

Abstract
Reactive oxygen species (ROS) react preferentially with certain atoms to modulate functions ranging from cell homeostasis to cell death. Molecular actions include both inhibition and activation of proteins, mutagenesis of DNA and activation of gene transcription. Cellular actions include promotion or suppression of inflammation, immunity and carcinogenesis. ROS help the host to compete against microorganisms and are also involved in intermicrobial competition. ROS chemistry and their pleiotropy make them difficult to localize, to quantify and to manipulate — challenges we must overcome to translate ROS biology into medical advances.
The term ‘reactive oxygen species’ (ROS) includes super-oxide, hydrogen peroxide, singlet oxygen, ozone, hypo-halous acids and organic peroxides1. ROS participate in phenomena that traverse all of biology, and their study has burgeoned for more than a century (TIMELINE). ROS are difficult to distinguish from each other by specific assays and are challenging to quantify. The diversity of their enzymatic sources has only recently become apparent, and tools for the identification of their subcellular localization are only now emerging. Many of their effects can be opposed to one another — for example, they can both promote and prevent cell death, inflammation or ageing.
Timeline.
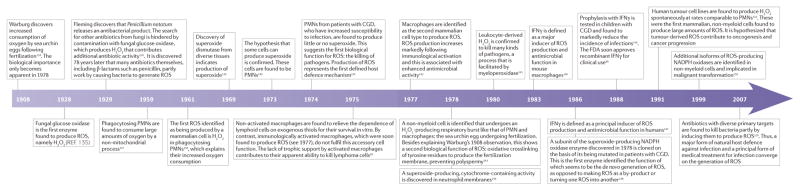
A sampling of milestones in ROS biology*
CGD, chronic granulomatous disease; FDA, US Food and Drug Administration; IFNγ, interferon-γ; PMN, polymorphonuclear leukocyte; ROS, reactive oxygen species. *This Timeline is an incomplete history, with only limited citations.
Further complicating their study, ROS are not the only class of endogenous small, reactive signalling molecules; other classes include reactive nitrogen species (RNS), such as nitric oxide (NO•) and NO2•; hydrogen sulphide (H2S) or its anion HS−; and carbon monoxide (CO). These other reactive molecules can have properties that both overlap with and are distinct from those of ROS. Scientists who study ROS and RNS organize separate conferences, but the molecules themselves interact and affect the production and targets of one another3–5.
Most medical interventions that target ROS have failed (discussed in REF. 6). This can be interpreted to mean that ROS have an unimportant role in pathophysiology; however, it might also be that the interventions tested were not based on an adequate understanding of ROS biochemistry and biology, which is still emerging7. Moreover, among immunologists who consider specificity the hallmark of the discipline, some labour under the misconception that ROS are nonspecific, and a few cling to the view that only phagocytes produce ROS and that the only function of ROS is to kill pathogens and host cells.
Thus, it is no surprise that until recently it was rare to find a diagram of signal transduction or a systems biology analysis that took ROS into account. However, in the past few years, many of the obstacles mentioned above have been overcome. Scientists are now aware that ROS routinely arise in most cells from defined sources, that they affect multiple targets in specific ways and that they exert considerable influence over cell function.
In this Review, we describe ROS in terms of their regulation, targets and actions. Because the topic is vast, our approach is illustrative rather than comprehensive. Given the conservation of ROS biology, we draw lessons from diverse fields to lend perspective to the role of ROS in immunology.
ROS homeostasis
Many authors state that ‘oxidative stress’ occurs when the production of ROS exceeds their catabolism. However, the term stress is an imprecise reference to a restricted range of ROS signalling that runs from adaptive to maladaptive (FIG. 1) and has a major role in cell and organismal biology1,8–10 (FIG. 1). Thus, oxidative stress is by no means a description of, nor a synonym for, the biology of ROS.
Figure 1. The broad range of ROS signalling is influenced by ROS production and catabolism, and by cellular adaptation.
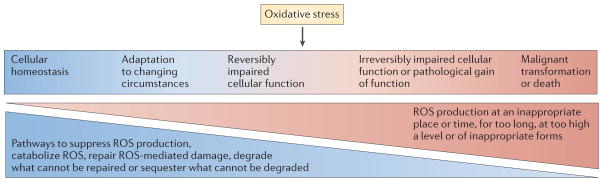
Restriction of reactive oxygen species (ROS) production to appropriate subcellular locations, times, levels, molecular species and for appropriate durations allows ROS to contribute to homeostasis and physiological cell activation. For example, brief pulses of H2O2 production at the plasma membrane or at the endosomal membrane mediate signalling in response to the engagement of receptors with cytokines, microbial products or antigens (left-hand side). When ROS production escapes these restrictions — for example, when there are high levels or sustained production of hydroxyl radicals — macromolecules are damaged (‘oxidative stress’). ROS-mediated damage can often be reversed by repair, replacement, degradation or sequestration of the damaged macromolecules (middle). However, damage that exceeds the capacity of the cell for these responses can lead to cell death (right-hand side). When damage to DNA results in mutagenesis without irreparable double-strand breakage, and when damage to other macromolecules is repaired, the consequence can be malignant transformation rather than death of the cell (right-hand side).
Sources of ROS
Endogenous sources of ROS in mammals (BOX 1) include seven isoforms of NADPH oxidases (NOXs)10,11 that are differentially expressed in diverse cells and species12; the mitochondrial respiratory chain; the flavoenzyme ERO1 in the endoplasmic reticulum; xanthine oxidase; lipoxygenases; cyclooxygenases; cyto-chrome P450s; a flavin-dependent demethylase; oxidases for polyamines and amino acids; and nitric oxide synthases. Free copper ions or iron ions that are released from iron–sulphur clusters, haem groups or metal storage proteins can convert O2•− and/or H2O2 to OH• (REF. 13).
