

Abstract
The V0 complex forms the proteolipid pore of a vesicular ATPase that acidifies vesicles. In addition, an independent function in membrane fusion has been suggested in vacuolar fusion in yeast and synaptic vesicle exocytosis in fly neurons. Evidence for a direct role in secretion has also recently been presented in mouse and worm. The molecular mechanisms of how the V0 components might act or are regulated are largely unknown. Here we report the identification and characterization of a calmodulin-binding site in the large cytosolic N-terminal region of the Drosophila protein V100, the neuron-specific V0 subunit a1. V100 forms a tight complex with calmodulin in a Ca2+-dependent manner. Mutations in the calmodulin-binding site in Drosophila lead to a loss of calmodulin recruitment to synapses. Neuronal expression of a calmodulin-binding deficient V100 uncovers an incomplete rescue at low levels and cellular toxicity at high levels. Our results suggest a vesicular ATPase V0-dependent function of calmodulin at synapses.
Calmodulin (CaM)4 is a small, ubiquitous Ca2+ -binding protein that has been implicated in the Ca2+ -dependent regulation of a plethora of cell biological processes. Characterized roles during cellular development and function include metabolic regulation, response to inflammation, apoptosis, intracellular trafficking, and membrane fusion (1–3). The vacuolar or vesicular ATPase (V-ATPase) is a multisubunit proton pump that acidifies mostly intracellular compartments and consists of two sectors: V1 (the cytoplasmic ATPase) and V0 (the transmembrane proteolipid pore) (4, 5). The largest (100 kDa) subunit of V0, subunit a, is encoded by four orthologous genes, named a1–a4, in worm, mouse, and human.
The same four orthologous genes exist in Drosophila, but a fifth gene encoding a less homologous variant is listed in the public database as vha100-3. In yeast, subunit a is the only V0 component encoded by more than one gene, namely the two homologs, vph1 and stv1. The two yeast versions or four animal versions of subunit a principally exhibit cell-specific and intracellular compartment-specific distribution; in many cases, they have been shown to target the complex to separate compartments (e.g. Vph1 to the vacuole and Stv1 to endosomal compartments (6, 7)). Of the four orthologous genes in animals, subunit a1 has consistently been shown to be highly enriched in, if not restricted to, neurons. In Drosophila, neuronal expression of subunit a1 in homozygous mutant animals fully rescues the lethality associated with the loss of a1, demonstrating its sole requirement in the nervous system (8).
Recently, subunit a has been implicated in membrane fusion or secretion independent of intracompartmental acidification. These roles include fusion of yeast vacuoles (9), synaptic vesicle exocytosis in fly neurons (8), exocytosis in apical secretion of exosomes and morphogens in worms (10, 11), and insulin secretion from pancreatic islets in mice (12). Moreover, in the endocytic degradative pathway, subunit a2 has recently been shown to exert another function independent of proton pumping; subunit a2 interacts with the guanine nucleotide-exchange factor ARNO in a pH-dependent manner, suggesting its function as a pH sensor for controlling the recruitment of the GTPase activated by ARNO, Arf6, which in turn directly interacts with the V0 sector (13). Finally, a recent analysis of another tissue-specific V0 sector component, subunit d2, revealed a defect of osteoclast fusion independent of acidification (14). How the putative multiple functions of the V0 sector, and subunit a in particular, are regulated is largely unknown.
Here, we report the identification and characterization of a CaM-binding site in subunit a1 (or V100) of Drosophila. CaM interacts with V100 in a Ca2+ -dependent, high affinity manner. Ablation of this binding site results in a loss of CaM recruitment to synapses. Expression of V100 with a mutated CaM-binding site at low levels leads to incomplete rescue of the neuronal function, and high levels of expression lead to cell death. Our data uncover a critical regulatory function of CaM that controls genetically separable V100 and possibly V-ATPase functions.
EXPERIMENTAL PROCEDURES
Cloning, Mutagenesis, and Purification of Proteins
The DNA sequence of the 407-residue N-terminal region (V100-N) of the fly neuronal a1 was amplified by PCR, cloned into pET44 plasmid, and expressed in Escherichia coli BL21 (DE3). The protein was purified initially using TALON beads (Clontech). After removing the His6-Nus tag by proteolysis, the V100-N was further purified onto a gel filtration column. Recombinant CaM was purified following a published procedure (15). Mutations of V100-N were introduced using QuikChange mutagenesis kit (Stratagene) and verified by DNA sequencing.
Pulldown and Sizing Exclusion Chromatography Assays
His-Nus-tagged native and mutant V100-N protein (0.2 mg) anchored on TALON beads (Clontech) was used as bait. The beads were incubated with CaM (0.22 mg) in the cold room for 3 h and washed extensively with 20 mM HEPES, pH 7.5, 0.5 M NaCl, 10% glycerol, 0.2% CHAPS, 5 mM CaCl2 and then subjected to SDS-PAGE for visualization. For the analytical gel filtration chromatography, the untagged wild type or mutant V100-N and CaM were mixed to a volume of 0.25 ml before loading onto a Superdex 200 HR 10/30 column (GE Healthcare).
