) a PGK1 promoter and an ADH2 terminator (P/A), and 3) a CUP1 promoter and an ADH1 terminator (C/A). The new expression vectors were named pRS31X-G/C, pRS31X-P/A, and pRS31X-C/A, where X designates 3, 5, or 6. All genes used in this study (Table 1) were cloned into expression cassettes of these vectors.
Construction and integration of yeast gene expression cassettes
Constructs for integration were prepared for the integration sites YORWΔ22 and YPRCΔ15 [37], and cloned in unique EcoRI and HindIII sites of a pUC19 vector backbone. The homologous regions were constructed from two PCR fragments, which were then combined by overlapping extension PCR. The PCR primers introduced the restriction sites AscI and NotI between the two original fragments, and SbfI sites at the outer ends. The KanMX cassette, flanked by loxP sites, was excised from pUG6 [38] and inserted into NotI. Two expression cassettes (described above) were inserted into the AscI site. The first cassette was amplified by PCR, changing one AscI site to a MluI. After cloning this fragment, the second cassette was inserted into the single regenerated AscI site. The entire construct was released by SbfI and used for integration. This approach was used to first integrate dmaW_Aj2 (G/C cassette) and easF_Af (P/A cassette) into the yeast genome at the YORWΔ22 site. After excision of the KanMX marker [38], the easC_Aj (G/C cassette) and easE_Aj (P/A) cassette were integrated into the YPRCΔ15 site. Yeast transformations were done using the LiAc method [39].
Yeast strain and culture conditions
Chanoclavine-I production was achieved in several S. cerevisiae strain backgrounds, including S288C, W303, and others (not shown). The strain used to prepare the results in this report had the genotype MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0. Engineered yeast strains were grown in standard SC broth with 2% glucose, minus leucine and histidine (ForMedium, Hunstanton, U.K.). When appropriate, CuSO4 was added to a final concentration of 300 μM for induction of gene expression. Cultures for analysis were started from re-streaked, single colonies. These were grown overnight in standard SC broth, and then diluted to an optical density, at 600 nm, of 0.1 to start the main culture. Main cultures were grown at 30°C with constant shaking at 150 rpm, 5 cm amplitude, for 72 hours in 250 ml shake flasks containing 25 ml medium. Final optical density was measured at 600 nm, typically reaching 12–14, although small variations were seen between individual experiments.
Analytical procedures
For analysis yeast cultures were spun down for 10 min at 1000 × g. The pellet and the supernatant were separated. Without further purification, 5 μl of supernatant were injected in a UPLC-TOF (Waters Acquity™ Ultra Performance LC, Waters, Milford, Mass., USA) coupled to a micrOTOF-Q II (Bruker Daltonik GmbH, Bremen, Germany). Stationary phase was an Acquity UPLC® Bridged Ethyl Hybrid (BEH) C18, 1.7 μm, 2.1 × 100 mm column. Liquid chromatography used mobile phases of H2O + 0.1% formic acid (A), and acetonitrile + 0.1% formic acid (B), in a linear gradient of 1% to 100% B in 5 min. The column was washed for 1 min in 100% B, and then equilibrated for 1.5 min in 1% B. Detection of compounds was done by a photo diode array using the following parameters: λ range 210 nm to 500 nm. Resolution 1.2 nm. Sampling rate 5 points/s. ELSD parameters: gain 50, gas pressure 40 psi, nebulizer mode heating, power level 80%, drift tube 80°C. TOF parameters Source: End Plate Offset −500 V. Capillary −4500 V. Nebulizer 1.6 bar. Dry gas 8.0 l/min. Dry temperature 180°C. TOF parameters Scan mode: MS Scan. Mass range from 80 to 1000 m/z. Quantification of chanoclavine-I was based on in-house, purified reference compound.
Preparative procedures
For compound purifications yeast cultures were spun down for 10 min at 1000 × g. The supernatant was adjusted to pH = 10 with 10 M NaOH and extracted by liquid/liquid extraction with an equal volume of ethyl acetate. The crude extract was dried under vacuum and reconstituted with dimethyl sulfoxide (DMSO) to a concentration of 100 mg/ml and then purified on a preparative HPLC system (Waters, Milford, Mass, USA). Stationary phase was an XBridge™ preparative C18, 5 μm, 19 × 250 mm column. Liquid chromatography used mobile phases of H2O + 0.1% trifluoroacetic acid (A), and acetonitrile + 0.1% trifluoroacetic acid (B), in a linear gradient of 1% to 30% B in 40 min. The column was washed for 5 min in 100% B, and then equilibrated for 5 min in 1% B. Fractions were collected every 2 min and analyzed as above. Fractions containing the purified analyte were pooled and dried under vacuum.