Box 1. Sources of ROS and mediators of their catabolism.
Exogenous sources of ROS
Smoke
Air pollutants
Ultraviolet radiation
γ-irradiation
Several drugs
Endogenous sources of ROS
NADPH oxidases
Mitochondria
ER flavoenzyme ERO1
Xanthine oxidase
Lipoxygenases
Cyclooxygenases
Cytochrome P450 enzymes
Flavin-dependent demethylase
Polyamine and amino acid oxidases
Nitric oxide synthases
Free iron or copper ions
Haem groups
Metal storage proteins
Catabolism by antioxidant systems
Superoxide dismutases
Catalases
Glutathione peroxidases
Glutathione reductase
Thioredoxins
Thioredoxin reductases
Methionine sulphoxide reductases
Peroxiredoxins or peroxynitrite reductases
Catabolism by small molecules that react with ROS non-enzymatically
Ascorbate
Pyruvate
α-ketoglutarate
Oxaloacetate
ER, endoplasmic reticulum; ROS, reactive oxygen species. Sources reviewed in REFS 10–12.
Regulation of ROS production
Several checkpoints restrict ROS production by the NOXs to times and locations that are appropriate for cellular functions. NOXs are transmembrane flavocytochrome proteins, the cytosolic domains of which transfer an electron from NADPH to a FAD cofactor. From there, the electron is passed to a haem group, which donates it to O2 on the extracellular side of the membrane, generating O2•−. One control step in this process is the loading of the apoprotein with the flavin cofactor14. In another level of regulation, Ca2+ signalling, phosphorylation cascades and the activation of small G proteins control the recruitment of accessory proteins from the cytosol to join the flavocytochrome at the membrane, forming the functional NOX complex8. NOXs are activated following the activation of receptors by ligands such as insulin, platelet-derived growth factor, nerve growth factor, fibroblast growth factor, chemokines that bind G protein-coupled receptors, tumour necrosis factor (TNF), granulocyte–macrophage colony-stimulating factor (GM-CSF), angiotensin, sphingosine-1-phosphate, lysophospholipids, complement component 5a (C5a) and leukotriene B4 (LTB4), as well as by cell adhesion and by phagocytosis1,8. The mechanisms linking receptor interaction to NOX activation are an important research area in immune signalling.
ROS production by mitochondria is regulated by diverse factors, including RNS, mammalian target of rapamycin (mTOR), p53, SHC-transforming protein 1 (SHC1), reactive oxygen species modulator 1 (ROMO1), B cell lymphoma 2 (BCL-2) family members8 and uncoupling proteins15. Another factor that promotes mitochondrial ROS generation is hypoxia (discussed below).
Nitric oxide synthases (NOS) can produce ROS when concentrations of their cofactor tetrahydrobiopterin or their cosubstrate L-arginine are limiting. For example, following L-arginine depletion by L-arginase, NOS donate some of their NADPH-derived electrons to O2 rather than passing all the electrons to the guanidino nitrogens of arginine, generating both O2•− and NO•. These species react with each other to produce peroxynitrite (OONO−), which decomposes to generate the strong oxidants OH− and NO2− (REF. 16), and can oxidize redox-sensitive cysteines 103-fold faster than peroxide17.
Transcriptional networks control proteins that sense and regulate the uptake and the storage of redox-active metal ions13. In addition, other mechanisms that affect the intracellular levels of ROS include the export of xenobiotics that generate ROS13 and the export of ROS themselves into neighbouring cells through connexin channels18.
Catabolism of ROS
Until recently, the antioxidant systems of a cell were thought to be superoxide dismutases, catalases and the enzymes of the glutathione redox cycle, which couples the reduction of peroxide to the oxidation and the regeneration of reduced glutathione. Many additional physiologically important ROS-regulating enzymes are now recognized, among them thioredoxins, thioredoxin reductases19, peroxiredoxins (which also function as peroxynitrite reductases) and methionine sulphoxide reductases8,20,21. Moreover, recent studies have increased our understanding of glutathione homeostasis. For example, the level of reduced glutathione is preserved in yeast not only through the action of glutathione reductase, but also through the actions of thioredoxin reductase and glutaredoxin, and the export of oxidized glutathione into the vacuole22.
Pyruvate kinase, an enzyme involved in glycolysis and gluconeogenesis, is crucial in the negative feedback regulation of ROS. When cells metabolize glucose, ROS inhibit pyruvate kinase by oxidizing Cys358. The resulting inhibition of glycolysis directs carbon flux into the pentose phosphate pathway. This increases the production of NADPH and sustains the reduction of oxidized glutathione and thioredoxin, returning ROS to homeostatic levels23.
In addition, many small molecules that react with ROS non-enzymatically can be recycled or replenished, giving them a ROS-buffering capacity. These include ascorbate and the α-ketoacids of central carbon metabolism, such as pyruvate, α-ketoglutarate and oxaloacetate24 (BOX 1). Just as some molecules that are better known for other functions can also function as antioxidants, so molecules that are mainly known as antioxidants can have other functions. For example, when released from cells, thioredoxin is a potent chemoattractant25 and peroxiredoxins can trigger inflammation26.
Repair of ROS-mediated damage
Cells maintain homeostasis despite ROS production not only by catabolizing ROS but also by repairing oxidative injury. For example, ROS can oxidize the sulphurs that hold the iron atoms in the iron–sulphur clusters. As a result, iron atoms are lost. The apoprotein then loses its original function and might have to be re-synthesized for a new iron–sulphur cluster to be attached. DNA oxidation by ROS activates the nucleotide excision repair and base excision repair pathways13.