Fluorescence Measurements
The peptide of the CaM-binding domain (CBD) of the fly V100 (314NLKNWFVKVRKIKAIYHTLNLFNLD338) was synthesized commercially (Bio-Synthesis, Inc., Lewisville, TX). Both CaM and CBD peptide were prepared in 5 mM NaCl, 5 mM CaCl2, and 20 mM HEPES, pH 7.5. The concentration of Drosophila CaM and CBD peptide were determined by the 280-nm absorption using extinction coefficients of 1280 and 6970 M−1 cm−1, respectively. For the titration, 1- or 2-μl aliquots of 15 or 45 μM Ca2+ ·CaM stock solution was added to 0.2 μM peptide in 3-ml volume at room temperature. The change in fluorescence was recorded on the SLM8000C fluorometer by tryptophan excitation at 295 nm and the emission spectrum from 300 to 400 nm. The titration experiments were repeated three times. The titration curve, using the normalized change in fluorescence at 340 nm, was fitted into a quadratic function (below) with SigmaPlot (SPSS) for a 1:1 stoichiometry of binding.
| (Eq. 1) |
where Kd is the apparent dissociation constant for the Ca2+ ·CaM (CaM) binding to V100 CBD peptide (Ppt).
Mutagenesis and Generation of V100-CaM-binding Deficient Transgenic Flies
V100 was subjected to site-directed mutagenesis using full-length cDNA in a pOT2 vector ((Berkeley Drosophila Genome Project) clone LD21248) and the following primers: 5′-CCAAGAATCTTAAGAACGCGGCCGTCAAGGTGCGC-3′ and its reverse complement. Introduction of the double mutation W318A and F319A was verified by sequencing. The resulting v100-WF cDNA was cloned into pUAST using EcoR1 and Xho1 restriction cutting sites as reported previously (8). DNA injection for the generation of transgenic flies was performed by Rainbow Transgenics, Inc. At least two independent transgene insertions per chromosome were isolated, mapped, and tested for expression in photoreceptors using GMR-Gal4 (16) and an anti-V100 polyclonal antibody (8).
In Vivo Expression Analysis and Immunohistochemistry
Rescue experiments by pan-neuronal or photoreceptor-specific expression of v100 and v100-WF in mutant backgrounds were performed as described previously (8). Immunohistochemistry was performed on whole mount larval and adult brains according to standard protocols described in the same reference. Antibodies used are: anti-V100 (1:2000 (8)); anti-CaM (1:500; Invitrogen); anti-Chaoptin (1:100 (17)). Images were obtained with a Leica SP5 confocal microscope and visualized using Amira 4.1.1 (Mercury, Inc.).
RESULTS
V100 Contains a Specific Site for Tight Binding of Ca2+ ·CaM
A critical regulatory role of CaM in V0-dependent membrane fusion has been shown only in yeast vacuolar fusion (18, 19). Reconstitution of the pore-forming V0 proteolipids into choline-loaded liposomes revealed that choline release from these liposomes is strictly Ca2+ ·CaM-dependent (19), resembling earlier reconstitution experiments of V0 proteolipids from neurons and acetylcholine release (20, 21). A search for the presence of a CBD in the primary sequences of all V0 sector protein components (subunits a, c, and d) of the Drosophila neuron V-ATPase hinted at the large N-terminal cytosolic region (V100-N) of subunit a1, suggesting that subunit a1 might be a direct target of CaM regulation. Therefore, purified recombinant V100-N was tested for its ability to bind CaM. Three different mixtures were subjected to analytical gel filtration (Fig. 1): A, the 46-kDa V100-N alone; B, a mixture of V100-N and the 16-kDa CaM with EDTA in the liquid mobile phase; and C, a mixture of V100-N and CaM in the identical ratio as in B onto the column equilibrated and eluted with a buffer containing Ca2+ (no EDTA). In the presence of EDTA (Fig. 1B), the peak fraction 1 is ascribed to V100-N because it matches exactly that of V100-N alone (Fig. 1A). The slower eluting peak fraction 2 represents CaM. Replacement of the EDTA by Ca2+ (Fig. 1C) caused the appearance of a faster eluting peak (peak 3), along with the disappearance of peak 1 shown in Fig. 1B. This finding indicates that peak 3 is associated with the formation of a tight complex between V100-N and Ca2+ ·CaM. To provide additional evidence, SDS-PAGE and Coomassie Blue staining were performed to determine the protein compositions of the various fractions (shown in Fig. 1, E and F). Peak 3, shown in Fig. 1C, is indeed composed of V100-N and Ca2+ ·CaM, which correspond to Fig. 1F, the two bands of lane 2. Peak 4 is unbound excess Ca2+ ·CaM (Fig. 1F, lane 3). For the gel filtration of V100-N and Ca2+ ·CaM in the presence of EDTA (Fig. 1B), the protein compositions (panel E) show that peak 1 is V100-N (lane 3), and peak 2 is a mixture of mostly Ca2+ ·CaM and some V100-N (lane 4) due to the inability to resolve overlapping peaks 1 and 2. Because of the formation of the V100-N-Ca2+ ·CaM complex (Fig. 1F, peak 3), the V100-N associated with peak 1 could no longer be detected (Fig.1B, cf. lanes 3 and 4). We conclude from the gel filtration results that the N-terminal cytosolic region of V100 forms a tight complex with CaM in a calcium-dependent manner and therefore must contain a specific CBD.
FIGURE 1. Characterization of binding of CaM to V100-N in a Ca2+ -dependent manner assessed by analytical gel filtration.