Acknowledgements
The authors wish to thank Federico Brianza for advice on the chemical analysis, Jens Klein for solving the compound structures (NMR), and Donna Williams and Louise Hunter for technical support.
Additional file
Primers for construction of dmaW variant. Table S2. N-terminal sequences of EasE_Aj and Pdi1_Sc. Table S3. Expression cassettes in pRS vectors. Figure S1. Alignment of in silico translated CDS predictions of DmaW enzymes. Figure S2. NMR data of Me-DMAT and chanoclavine-I. Figure S3. Alignment of EasE proteins, showing conserved cysteine residues. Figure S4. Alignment of EasE proteins with two BBE proteins, indicating putative FAD binding. Figure S5. Alignment of EasE protein sequences derived from GenBank.
Footnotes
Competing interests
CAFN, CF, AH, and MN are employed by Evolva SA, and AM and HS by BASF SE. Both companies may eventually benefit commercially from the results presented here. SOC has no conflict of interest.
Authors’ contributions
HS, AM, and MN conceived and initiated the project. CAFN, AH, CF, and MN did the experimental design. CAFN did essentially all the molecular biology work including the yeast genetic engineering. AH and CF set up analytical methods, and carried out the analytical work. CF, AH, CAFN, and MN interpreted the experimental data. MN did DNA and protein sequence analysis. SOC provided field expertise and critical discussion. MN coordinated the study and drafted the manuscript, with critical contributions from SOC, CAFN, AH, and AM. All authors read and approved the final manuscript.
Contributor Information
Curt AF Nielsen, Email: curtn@evolva.com.
Christophe Folly, Email: christophef@evolva.com.
Anaëlle Hatsch, Email: anaelleh@evolva.com.
Andrea Molt, Email: andrea.molt@basf.com.
Hartwig Schröder, Email: hartwig.schroeder@basf.com.
Sarah E O’Connor, Email: sarah.oconnor@jic.ac.uk.
Michael Naesby, Email: naesby@evolva.com.
References
- 1.Schardl CL, Panaccione DG, Tudzynski P. Ergot alkaloids - biology and molecular biology. Alkaloids Chem Biol. 2006;63:45–86. doi: 10.1016/S1099-4831(06)63002-2. [DOI] [PubMed] [Google Scholar]
- 2.Haarmann TY, Rolke Y, Giesbert S, Tudzynski P. Ergot: from witchcraft to biotechnology. Mol Plant Pathol. 2009;10:563–577. doi: 10.1111/j.1364-3703.2009.00548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallwey C, Li SM. Ergot alkaloids: structure diversity, biosynthetic gene clusters and functional proof of biosynthetic genes. Nat Prod Rep. 2011;28:496–510. doi: 10.1039/c0np00060d. [DOI] [PubMed] [Google Scholar]
- 4.Tudzynski P, Correia T, Keller U. Biotechnology and genetics of ergot alkaloids. Appl Microbiol Biotechnol. 2001;57:593–605. doi: 10.1007/s002530100801. [DOI] [PubMed] [Google Scholar]
- 5.Tudzynski P, Hölter K, Correia T, Arntz C, Grammel N, Keller U: Evidence for an ergot alkaloid gene cluster inClaviceps purpurea.Mol Gen Genet 1999, 261:133–141. [DOI] [PubMed]
- 6.Unsöld IA, Li SM: Overproduction, purification and characterization of FgaPT2, a dimethylallyltryptophan synthase fromAspergillus fumigatus.Microbiology 2005, 151:1499–1505. [DOI] [PubMed]
- 7.Coyle CM, Panaccione DG: An ergot alkaloid biosynthesis gene and clustered hypothetical genes fromAspergillus fumigatus.Appl Environ Microbiol 2005, 71:3112–3118. [DOI] [PMC free article] [PubMed]
- 8.Fleetwood DJ, Scott B, Lane GA, Tanaka A, Johnson RD: A complex ergovaline gene cluster inepichloeendophytes of grasses.Appl Environ Microbiol 2007, 73:2571–2579. [DOI] [PMC free article] [PubMed]
- 9.Tsai HF, Wang H, Gebler JC, Poulter CD, Schardl CL: TheClaviceps purpureagene encoding dimethylallyltryptophan synthase, the committed step for ergot alkaloid biosynthesis.Biochem Biophys Res Commun 1995, 216:119–125. [DOI] [PubMed]