Some oxidative damage cannot be repaired, such as the formation of carbonyl groups on amino acid side chains in proteins. This results in cells degrading irreversibly oxidized macromolecules using the proteasome27 or autophagosomes28. Some macromolecules might be so extensively oxidized that they can be neither repaired nor degraded; cells might respond by sequestering these macromolecules with chaperones. Thus, it is not surprising that a genetic screen of viable deletion strains of Saccharomyces cerevisiae for hypersensitivity to ROS identified 456 genes29. Many of these genes encoded proteins with additional functions to catabolism of ROS, repair of ROS-dependent damage and the other mechanisms discussed above.
The fact that regulators of ROS are encoded by such a large proportion of the genome and are distributed across so many functional classes reflects the widespread functional effects of ROS. It seems unlikely that evolution selected for fitness by ascribing such a widespread role to molecules that react nonspecifically. We argue below that the biological effects of ROS are in fact highly specific.
Targets of ROS
Atomic targets
ROS display a type of specificity that is atomic rather than molecular: ROS react covalently, often reversibly, with only certain atomic elements in macromolecules, and with only a subset of those atoms30 (FIG. 2). Therefore, ROS only seem to react nonspecifically if we limit our ideas of specificity in signalling to molecular ‘handshakes’ that depend on complementarity, as in the case of insulin binding to its receptor. Such handshakes are well suited to the transmission of a signal along a discrete pathway. By contrast, reactions of ROS with specific atoms that are present in many macromolecules might transmit signals to multiple pathways at once. Depending on the origin and level of ROS, this might occur in discrete subcellular locations or across a large proportion of the cell. ROS that are produced by a local source in small enough amounts to be confined to a restricted subcellular location can function as a rheostat for discrete signalling pathways in that location. ROS that are produced in large enough amounts to diffuse across more of the cell can function as coordinators of global signalling in the cell. After ROS have accumulated to a certain level, their activating effects might give way to inhibitory effects. As a result, the diverse signalling pathways that are simultaneously regulated by cellular ROS levels are associated with the metabolic state of the cell30.
Figure 2. ROS and their atomic specificity.

During the reduction of oxygen to water, sequential one-electron reductions can produce reactive oxygen intermediates (ROIs) — superoxide, hydrogen peroxide and hydroxyl radicals — along with singlet oxygen and ozone. ROIs comprise a subset of reactive oxygen species (ROS). Additional ROS are the hypochlorous (HOCl), hypobromous (HOBr) and hypoiodous acids (HOI) that arise when peroxidases catalyse the oxidation of halides by H2O2, as well as important products of the reaction of ROS with other molecules that retain strong oxidizing potential, such as lipid peroxides (included as ROOH in the figure). ROS at low levels tend to react reversibly with a limited number of atoms — for example, selenium or sulphur atoms in a subset of cysteine and methionine residues — conferring atomic specificity in reactions involving diverse macromolecules. At higher levels ROS are likely to react irreversibly with certain iron and carbon atoms. e−, electron.
Consistent with the idea of ‘atomic specificity’, one of the atoms with which ROS most often reversibly reacts in cell signalling — sulphur — is one of the least abundant atoms in biological macromolecules. Even then, ROS do not react with all sulphur atoms, but mostly with a subset of sulphur atoms in the side chains of cysteine or methionine residues in peptides or proteins. Many of the most ROS-reactive cysteines in proteins are located in environments that are conducive to the participation of the thiolate in active site chemistry. The reactivity of specific cysteinyl thiols is partly influenced by neighbouring amino acid side chains that confer an acidic dissociation constant (low pKa), but additional factors are also involved that remain to be identified31. Much remains to be learned about this important form of intracellular signalling32.
Molecular targets: proteins
A partial list of proteins that have been reported to be physiologically regulated by ROS includes tyrosine and serine/threonine phosphatases, such as phosphatase and tensin homologue (PTEN)33; tyrosine and serine/threonine kinases, such as epidermal growth factor receptor34, protein kinase B35, ataxia-telangiectasia mutated (ATM) kinase36, calmodulin-dependent kinase II37 and protein kinase G-Iα38; zinc-finger proteins39; other transcription factors, including those of the forkhead box O (FOXO) family40; histone deacetylases41; signal-regulating binding proteins, such as heat-shock proteins41; caspases41; metalloproteinases42; protease inhibitors, such as α1-antitrypsin43, α2-macroglobulin44 and secretory leukocyte protease inhibitor45; metabolic enzymes; prolyl hydroxylase46 and glucose uptake regulator47, which both depend on α-ketoglutarate as a cofactor; GTP cyclohydrolase48; guanylyl cyclase49; and ion channels, such as the ryanodine receptor50.
An important role of ROS in signal transduction is to transiently oxidize the cysteine sulphydryl that contributes to the active site of most phosphatases. The phosphorylation that follows the binding of a ligand to its receptor is usually attributed to the activation of a kinase, but can also be the result of a burst of ROS formation and the transient inactivation of cognate phosphatases. Proteome-wide assessment has shown that ROS regulation of phosphatases, and hence of phosphorylation, is widespread51.
In other proteins, ROS sensing by cysteine residues can provide feedback control to regulate the levels of ROS. For example, the intermolecular oxidation of conserved cysteines in the transcription factor FOXO4 allows for the binding of this protein to the p300/CBP acetylase, which leads to lysine acetylation of FOXO4. Such a phenomenon might be related to the ROS-dependent transcriptional induction of manganese superoxide dismutase, catalase, peroxiredoxin and sulphiredoxin, and to the reduction in levels of ROS40,52. Similarly, oxidation of important cysteine residues in ATM kinase promotes glucose flux through the pentose phosphate shunt, increasing the levels of NADPH. NADPH is the physiological reductant for oxidized glutathione and thioredoxin, and thus, the ultimate reductant for many ROS-catabolizing enzymes, such as glutathione reductase, peroxiredoxins and methionine sulphoxide reductases53. Regulation of ROS seems to be important for the functions of ATM in haematopoiesis54 and neoangiogenesis55.