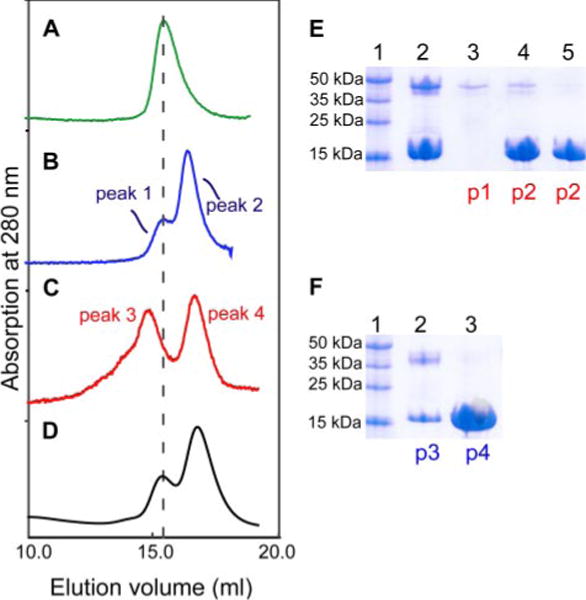
Left row shows the following chromatograms: A, V100-N (0.06 mM); B, V100-N (0.06 mM) and CaM (0.15 mM) eluted with a buffer containing 2 mM EDTA; C, V100-N and CaM (same molar ratio as in B) with the mobile phase containing 5 mM Ca2+; D, identical to C, but with V100-N F319A mutant. In B, C, and D chromatograms, the molar ratio of V100-N to CaM is 1:2.5 with identical volume (0.25 ml) of each protein mixture loaded onto the column. Right row (E and F) shows the results of the SDS-PAGE and Coomassie Blue staining of the different fractions from the gel filtrations. The fractions were concentrated to roughly the same volume. p1–p4 correspond to the different elution peaks identified in the gel filtrations (B and C profiles). E, lane 1, protein markers; lane 2, a mixture of Ca2+ ·CaM and V100-N before loading; lane 3, peak 1; lane 4, first fraction from the peak of peak 2; lane 5, second fraction from the descending slope of peak 2. F, lane 1, protein markers; lane 2, peak 3; lane 3, peak 4.
CBDs of target proteins are generally small, contiguous amino acid sequences of about 14–26 amino acids in length, containing a number of important hydrophobic and basic residues and having the propensity to form an amphiphilic α helix (22, 23). A key feature of most of these domains is the presence of two large hydrophobic residues that are located at position 1 close to the amino end and position 10 or 14 near the C-terminal end (24). Each pair (1–10 or 1–14) of hydrophobic residues serves as anchors to dock the helical peptide in the hydrophobic pockets of the two globular lobes of Ca2+ ·CaM, often in a manner in which the N- and C-terminal hydrophobic residues bind to the pockets in the C- and N-terminal lobes, respectively, of Ca2+ ·CaM (25, 26). The middle portion of the long central helix linking the two lobes is capable of uncoiling into loops of varying sizes for optimal positioning of the pair of hydrophobic residues to the lobes (25–27). In searching the presence of CBDs in the entire primary sequence of the fly V100 using an online search program (28), only two segments, located in the V100-N region, were identified to be potential CBDs: Segment 1, 245FIIFFQGDQLKTRVKKICEGFRATLYP271, and Segment 2, 314NLKNWFVKVRKIKAIYHTLNLFNLD338. Bold and underlined aromatic/large aliphatic residues represent either 1–10 or 1–14 pairs of hydrophobic residues.
Segment 1 has the 1–14 (Phe-248 and Ile-259) type pair of anchor hydrophobic residues, but secondary structure prediction suggests that only a portion of the segment (252–265) may assume an α helical configuration. Segment 2, which is predicted to be entirely α helical, contains an unusual combination of both 1–10 (Phe-319–Ile-328) and 1–14 (Phe-319–Ile-332) hydrophobic motifs. To identify which of the two segments is the CBD, single or double mutations of the potentially important hydrophobic residues in either segment, especially those confined to the N termini, were introduced in V100-N. The effects of the mutations on binding of Ca2+ ·CaM were first evaluated by pulldown assays, with the mutated His6-tagged V100-N bound to nickel beads serving as the bait (supplemental Table S1). The single mutation F248A or F249A and double mutations F248A and I261A in Segment 1 had no effect on Ca2+ ·CaM binding. However, mutations of either Trp-318 or Phe-319 in Segment 2 abrogated Ca2+ ·CaM binding. The effects of the double mutations in Segment 2 (F319A+I328 and F319+L332) could not be tested because the mutations resulted in insoluble V100-N proteins. Based on these preliminary results, we further examined the effects of the mutations using analytical gel filtration as described above, and the results are consistent with the pulldown assays (supplemental Table S1 and Fig. S1). For example (Fig. 1D), the gel filtration profile of the mixture of V100-N with the F319A mutation in Segment 2 and Ca2+ ·CaM is virtually identical to that shown in Fig. 1B, which portrays no complex formation between wild type V100-N and CaM in the presence of EDTA. Taken together, our binding studies and mutagenesis experiments indicate the existence of a CBD in V100, which is confined to Segment 2.