- 10.Rigbers O, Li SM. Ergot alkaloid biosynthesis in Aspergillus fumigatus. Overproduction and biochemical characterization of a 4-dimethylallyltryptophan N-methyltransferase. J Biol Chem. 2008;283:26859–26868. doi: 10.1074/jbc.M804979200. [DOI] [PubMed] [Google Scholar]
- 11.Lorenz N, Olsovská J, Sulc M, Tudzynski P: Alkaloid cluster gene ccsA of the ergot fungusClaviceps purpureaencodes chanoclavine I synthase, a flavin adenine dinucleotide-containing oxidoreductase mediating the transformation of N-methyl-dimethylallyltryptophan to chanoclavine I.Appl Environ Microbiol 2010, 76:1822–1830. [DOI] [PMC free article] [PubMed]
- 12.Goetz KE, Coyle CM, Cheng JZ, O’Connor SE, Panaccione DG: Ergot cluster-encoded catalase is required for synthesis of chanoclavine-I inAspergillus fumigatus.Curr Genet 2011, 57:201–211. [DOI] [PubMed]
- 13.Ryan KL, Moore CT, Panaccione DG: Partial reconstruction of the ergot alkaloid pathway by heterologous gene expression inAspergillus nidulans.Toxins 2013, 22:445–455. [DOI] [PMC free article] [PubMed]
- 14.Inglis DO, Binkley J, Skrzypek MS, Arnaud MB, Cerqueira GC, Shah P, Wymore F, Wortman JR, Sherlock G. Comprehensive annotation of secondary metabolite biosynthetic genes and gene clusters of Aspergillus nidulans, A. fumigatus, A. niger and A. oryzae. BMC Microbiol. 2013;13:91. doi: 10.1186/1471-2180-13-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thoms S, Debelyy MO, Connerth M, Daum G, Erdmann R: The putativeSaccharomyces cerevisiaehydrolase Ldh1p is localized to lipid droplets.Eukaryot Cell 2011, 10:770–775. [DOI] [PMC free article] [PubMed]
- 16.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 17.Haarmann T, Machado C, Lübbe Y, Correia T, Schardl CL, Panaccione DG, Tudzynski P: The ergot alkaloid gene cluster inClaviceps purpurea: extension of the cluster sequence and intra species evolution.Phytochemistry 2005, 66:1312–1320. [DOI] [PubMed]
- 18.Nevoigt E: Progress in metabolic engineering ofSaccharomyces cerevisiae.Microbiol Mol Biol Rev 2008, 72:379–412. [DOI] [PMC free article] [PubMed]
- 19.Chen Y, Daviet L, Schalk M, Siewers V, Nielsen J. Establishing a platform cell factory through engineering of yeast acetyl-CoA metabolism. Metab Eng. 2013;15:48–54. doi: 10.1016/j.ymben.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Borodina I, Nielsen J: Advances in metabolic engineering of yeastSaccharomyces cerevisiaefor production of chemicals.Biotechnol J 2014, 9:609–20. [DOI] [PubMed]
- 21.Mattanovich D, Sauer M, Gasser B. Yeast biotechnology: teaching the old dog new tricks. Microb Cell Fact. 2014;13:34. doi: 10.1186/1475-2859-13-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schröder H, Hoff B. Gene Cluster for Biosynthesis of Cycloclavine. US Patent Application. Geneva, Switzerland: WIPO; 2012. [Google Scholar]
- 23.Facchini FD, Vici AC, Benassi VM, Freitas LA, Reis RA, Jorge JA, Terenzi HF, Polizeli Mde L: Optimization of fibrolytic enzyme production byAspergillus japonicusC03 with potential application in ruminant feed and their effects on tropical forages hydrolysis.Bioprocess Biosyst Eng 2011, 34:1027–1038. [DOI] [PubMed]
- 24.Pradeep S, Faseela P, Josh MK, Balachandran S, Devi RS, Benjamin S: Fungal biodegradation of phthalate plasticizerin situ.Biodegradation 2013, 24:257–267. [DOI] [PubMed]
- 25.Bel-Rhlid R, Pagé-Zoerkler N, Fumeaux R, Ho-Dac T, Chuat JY, Sauvageat JL, Raab T: Hydrolysis of chicoric and caftaric acids with esterases andLactobacillus johnsoniiin Vitro and in a gastrointestinal model.J Agric Food Chem 2012, 60:9236–9241. [DOI] [PubMed]
- 26.Furuta T, Koike M, Abe M: Isolation of Cycloclavine from the Culture Broth ofAspergillus japonicusSAITO.Agric Biol Chem 1982, 46:1921–1922.