A main function of ROS is their contribution to the activation of transcription by several mechanisms. For example, the bacterial transcription factor SoxR is activated by the superoxide anion, which interacts with the iron–sulphur redox centre of SoxR, whereas another transcription factor, OxyR, is activated by H2O2, which oxidizes the cysteinyl thiol of OxyR. Both events lead to the expression of antioxidant enzymes56. Moreover, ROS-facilitated protein phosphorylation can lead to the kinase-mediated activation of a transcription factor such as JUN, or to the translocation to the nucleus of a cytosolic transcription factor such as nuclear factor-κB (NF-κB) following the kinase-triggered ubiquitylation and subsequent degradation of its inhibitor (FIG. 3).
Figure 3. Examples of transcriptional regulation by ROS acting at the plasma membrane or in the cytosol.

Activation of tumour necrosis factor receptor (TNFR) triggers reactive oxygen species (ROS) production, which enhances the phosphorylation (P) of inhibitor of NF-κB (IκB), probably through the oxidative inhibition of a phosphatase. This leads to the ubiquitylation (Ub) of IκB and its subsequent degradation by the proteasome. Nuclear factor-κB (NF-κB) is then released and translocates to the nucleus to initiate transcription. ROS production can also trigger oxidative glutathionylation of NF-κB at its redox sensitive cysteine, which reduces its DNA binding affinity133. Under normoxia, prolyl hydroxylases (PHs) hydroxylate hypoxia-inducible factor 1α (HIF11α), which allows it to be recognized by the E3 ligase von Hippel–Lindau tumour-suppressor protein (VHL) and promotes its degradation by the proteasome. Under hypoxia there is increased production of ROS (FIG. 4), which inhibits prolyl hydroxylases, leading to the accumulation of HIF11α. HIF11α then translocates to the nucleus to mediate gene transcription. ROS production by NADPH oxidases (NOXs) following receptor activation by specific ligands, for example, epidermal growth factor receptor (EGFR), inhibits protein tyrosine phosphatases (PTPs), which promotes the phosphorylation of tyrosine kinases (TKs) and the subsequent signal transduction. By contrast, ataxia-telangiectasia mutated (ATM) kinase is activated directly by ROS, through disulphide bond-mediated homodimerization, which leads to the phosphorylation of heat shock protein 27 (HSP27) and the subsequent activation of glucose-6-phosphate-dehydrogenase (G6PD). The resulting increase in NADPH levels contributes to the maintenance of cellular redox homeostasis. ROS-mediated disulphide bonding can also lead to heterodimerization, as seen between forkhead box O (FOXO) transcription factors and p300/CBP acetyltransferase, which leads to the acetylation of FOXO proteins and the activation of specific gene transcription. ERK, extracellular signal-regulated kinase.
ROS can also promote transcription by increasing the accumulation of transcription factors via inhibition of their degradation. For example, increased production of ROS by hypoxic mitochondria (FIG. 4) has an important role in preventing the degradation of hypoxia-inducible factor 1α (HIF1α), which is the main transcription factor that cells use to adapt to the hypoxic state57–59. In turn, HIF1a mediates hypoxic transition to glycolytic metabolism, reducing electron flow through mitochondria and reducing the associated production of ROS. Thus, the generation of reactive oxygen intermediates (ROIs; a subset of ROS) under hypoxic conditions should not be viewed as a wasteful process carried out by a poorly evolved system, but rather as an elegant network, through which the reduction in the availability of a crucial molecule triggers transcriptional adaptation.
Figure 4. Regulation of HIF1α by mitochondrial ROS production during hypoxia.

In the presence of O2 and its cofactor α-ketoglutarate (αKG), HIF-prolyl hydroxylase 2 (HIF-PH2) hydroxylates two proline residues in hypoxia-inducible factor 1α (HIF1α). Hydroxylated HIF1α is then ubiquitylated (Ub) by the E3 ligase von Hippel–Lindau tumour-suppressor protein (VHL) and is subsequently degraded by the proteasome. Because O2 is a substrate for HIF-PH2, hypoxia limits HIF-PH2 activity. This limitation is enhanced by the negative effect of hypoxia-driven mitochondrial reactive oxygen species (ROS) on HIF-PH2 function. Complex III in the mitochondrial electron transport chain (METC) receives two electrons from ubiquinone but can only transfer one electron at a time to cytochrome c (Cyt c). Complex III therefore transfers one electron to a quasi-stable ubisemiquinone radical. If this radical accumulates, O2 that is dissolved in the mitochondrial membrane can capture the electron before cytochrome c accepts it, generating superoxide (O2•−). If cellular O2 is low, complex IV at the end of the METC is slow to transfer pairs of electrons to O2 (making water), and the METC is blocked between complex III and IV, which favours the electron leak described above. The newly formed superoxide dismutates to H2O2. H2O2 can inhibit HIF-PH2 and oxidatively decarboxylate its cofactor, αKG. The resulting decrease in the hydroxylation of HIF1α and its subsequent proteasomal degradation supports HIF1α accumulation. Dashed arrows indicate the reactions that are slowed in hypoxia.
Molecular targets: DNA
Mitochondrial ROS can promote transcription by oxidizing DNA itself, rather than by oxidizing DNA-binding regulatory proteins (FIG. 5a). For example, ROS that are produced by hypoxic mitochondria oxidize specific bases in the promoter of vascular endothelial growth factor (VEGF). This enhances binding of HIF1α to the VEGF promoter60. Evidence for the physiological relevance of this response comes from the observation in pulmonary artery endothelial cells that hypoxia triggers dynein-dependent movement of mitochondria along microtubules so that the mitochondria cluster around the nucleus. As a result, mitochondrial-dependent ROS promote oxidative modifications of, and HIF1α binding to, the VEGF promoter61. Thus, ROS are physiological mediators in signalling between mitochondria and the nucleus9.