To simplify the study of the CaM interactions, synthetic peptides comprising the CBDs are normally used instead of the large intact target enzymes or proteins. This system is in most cases an excellent mimic for the interaction of CaM with the whole target proteins. For example, the Kd values, many in the nM range, of the complexes of Ca2+ ·CaM with intact targets and with their corresponding CBD peptides are very similar (22, 29). To provide further evidence that Segment 2 is a bona fide CBD, we determined the binding affinity of a synthetic peptide of the segment (residues 314–338) using intrinsic fluorescence measurements. Because CaM is devoid of a tryptophan residue, we surmised that the tryptophan fluorescence of Trp-318 of the peptide (emission maximum at 340 nm) could be exploited to follow the peptide binding to Ca2+ ·CaM. The addition of Ca2+ ·CaM to the peptide did cause an ~50% increase in fluorescence intensity and an ~10-nm blue shift (Fig. 2A), consistent with a hydrophobic interaction between the tryptophan and CaM. Importantly, these spectral changes do not occur in the presence of EDTA. Titration experiments showed that the intensity at the maximum emission wavelength reached a plateau when about 1 molar eq. of Ca2+ ·CaM was added to the peptide solution (Fig. 2B). Fitting of the titration curve yielded a Kd of 21.5±3.6 nM, a value indicative of a CBD with very tight affinity for Ca2+ ·CaM.
FIGURE 2. Binding of CaM to V100 CBD peptide assessed by Trp fluorescence.
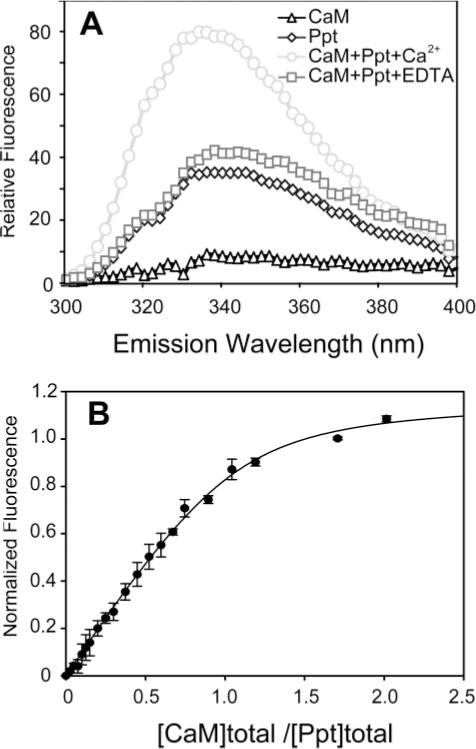
A, fluorescence emission spectra of CaM (0.3 μM, triangle) and the CBD peptide (314NWFVKVRKIKAIYHTLNLFNLD338) of segment 2 (named Ppt) (0.3 μM, diamond) and their complex in the presence of 5 mM Ca2+ (circle) or 10 mM EDTA (square). The spectra were obtained in 5 mM NaCl and 20 mM HEPES, pH 7.5 (see “Experimental Procedures”). The fluorescence emission was measured from 300 to 400 nm with the excitation wavelength set at 295 nm. B, fluorescence titration of Ca2+ ·CaM to V100 CBD peptide. The titration curve, using the normalized change in fluorescence at 340 nm, was fitted as described in “Experimental Procedures.” The fitting correlation coefficient (R2) is 0.98.
In summary, our biochemical data demonstrate that Segment 2 is a Ca2+ -dependent, high affinity CBD in V100. Comparison of the orthologous regions in other subunit a1 genes reveals a high conservation of the CBD (Fig. 3A). In contrast, all a2–a4 variants across species have a less conserved putative CBD (Fig. 3B). Whether these weaker a2–a4 CBD-homologous regions are capable of binding CaM remains to be tested for each individual case. The strong a1-specific conservation of the CBD tested here suggests that Ca2+ ·CaM regulation is employed for specific subunit a1 function, most likely having critical regulatory function in the nervous system.
FIGURE 3. Alignment of the amino acid sequences of the CBD segments in the V-ATPase V0 subunit a in fly, human, mouse, worm, and yeast.
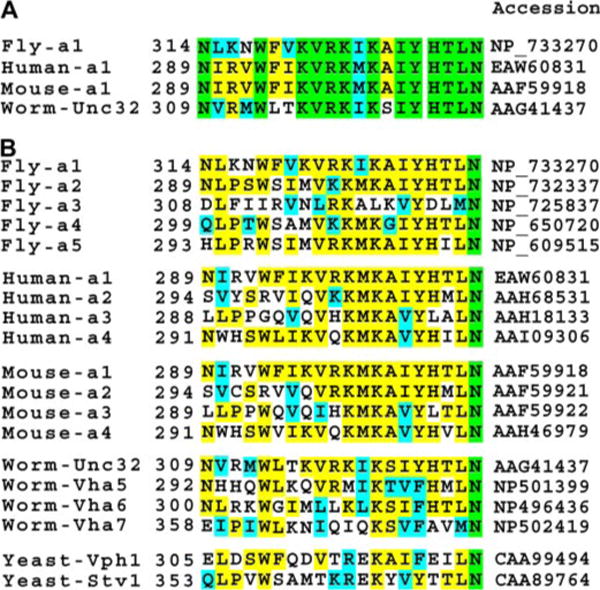
The alignment is based on the alignment of the entire sequences of the various homologues. Residues in green background are invariant; yellow background, identical residues; cyan, similar residues. A, alignment of CBDs of only the a1 orthologs. B, alignment of the different subunit a orthologs. Sequence comparison of the fly subunit a homologues shows significantly high sequence identity among a1, a2, a4, and a5 (>50%), whereas a3 shares only 36% identity. Thus, there are only four major homologues (a1, a2, a4, and a5) in fly, which corresponds to the usual four orthologs present in other mammalian species. Worm Unc32, Vha5, Vha6, and Vha7 correspond to subunits a1, a2, a3, and a4, respectively. The yeast Vph1 segment is similar to those of subunit a1s largely due to the presence of the 1–10 and 1–14 pairs of hydrophobic residues, as well as Trp preceding the residue at position 1. However, the unusual presence of several acidic residues sets Vph1 apart from the other subunit a segments.