- 27.Delic M, Valli M, Graf AB, Pfeffer M, Mattanovich D, Gasser B. The secretory pathway: exploring yeast diversity. FEMS Microbiol Rev. 2013;37:872–914. doi: 10.1111/1574-6976.12020. [DOI] [PubMed] [Google Scholar]
- 28.Gasser B, Saloheimo M, Rinas U, Dragosits M, Rodríguez-Carmona E, Baumann K, Giuliani M, Parrilli E, Branduardi P, Lang C, Porro D, Ferrer P, Tutino ML, Mattanovich D, Villaverde A. Protein folding and conformational stress in microbial cells producing recombinant proteins: a host comparative overview. Microb Cell Fact. 2008;7:11. doi: 10.1186/1475-2859-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alcantara J, Bird DA, Franceschi VR, Facchini PJ. Sanguinarine biosynthesis is associated with the endoplasmic reticulum in cultured opium poppy cells after elicitor treatment. Plant Physiol. 2005;138:173–183. doi: 10.1104/pp.105.059287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winkler A, Motz K, Riedl S, Puhl M, Macheroux P, Gruber K. Structural and mechanistic studies reveal the functional role of bicovalent flavinylation in berberine bridge enzyme. J Biol Chem. 2009;284:19993–20001. doi: 10.1074/jbc.M109.015727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petrova VY, Drescher D, Kujumdzieva AV, Schmitt MJ. Dual targeting of yeast catalase A to peroxisomes and mitochondria. Biochem J. 2004;380:393–400. doi: 10.1042/BJ20040042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu Q, McAlister-Henn L. Peroxisomal localization and function of NADP + −specific isocitrate dehydrogenases in yeast. Arch Biochem Biophys. 2010;493:125–134. doi: 10.1016/j.abb.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kramer G, Boehringer D, Ban N, Bukau B. The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat Struct Mol Biol. 2009;16:589–597. doi: 10.1038/nsmb.1614. [DOI] [PubMed] [Google Scholar]
- 34.Feige MJ, Hendershot LM. Disulfide bonds in ER protein folding and homeostasis. Curr Opin Cell Biol. 2011;23:167–175. doi: 10.1016/j.ceb.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sikorski RS, Hieter P: A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA inSaccharomyces cerevisiae.Genetics 1989, 122:19–27. [DOI] [PMC free article] [PubMed]
- 36.Naesby M, Nielsen SV, Nielsen CA, Green T, Tange TO, Simón E, Knechtle P, Hansson A, Schwab MS, Titiz O, Folly C, Archila RE, Maver M, van Sint Fiet S, Boussemghoune T, Janes M, Kumar AS, Sonkar SP, Mitra PP, Benjamin VA, Korrapati N, Suman I, Hansen EH, Thybo T, Goldsmith N, Sorensen AS. Yeast artificial chromosomes employed for random assembly of biosynthetic pathways and production of diverse compounds in Saccharomyces cerevisiae. Microb Cell Fact. 2009;8:45. doi: 10.1186/1475-2859-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flagfeldt DB, Siewers V, Huang L, Nielsen J: Characterization of chromosomal integration sites for heterologous gene expression inSaccharomyces cerevisiae.Yeast 2009, 26:545–551. [DOI] [PubMed]
- 38.Güldener U, Heck S, Fielder T, Beinhauer J, Hegemann JH. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 1996;24:2519–2524. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]