Figure 5. Regulation of transcription through DNA targeting by intranuclear ROS.

a | The induction of the transcription of the gene encoding vascular endothelial growth factor (VEGF) by hypoxia-inducible factor 1α (HIF1α) is enhanced through the dynein-mediated, perinuclear localization of mitochondria. Mitochondria-derived reactive oxygen species (ROS) diffuse into the nucleus, where they promote the oxidation of guanine nucleotides, forming 8-oxoguanine (8OG). b | Transcriptional regulation downstream of the activation of androgen receptors (ARs) or oestrogen receptors (ORs) and other nuclear receptors also involves DNA modifications by ROS. Engagement of ORs or ARs promotes the phosphorylation (P) of lysine-specific histone demethylase 1A (LSD1) by cAMP-dependent protein kinase (PKA). Active LSD1 not only demethylates histone 3 (H3) but also produces ROS, which then promote the formation of 8OG in the DNA. The altered DNA bases recruit base excision repair machinery, and the DNA breaks that are generated by 8OG DNA glycosylase 1 (OGG1) enable the activation of transcription by AR, OR and possibly also MYC.
In another example of gene regulation through DNA oxidation (FIG. 5b), transcriptional activation by the oestrogen receptor, the androgen receptor, the retinoic acid receptor, the thyroxin receptor and activator protein 1 all require topoisomerase IIb-mediated, site-specific DNA breaks in target gene-regulatory regions62. ROS are involved in the formation of these breaks. The oestrogen receptor-activated flavin-dependent lysine-specific histone demethylase 1 (LSD1; also known as KDM1A) produces H2O2 (REF. 63). This locally produced H2O2 oxidizes bases in target gene-regulatory regions, forming 8-oxoguanine. 8-oxoguanine recruits 8-oxoguanine DNA glycosylase 1 (OGG1), which causes single-stranded DNA breaks. These breaks recruit topoisomerase IIb, which bends DNA while repairing the lesion, thereby enabling the transcription initiation complex to access the targeted promoters63. The same process seems to be required for MYC-mediated transcription64.
Thus, ROS arising from NOXs at the plasma, endosomal and phagolysosomal membranes, and from mitochondria in the cytosol, can influence transcription by regulating the phosphorylation of transcription factors; whereas ROS arising from perinuclear mitochondria or from a nuclear flavoenzyme can participate in transcriptional control by targeting DNA directly. This recent understanding of ROS–DNA interactions complements the long-standing recognition of the mutagenic potential of ROS.
Effects of ROS in the immune system
In the immune system, ROS are neither unique products of one subset of cells, such as phagocytes, nor do they have one action, such as to kill other cells. Instead, ROS have a physiological role in signalling that probably extends to every cell type in immunology. As with any signalling mechanism, ROS can become cytotoxic if the signal is too strong, if it lasts too long or if it arises at the wrong time or place.
ROS in innate immunity
The first functional role ascribed to the production of ROS by mammalian cells was the killing of microorganisms by phagocytes (TIMELINE). This was confirmed by the discovery that individuals with chronic granulomatous disease (CGD), a disorder characterized by heightened susceptibility to infection, have disease-causing mutations in NOX2, the main source of ROS in polymorphonuclear leukocytes (PMNs) and mononuclear phagocytes.
PMNs migrate to sites of tissue damage in response to chemotactic factors such as interleukin-8 (IL-8), C5a, LTB4, and formyl peptides released by mitochondria or bacteria. All of these can trigger NOX2-dependent H2O2 production. It was shown in zebrafish larvae, where endothelial cells near wounded tissue activate the Nox isoform dual oxidase (Duox), that H2O2 itself is a chemotactic factor65. One mechanism through which H2O2 affects the migration of leukocytes in fish and humans involves the oxidative activation of the tyrosine kinase LYN and perhaps other SRC family kinases66. Thus, H2O2 might help to direct PMN movement in an autocrine or a paracrine manner.
Moreover, H2O2 that is produced in PMNs can contribute to PMN migration by promoting the accumulation of phosphatidylinositol-3,4,5-trisphosphate at the leading edge of the plasma membrane. NOX2 that is localized at the plasma membrane generates H2O2, which transiently inactivates the phosphatase PTEN, allowing inositol polyphosphates to accumulate33. At the peak of a chemotactic gradient, where migrating PMNs congregate and begin to ingest bacteria, ROS levels are probably much higher and more sustained than near individual PMNs that are beginning to emigrate from the bloodstream. High ROS levels can suppress cell motility by promoting the accumulation of glutathionylated actin, which is impaired for polymerization67. Thus, ROS can coordinate the migration of PMNs towards the pathogens that wounding will probably introduce, and then ROS can promote the retention of the PMNs at that site.
Consistent with the potential role of NOX2 in the migration of mammalian PMNs, NOX2-deficient PMNs failed to migrate up a chemotactic gradient in vitro or to sites of inflammation in vivo68. Thus, the immuno-deficiency of CGD not only involves defective bacterial killing, but might also be a result of delayed PMN accumulation. Therefore, it might seem paradoxical that CGD was named for the tendency to form excessive granulomas; however, this might reflect an impaired oxidative inactivation of chemotactic factors, such as C5a, formylated peptides and LTB4 (REFS 69,70). This indicates that ROS might help not only to initiate but also to terminate inflammation.