CaM Is a Critical Regulator of V100 in Drosophila Neurons
In Drosophila, the V0 subunit a1 V100 is required for synaptic vesicle exocytosis independent of vesicle acidification (8). We therefore wondered whether CaM regulation is required for V100 function in vivo. To test this possibility, we mutated two key residues of the CaM-binding site, Trp-318 and Phe-319, to alanine and generated transgenic flies for tissue-specific expression of v100WF. Neuronal expression (using the neuron-specific elav-Gal4 driver and UAS wild type v100 (30)) in homozygous mutants is sufficient to rescue viability to adulthood in flies (8). In contrast, expression of v100WF is not sufficient to rescue mutant animals to adulthood. However, in contrast to the mutants, embryos with neuronal v100WF expression hatch as L1 larvae and exhibit contraction waves and movement. The larvae are progressively sluggish and die within 2–3 days. Interestingly, we found a dosage-dependent effect of v100WF expression; high levels of neuronal v100WF in mutant and importantly also wild type backgrounds lead to increased lethality, suggesting a dominant negative effect. To investigate whether this effect is cell-autonomous, we performed analogous rescue experiments in photoreceptors, which are not required for the viability of the organism. Surprisingly, photoreceptor-specific expression of v100WF causes cell lethality in a dosage-dependent manner (Fig. 4, D–F). In contrast, neither overexpression of wild type v100 nor the complete loss of v100 in photoreceptors causes obvious developmental defects (Fig.4, B and C). Hence, v100WF does not act as a classical dominant negative but exerts a neomorphic toxic function. These results suggest that interference with CaM binding releases V100 from a critical CaM-dependent regulation.
FIGURE 4. Expression of calmodulin-binding deficient V100 exhibits dosage-dependent cellular toxicity in Drosophila photoreceptors.
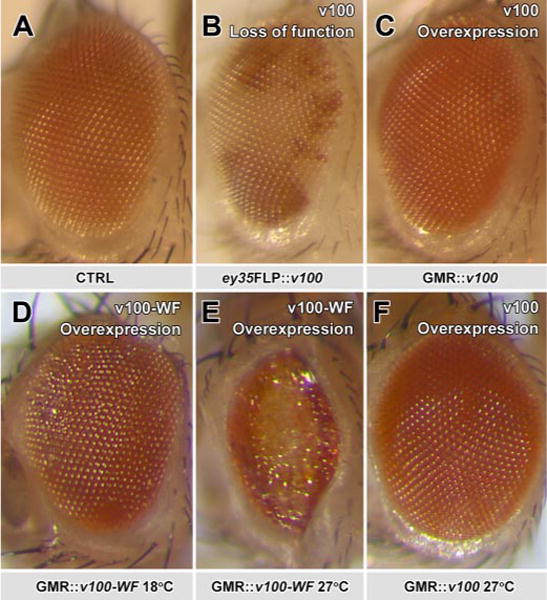
A–C, pictures of a fly eye for control, loss, and gain of V100. All are morphologically normal. D–F, in contrast, overexpression of V100-WF causes mild developmental defects when expressed at low levels (by raising flies at 18 °C) and loss of photoreceptor neurons at high levels (27 °C). At the same high temperature, wild type v100 does not affect eye morphology (F). All effects were verified for three independent insertions of UAS-v100 and UAS-V100-WF.
CaM-binding Deficient V100 Correctly Localizes to Synapses but Fails to Recruit CaM
If expressions of V100 and V100WF have different activities because of altered CaM binding, this difference could be reflected in mislocalized V100, CaM, or both. We therefore investigated CaM and V100 localization when V100 or V100WF are expressed in v100 mutant photoreceptors, a replacement that fully rescues when wild type V100 is expressed. Both V100 and V100WF exhibit very similar expression profiles with strong enrichment of the protein in synaptic terminals (Fig. 5, A and B, red; arrows). Hence, the V100WF is correctly made and intracellularly sorted. Importantly, these data further indicate that CaM binding is not required for the correct localization of V100. CaM is predominantly and ubiquitously found in the cell bodies (Fig. 5, A and B, cb). However, as shown in Fig. 5, C and D, a weaker, punctate CaM labeling is also apparent in high resolution confocal scans of photoreceptor synapses. Importantly, this CaM labeling (green) is strongly enriched only in v100 mutant synaptic terminals rescued with wild type V100 expression (Fig. 5, C and E) but not V100WF expression (Fig. 5, D and F). These data indicate that V100 can recruit CaM to the synapse but that this recruitment is abolished when V100 lacks its CaM-binding site. In contrast, we found no obvious mislocalization of CaM in the cell bodies or axons of developing photoreceptors (supplemental Fig. S2), suggesting that recruitment of CaM occurs specifically at synapses. Taken together, our in vivo data indicate that CaM critically regulates V100 in neurons and that CaM binding is not required for the correct localization of V100 but for the recruitment of CaM to synapses.