As mentioned above, host-derived factors, such as IL-8 and C5a, which are induced in response to bacterial products, can trigger ROS production from leukocytes. However, bacteria can also trigger ROS production directly through diverse types of receptors on leukocytes. For example, Toll-like receptor (TLR) engagement can induce ROS production by NOX2 and by other sources that contribute to signalling. NOX2-derived ROS were shown to be required for X-box-binding protein 1 (XBP1)-mediated transcriptional responses downstream of TLR2 and TLR4 that led to control of Franciscella tularensis71. In macrophages, TLR1, TLR2 and TRL4 engagement recruited mitochondria to phagosomes and promoted mitochondrial H2O2 production, which contributed to the control of Salmonella enterica subsp. enterica serovar Typhimurium infection in vitro. Mice expressing mitochondrial catalase had a twofold to threefold greater burden of S. Typhimurium 5 days after infection compared with wild-type mice72. However, it is not clear whether TLR-induced mitochondrial ROS are required for mice to clear infection.
ROS and inflammasomes
An important question in innate immunity is how structurally diverse agonists activate the same inflammasomes. ROS seem to mediate inflammasome activation in a range of circumstances, although the molecular steps involved and the sources of ROS remain controversial. The late Jurg Tschopp and his colleagues advanced a theory based on the premise that all known activators of the NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome induce ROS, which lead to the dissociation of thioredoxin-interacting protein (TXNIP) from thioredoxin. They proposed that mitochondria are the source of ROS and that the association of TXNIP with NLRP3 is the basis of its activation73. Other work indicates that diverse NLRP3-activating stimuli might converge to promote mitochondrial damage. Damaged mitochondria produce excess ROS, which attack the mitochondrial DNA. Oxidized mitochondrial DNA enters the cytosol, and binds to and activates NLRP3 (REF. 74).
Further studies might help to clarify the links between diverse NRLP3-activating stimuli, mitochondrial damage and the resulting ROS overproduction. It is possible that another source of ROS might initiate the mitochondrial damage, in effect beginning a process that then becomes self-sustaining. This might explain why knock down of the common NOX2 subunit p22phox (also known as CY24A) decreased IL-1β secretion in response to a range of inflammasome activators75. It remains to be explained what terminates this process that risks initiating a positive feedback loop that could be lethal for the cell. In fact, production of mature IL-1β is closely associated with apoptosis76.
ROS, the intestinal microbiota and enteric pathogens
In contrast to patients with CGD, mice that are only deficient in NOX2 are not susceptible to spontaneous infections during standard husbandry, which is similar to mice that are only deficient in inducible nitric oxide synthase (NOS2; also known as iNOS). However, mice lacking both NOX2 and NOS2 succumb to spontaneous, invasive infections by their own microbiota in the form of massive abscesses filled with PMNs and monocytes77. It seems that the antibiotic proteins and peptides, the acidification pumps, the lysosomal hydrolases and the autophagic mechanisms of PMNs and monocytes are not effective in the infected organs in the absence of both NOX2 and NOS2. This might indicate that both ROS and RNS are required to integrate with the other mechanisms listed above. Indeed, ROS are required for antibacterial autophagy in PMNs, macrophages and some epithelial cells78. The partial mutual redundancy of NOX2 and NOS2 obscures the full importance of ROS in controlling infection. Taken together, these data show that NOX2 and NOS2 are essential for mice to coexist with their microbiota77.
DUOX in colonic epithelial cells also contributes to coexistence of the host with the microbiota. This enzyme produces submicrobicidal levels of ROS in response to commensal bacteria, such as Lactobacilli spp., which promote epithelial repair and suppress inflammatory responses79. One mechanism of suppression of inflammation by ROS involves the reversible, oxidative inactivation of a component of the neddylation pathway, which prevents the ubiquitylation and the proteasomal degradation of inhibitor-α of NF-κB (IκBα) and the activation of NF-κB80. By contrast, high levels of ROS that are produced during intestinal inflammation, along with high levels of RNS, contribute not only to the suppression of microbial growth but also to epithelial injury79.
Some intestinal pathogens capitalize on inflammatory ROS production. For example, ROS can convert endogenous thiosulphate into tetrathionate, which S. Typhimurium can use as an electron acceptor81 to achieve a growth advantage in regions of the intestinal lumen that are nearly anoxic79.
ROS and the regulation of lymphocyte function
ROS were discovered when phagocytosing leukocytes were found to consume large amounts of oxygen by a non-mitochondrial process (TIMELINE). B cells and T cells neither phagocytose nor show large increases in mitochondrial-independent oxygen consumption following activation. Thus, it was assumed that ROS have no role in adaptive immunity, and little notice was paid when superoxide production by the ‘phagocyte oxidase’ was discovered in transformed82 and primary83 human B cells at about one-tenth of the levels of ROS that are released by phagocytes. However, later studies showed that ROS production is functionally important in lymphocytes. Following B cell receptor (BCR) ligation, ROS transiently inactivate BCR-associated phosphatases and function synergistically with calcium transients to potentiate signal transduction84. Similarly, T cell receptor (TCR) engagement triggers ROS production, with superoxide and peroxide regulating pro-apoptotic and proliferative pathways, respectively85. NOX-derived ROS that are released outside of the plasma membrane or into the lumen of a plasma membrane-derived vesicle enter the T cell via aquaporin 3 to affect cytosolic phosphatases86. T cell-derived peroxide also activates NF- B, which leads to the production of IL-2 (REF. 87). T cell activation also indirectly depends on ROS, in that the consumption of protons by NOX2 in the phagosomes of dendritic cells (DCs) retards phagosomal acidification, impedes the action of proteases and preserves peptides of sufficient length to be presented on MHC molecules88.
Lymphocytes could not be cultured until Mishell and Dutton89 discovered the trophic effect of supplementing the medium with the reducing agent 2-mercapto-ethanol89. Using transformed lymphocytes, Nathan and Terry90 showed that non-activated macrophages could replace the reducing agent to sustain lymphocyte growth, but that activated, ROS-producing macrophages could not90 (TIMELINE). Subsequent work established that macrophages and DCs can respond to various stimuli, including contact with antigen-specific T cells, by secreting glutathione, which is broken down to release cysteine, an amino acid that is otherwise scarce in extra-cellular fluid. Cysteine uptake allows the lymphocytes to synthesize their own glutathione, to maintain redox homestasis, and to undergo antigen-specific activation and proliferation91. Consistent with the observation that mouse macrophages differ in their secretion of lymphocyte-trophic thiols depending on their activation state90, lamina propria-resident human macrophages secrete little cysteine, and neighbouring T cells are fairly glutathione-deficient and hyporeactive, whereas the lamina propria is infiltrated by innate immune cells with higher cysteine-releasing capacity during inflammatory bowel disease92. Moreover, regulatory T (TReg) cells can suppress glutathione release by DCs, thereby indirectly regulating the activation potential of other T cells93.
As noted above, even though ROS help to mediate T cell activation, T cell activation also partly depends on help from accessory cells to maintain T cell levels of the antioxidant glutathione. This indicates that ROS can be immunosuppressive. Indeed, ROS were the first molecules found to suppress T cell function94. Some TReg cells can suppress other T cells through their release of ROS, and TReg cells can be induced by macrophage-derived ROS95,96. In keeping with these observations, TReg cells were found to be more resistant to ROS than effector T cells; their enhanced resistance was associated with greater secretion of thioredoxin97.
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells that are temporarily restricted from further differentiation. MDSC numbers can increase tenfold in the blood of patients with cancer and inflammatory disorders98–100. During cancer progression, the recruitment of MDSCs to the tumour site can suppress immune reactivity101. MDSCs produce large quantities of ROS. ROS production not only underlies the immunosuppressive properties of MDSCs but it is also thought to maintain them in an undifferentiated state99,102. Nitration of CD8, TCR, and CC-chemokine ligand 2 (CCL2) by MDSC-derived peroxynitrite prevents the binding of CD8+ T cells to peptide-loaded MHC class I, promotes antigen-specific tolerance and impairs cytotoxic T cell recruitment to tumours101,103. MDSCs from mice lacking NOX2 demonstrated little or no ROS production and they lacked the ability to suppress T cell proliferation and the production of IFNγ101,103.
ROS and tumour cells
Once ROS were discovered to be a principal mechanism by which the immune system controls pathogens, investigators asked whether ROS also contribute to the ability of activated macrophages to selectively kill tumour cells. In several mouse models ROS seemed to account for much of the antitumour activity of immunologically activated macrophages104–106. Killing could be greatly increased by pharmacologically inhibiting tumour cell glutathione synthesis or glutathione reductase, or by restricting dietary selenium, which is required for the function of glutathione peroxidase107. It was even possible to mimic activated macrophages with an artificial, particulate H2O2-generating system and to cure mice of an advanced lymphoma108. However, human cells proved to be about 100-fold more resistant to ROS than their mouse counterparts109. Moreover, cell lines derived from metastatic human tumours were found to produce ROS at high levels110.
The phenomenon of ROS overproduction by tumour cells has been widely confirmed111. In some cases, excessive ROS production has been attributed to mutations in a mitochondrial gene that encodes a component of the mitochondrial electron transport chain. Introduction of those mutations into other tumour cells was sufficient to confer both enhanced ROS generation and ROS-dependent metastatic potential112. ROS release by tumour cells might sensitize, or even self-activate, their growth factor receptors by inhibiting the associated tyrosine phosphatases51,113. Tumour cell ROS production might also help to explain how tumour cells alter their central carbon metabolism114 — for example, towards synthesis of nucleic acid precursors23 — and it might also contribute to immunosuppression. Moreover, the mutagenic actions of ROS that are derived from inflammatory cells can contribute to the initial stages of tumorigenesis115. After a tumour is established, ROS derived from radiotherapy, from chemotherapy or from the tumour cells themselves might contribute to the genomic instability of tumour cells116, fostering drug resistance, just as ROS production that is induced in bacteria by antibiotics can cause mutations that promote antibiotic resistance117. Finally, tumour cell-derived ROS can trigger the tumour stroma to produce angiogenic factors111. In contrast to more differentiated cancer cells, cancer stem cells exhibited lower levels of intracellular ROS, greater levels of antioxidant pathways and more efficient DNA repair responses to ionizing radiation118–120. Thus, the effects of ROS on tumours can range from tumour-promoting effects to tumour-destroying effects, which means that ROS-producing phagocytes are potentially dangerous cells for both the tumour and the host.
Looking ahead
Two sets of questions loom large over ROS biology: the mechanisms of ROS production and action, and the translational potential of this information in medicine.
Mechanistic mysteries
It remains unclear exactly how cytokines, TLR ligands, inflammasome agonists and lymphocyte antigen receptors trigger ROS production and how the subcellular localization, the magnitude and the duration of ROS production determine their specific functions. Systems biology has yet to integrate ROS biology fully into the ‘wiring’ diagrams that reflect our understanding of cellular behaviour.
These advances will partly depend on the development of tools that would help to identify ROS and their subcellular localization and to quantify them at the level of single cells and single molecular species in real time. These tools are beginning to be developed but the challenges are considerable. Not all redox couples in a cell are maintained at the same equilibrium, and it is not well understood what insulates one couple from another. Some of the older organic dyes that react with ROS to produce or suppress fluorescence lack specificity for individual ROS121. Newer approaches that allow for sensitive and specific measurement of ROS include small compounds122; novel delivery systems, such as nanotubes123 and peroxalate micelles124; and genetically encoded redox-sensitive fluorescent proteins, such as redox-oxidation-sensitive green fluorescent proteins (roGFPs)125. The use of some genetically encoded bio-sensors is limited by their slow or irreversible response to changing redox levels. However, roGFPs that are fused with glutaredoxin can capture real-time changes in the aspect of cellular redox potential that is linked to the redox state of glutathione126. Another challenge is to determine the extent of oxidation of specific targets, both per single molecule and as a proportion of the molecules of a given molecular species. Quantitative redox proteomic techniques are struggling with this challenge121.