FIGURE 5. The CaM-binding domain in V100 is not required for V100 localization, but CaM is recruited to synapses.
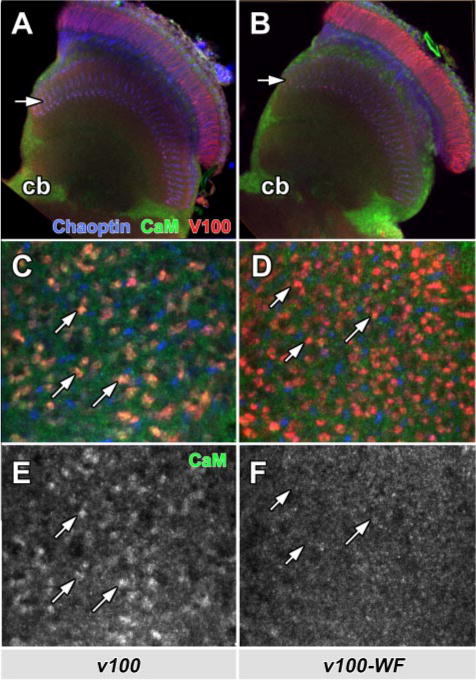
Shown are adult optic lobes from whole mount brain dissection showing photoreceptor projections in the brain. Photoreceptors are rendered mutant using the ey35FLP system and replaced with wild type (A, C, and E) or mutant (B, D, and F) V100. Blue, Chaoptin (24B10, a photoreceptor-specific marker); red, anti-V100; green, anti-CaM. A and B, in the optic lobes, V100 staining reveals the overexpressed V100 as well as V100-WF protein strongly enriched in synaptic layers (arrows), whereas Chaoptin (blue) primarily marks the axons. CaM is mostly detected in neuronal cell bodies (cb). C–F, high resolution lamina cross-section (upper arrows in A and B). V100 labels individual synaptic photoreceptor terminals. Only synaptic terminals enriched for wild type V100, but not V100-WF, exhibit increased levels of CaM at synapses (arrows).
DISCUSSION
We have identified a high affinity CBD that is specific to the neuronal variant of the V-ATPase V0 subunit a1 (V100) in Drosophila. Disruption of this CBD impairs recruitment of CaM to synapses as well as the V100 function required for viability. High levels of CBD-defective V100 reveal cellular toxicity, a phenomenon that is not associated with loss or gain of wild type v100. Our data therefore indicate that V100 is critically regulated by CaM in neuronal system.
There are important differences between yeast homotypic vacuolar fusion and tightly regulated exocytosis at synapses, especially with respect to speed (31, 32). Nevertheless, intriguingly, it has been shown previously that Ca2+ ·CaM regulates yeast vacuolar fusion by binding tightly to the 16-kDa proteolipid V0 subunit c, Vma3, which forms a hexameric channel (19). A problem with this model is that the primary sequence of subunit c does not contain a CaM-binding segment that is even remotely similar to that found in several V0 subunit a proteins or in many other enzymes and structural proteins that bind tightly and are regulated by Ca2+ ·CaM (29). Moreover, the subunit c has been predicted to fold into four transmembrane helices connected by loops, which are presumably exposed but too short to allow the formation of helical segments for any CaM binding. Indeed, the crystal structure of a ring containing 10 proteolipid subunits of a bacterial V-type Na+ -ATPase (Protein Data Bank code 2BL2) does show that the two loops of a subunit connecting two pairs of helices and lying on the cytoplasmic side are composed of no more than four residues (33). On the other hand, the yeast subunit a homolog Vph1, which has been shown to be involved in vacuolar fusion downstream of trans-SNARE docking (9, 18, 19), has a CBD, largely on account of the presence of the 1–10 (Phe-310–Ile-319) or 1–14 (Phe-310–Ile-323) pairing of hydrophobic residues (Fig. 3B). Thus, if Ca2+ ·CaM is involved in yeast vacuolar fusion, its target may indeed be Vph1.
Like the Drosophila subunit a1 studied here, the worm ortholog, Unc32, exhibits both a highly conserved CBD (Fig. 3B) and predominantly neuronal expression (34). The apical secretion of exosomes that is mediated by Vha5, another worm subunit a homolog (10), may also be regulated by Ca2+ ·CaM. In contrast, the absence of the hydrophobic residue at position 1, as well as the preceding tryptophan, in the putative CBD of the human a2 suggests little or no involvement of Ca2+ ·CaM. Mouse a3 similarly lacks the large hydrophobic residue at position 1 but has the preceding tryptophan. The fact that the neuronal type CBD is not a universal feature of “subunit a” proteins suggests that only a subset of V-ATPases (or V0 sectors) is regulated by Ca2+ ·CaM. It further suggests that the general function of V-ATPase as a proton pump may not require Ca2+ ·CaM.
CaM regulation of V100 may represent regulation of only subunit a, the V-ATPase V0 sector, or the complete V-ATPase. Does CaM binding separate a function in membrane fusion from a function in proton pumping? Evidences from yeast and Drosophila support this idea. First, Vph1, but not Stv1 (which lacks CBD), is required for yeast vacuolar fusion independent of intracompartmental acidification (9). Second, the Drosophila subunit a1 V100 (which has the highly conserved a1-type CBD) is required for synaptic vesicle exocytosis independent of vesicle acidification (8). In the case of the C. elegans subunit a2, Vha-5, it is unclear at this point whether it is regulated by Ca2+ ·CaM. However, Vha-5 is required for two genetically separable functions, one most consistent with secretion and the other most consistent with acidification (10, 11). Similarly, subunit a3 in mouse has also been implicated in a secretory defect independent of acidification (12). Taken together with a recent mouse knock-out of the V0 subunit d2, which resulted in a cell fusion defect of osteoclasts independent of acidification (14), evidence is mounting for a secretory function of the V-ATPase V0 sector. However, whether only one function of V0 is selectively regulated by CaM binding, and if so which, or whether CaM exerts different functions depending on the context a particular V-ATPase is employed in, remains to be determined.
Importantly, neither loss nor gain of v100 in Drosophila neurons causes cell lethality. In contrast, V100WF is toxic for the cell in a dosage-dependent manner. Taken together with the failure of V100WF to recruit CaM to synapses, the neomorphic toxicity may be caused either by the mislocalization of CaM or by the mutant V100 protein itself. As complete loss of v100 (and thus assumedly a corresponding loss of V100-dependent CaM recruitment to the synapse) does not cause cellular toxicity, we conclude that at least this phenotype cannot be caused by a V100-independent function of CaM. The V100WF protein itself may be partially misfolded; however, the mutant and wild type proteins exhibit identical localization and no obvious aggregation when expressed in photoreceptors (cf. Fig. 5 and supplemental Fig. S2). These data indicate that the mutant protein is stable and intracellularly sorted like the wild type protein. We can envision two scenarios as to how v100WF expression might lead to cell lethality; both are based on the idea that interference with CaM binding releases V100 from normally tight Ca2+ ·CaM-dependent regulation. In the first model, overexpression of v100WF leads to more promiscuous engagement in V-ATPase V0 complexes that are normally serviced by subunit a2–a4 and required for cell viability. Removal of V-ATPase activity by loss of at least one V1 subunit (vha55) indeed causes cell lethality in photoreceptors5. In the second model, release of V100 from Ca2+ ·CaM regulation allows V100 to function similarly to overexpression of the t-SNARE syntaxin (35). In photoreceptors, both V100WF as well as syntaxin exert dosage-dependent toxicity when overexpressed (8). In view of this, the neomorphic toxicity of v100WF would be caused by an “overactive” protein due to lack of CaM-constrained regulation (35). We cannot distinguish between these possibilities currently, as the molecular mechanisms of both syntaxin and V100WF overexpression require further investigation.
In conclusion, our studies have shown that CaM not only binds tightly to the V0 sector subunit a1 in a calcium-dependent manner, but also critically regulates V100 in neurons. The initial work presented here paves the way for further in vitro and in vivo investigations to deepen our understanding of the biological roles of the interaction between CaM and V0.
Supplementary Material
Acknowledgments
We thank Jing He and Robert L. Welschhans in the F. A. Q. laboratory and Victor Galanis in the P. R. H laboratory for technical assistance. We greatly appreciate the gifts of plasmid of the Drosophila CaM and anti-CaM antibody from Dr. Kathleen M. Beckingham (Rice University).
Footnotes
This work was supported by the Welch Foundation (Grants Q581 to F. A. Q. and I-1657 to P. R. H.) and Eugene McDermott Endowed Scholarship (to P. R. H.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1 and S2.
The abbreviations used are: CaM, calmodulin; CBD, CaM-binding domain; V-ATPase, vesicular ATPase; V0, the transmembrane proteolipid complex of V-ATPase; V100, Vha100-1 in Drosophila; V100-N, N-terminal cytoplasmic region of V100; SNARE, soluble NSF attachment protein receptors; NSF, N-ethylmaleimide-sensitive factor; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid.
P. Robin Hiesinger, unpublished data.
References
- 1.Chin D, Means AR. Trends Cell Biol. 2000;10:322–328. doi: 10.1016/s0962-8924(00)01800-6. [DOI] [PubMed] [Google Scholar]
- 2.Saimi Y, Kung C. Annu Rev Physiol. 2002;64:289–311. doi: 10.1146/annurev.physiol.64.100301.111649. [DOI] [PubMed] [Google Scholar]
- 3.Steinhardt RA, Alderton JM. Nature. 1982;295:154–155. doi: 10.1038/295154a0. [DOI] [PubMed] [Google Scholar]
- 4.Nelson N, Harvey WR. Physiol Rev. 1999;79:361–385. doi: 10.1152/physrev.1999.79.2.361. [DOI] [PubMed] [Google Scholar]
- 5.Nishi T, Forgac M. Nat Rev Mol Cell Biol. 2002;3:94–103. doi: 10.1038/nrm729. [DOI] [PubMed] [Google Scholar]
- 6.Manolson M, Wu B, Proteau D, Taillon B, Roberts B, Hoyt M, Jones E. J Biol Chem. 1994;269:14064–14074. [PubMed] [Google Scholar]
- 7.Perzov N, Padler-Karavani V, Nelson H, Nelson N. J Exp Biol. 2002;205:1209–1219. doi: 10.1242/jeb.205.9.1209. [DOI] [PubMed] [Google Scholar]
- 8.Hiesinger PR, Fayyazuddin A, Mehta SQ, Rosenmund T, Schulze KL, Zhai RG, Verstreken P, Cao Y, Zhou Y, Kunz J, Bellen HJ. Cell. 2005;121:607–620. doi: 10.1016/j.cell.2005.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bayer MJ, Reese C, Buhler S, Peters C, Mayer A. J Cell Biol. 2003;162:211–222. doi: 10.1083/jcb.200212004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liegeois S, Benedetto A, Garnier JM, Schwab Y, Labouesse M. J Cell Biol. 2006;173:949–961. doi: 10.1083/jcb.200511072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liegeois S, Benedetto A, Michaux G, Belliard G, Labouesse M. Genetics. 2007;175:709–724. doi: 10.1534/genetics.106.066035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun-Wada GH, Toyomura T, Murata Y, Yamamoto A, Futai M, Wada Y. J Cell Sci. 2006;119:4531–4540. doi: 10.1242/jcs.03234. [DOI] [PubMed] [Google Scholar]
- 13.Hurtado-Lorenzo A, Skinner M, Annan JE, Futai M, Sun-Wada GH, Bourgoin S, Casanova J, Wildeman A, Bechoua S, Ausiello DA, Brown D, Marshansky V. Nat Cell Biol. 2006;8:124–136. doi: 10.1038/ncb1348. [DOI] [PubMed] [Google Scholar]
- 14.Lee SH, Rho J, Jeong D, Sul JY, Kim T, Kim N, Kang JS, Miyamoto T, Suda T, Lee SK, Pignolo RJ, Koczon-Jaremko B, Lorenzo J, Choi Y. Nat Med. 2007;12:1403–1409. doi: 10.1038/nm1514. [DOI] [PubMed] [Google Scholar]
- 15.Putkey J, Slaughter G, Means A. J Biol Chem. 1985;260:4704–4712. [PubMed] [Google Scholar]
- 16.Freeman M. Cell. 1996;87:651–660. doi: 10.1016/s0092-8674(00)81385-9. [DOI] [PubMed] [Google Scholar]
- 17.Zipursky SL, Venkatesh TR, Teplow DB, Benzer S. Cell. 1984;36:15–26. doi: 10.1016/0092-8674(84)90069-2. [DOI] [PubMed] [Google Scholar]
- 18.Peters C, Mayer A. Nature. 1998;396:575–580. doi: 10.1038/25133. [DOI] [PubMed] [Google Scholar]
- 19.Peters C, Bayer MJ, Buhler S, Andersen JS, Mann M, Mayer A. Nature. 2001;409:581–588. doi: 10.1038/35054500. [DOI] [PubMed] [Google Scholar]
- 20.Israel M, Meunier FM, Morel N, Lesbats B. J Neurochem. 1987;49:975–982. doi: 10.1111/j.1471-4159.1987.tb00989.x. [DOI] [PubMed] [Google Scholar]
- 21.Morel N, Dunant Y, Israel M. J Neurochem. 2001;79:485–488. doi: 10.1046/j.1471-4159.2001.00611.x. [DOI] [PubMed] [Google Scholar]
- 22.Van Eldik LJ, Watterson M. Calmodulin and Signal Transduction. Academic Press; New York: 1998. [Google Scholar]
- 23.O’Neil KT, DeGrado WF. Trends Biochem Sci. 1990;15:59–64. doi: 10.1016/0968-0004(90)90177-d. [DOI] [PubMed] [Google Scholar]
- 24.Rhoads A, Friedberg F. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- 25.Meador WE, Means AR, Quiocho FA. Science. 1992;257:1251–1255. doi: 10.1126/science.1519061. [DOI] [PubMed] [Google Scholar]
- 26.Meador WE, Means AR, Quiocho FA. Science. 1993;262:1718–1721. doi: 10.1126/science.8259515. [DOI] [PubMed] [Google Scholar]
- 27.Meador WE, George SE, Means AR, Quiocho FA. Nat Struct Biol. 1995;2:943–945. doi: 10.1038/nsb1195-943. [DOI] [PubMed] [Google Scholar]
- 28.Yap KL, Kim J, Truong K, Sherman M, Yuan T, Ikura M. J Struct Funct Genomics. 2000;1:8–14. doi: 10.1023/a:1011320027914. [DOI] [PubMed] [Google Scholar]
- 29.Cohen P, Klee BC. Calmodulin (Molecular Aspects of Cellular Regulation) Elsevier; Amsterdam: 1988. [Google Scholar]
- 30.Brand A, Perrimon N. Development (Camb) 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 31.Wickner W, Haas A. Annu Rev Biochem. 2000;69:247–275. doi: 10.1146/annurev.biochem.69.1.247. [DOI] [PubMed] [Google Scholar]
- 32.Murthy VN, De Camilli P. Annu Rev Neurosci. 2003;26:701–728. doi: 10.1146/annurev.neuro.26.041002.131445. [DOI] [PubMed] [Google Scholar]
- 33.Murata T, Yamato I, Kakinuma Y, Leslie AGW, Walker JE. Science. 2005;308:654–659. doi: 10.1126/science.1110064. [DOI] [PubMed] [Google Scholar]
- 34.Pujol N, Bonnerot C, Ewbank JJ, Kohara Y, Thierry-Mieg D. J Biol Chem. 2001;276:11913–11921. doi: 10.1074/jbc.M009451200. [DOI] [PubMed] [Google Scholar]
- 35.Schulze KL, Broadie K, Perin MS, Bellen HJ. Cell. 1995;80:311–320. doi: 10.1016/0092-8674(95)90414-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.