Medical advances
The disappointing history of efforts to prevent or to treat disease with exogenous anti-oxidants does not reflect the important role of ROS in pharmacology. Many drugs partly work by generating ROS, by inducing intracellular production of ROS, by sensitizing cells to ROS107, by diminishing the cellular production of ROS or by increasing the catabolism of ROS111,127,128.
For example, many antibiotics kill bacteria partly by inducing them to make ROS. β-lactams, which target peptidoglycan synthesis in the bacterial cell wall, and aminoglycosides, which inhibit the bacterial ribosome, both create metabolic stress that results in NADH auto-oxidation and O2•− production in bacteria. O2•− can dislodge Fe2+ from iron–sulphur clusters, and Fe2+ together with O2 or H2O2 can generate the most potent oxidant known, OH•, which contributes to bacterial death129,130. In short, a principal biochemical mechanism of host defence that evolved in the immune system over millions of years matches a major form of artificial host defence that was engineered by scientists over the past few decades. This is not a coincidence; it is a consequence of convergent evolution. Most antibiotics in clinical use are, or mimic, natural microbial products — signalling molecules that microorganisms use to communicate with each other. Apparently, similar advantages in fitness against microbial competitors supported the evolution of small, secreted, ROS-inducing molecules in bacteria and large, ROS-generating intra-cellular enzymes in eukaryotes. A better understanding of how antibiotics lead to ROS production as part of their mechanism of action129 could help the urgent need to revitalize antibiotic research and discovery131.
Some anti-infectives, such as clofazimine151, and anti-cancer agents that also have antibiotic actions, such as adriamycin152 and bleomycin153, produce ROS directly. In the case of the anticancer agents, ROS production contributes both to their efficacy and to their toxicity.
Among the unanticipated anti-inflammatory actions of statins is their ability to decrease ROS production by endothelial cells. The synthetic oleanoid triterpenoids, one of the newest classes of anti- inflammatory agents, partly work by binding to KEAP1 (Kelch-like ECH-associated protein 1), which releases nuclear-related factor 2 (NRF2; also known as NF2L2) to induce a panoply of endogenous antioxidant defences132.
Substantial medical advances could result from an increased understanding of how to foster or to inhibit ROS production, and how to monitor the effects of these interventions.
Acknowledgments
A.C.-B. is a member of the Weill Cornell/Rockefeller/Sloan-Kettering Tri-Institutional MD-PhD Programme, which is supported by the Medical Scientist Training Program grant (GM07739) from the National Instiute of General Medical Sciences, USA. The Department of Microbiology and Immunology is supported by the William Randolph Hearst Trust.
Glossary
- Iron–sulphur clusters
Prosthetic groups that are required for the function of some enzymes. In iron–sulphur clusters two, three or four atoms of iron are attached to the protein through two or four sulphydryl groups
- Uncoupling proteins
Proteins in the mitochondrial inner membrane that can divert the proton gradient away from the formation of ATP, resulting in the generation of heat instead
- Xenobiotics
Small chemical compounds that enter an organism unnaturally, such as drugs or pollutants
- Acidic dissociation constant (pKa)
The equilibrium constant for the dissociation of an acid into its conjugate base and hydrogen ion, expressed as the negative logarithm. The lower the pKa of a sulphydryl group, the greater the likelihood that the sulphur will be anionic at ambient pH
- Chronic granulomatous disease (CGD)
An immunodeficiency state manifested by recurrent, often life-threatening, infections and the excessive formation of granulomas, caused by mutations in any one of four subunits of NADP oxidase 2
- Granulomas
Histological collections of macrophages, usually surrounded by lymphocytes and sometimes fibrocytes. Some of the macrophages might seem to be ‘epithelioid’ or fuse to become multinucleated giant cells. Granuloma formation is a chronic inflammatory response to various infectious and non-infectious agents
- Neddylation
A process that is analogous to ubiquitylation, in which ubiquitin-like protein NEDD8 is conjugated to a protein substrate
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Note added in proof
Two recent reports154,155 demonstrated that killing of Escherichia coli by norfloxacin, ofloxacin, kanamycin or ampicillin does not involve ROS.
FURTHER INFORMATION
Carl Nathan’s homepage: http://weill.cornell.edu/research/cnathan/
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Nathan C, Ding A. Snapshot: reactive oxygen intermediates (ROI) Cell. 2010;140:951–951.e2. doi: 10.1016/j.cell.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 2.Nishida M, et al. Hydrogen sulfide anion regulates redox signaling via electrophile sulfhydration. Nature Chem Biol. 2012;8:714–724. doi: 10.1038/nchembio.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finkel T. From sulfenylation to sulfhydration: what a thiolate needs to tolerate. Sci Signal. 2012;5:pe10. doi: 10.1126/scisignal.2002943. [DOI] [PubMed] [Google Scholar]
- 4.Paul BD, Snyder SH. H2S signalling through protein sulfhydration and beyond. Nature Rev Mol Cell Biol. 2012;13:499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 5.Wink DA, et al. Nitric oxide and redox mechanisms in the immune response. J Leuk Biol. 2011;89:873–891. doi: 10.1189/jlb.1010550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinhubl SR. Why have antioxidants failed in clinical trials? Am J Cardiol. 2008;101:14D–19D. doi: 10.1016/j.amjcard.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Brennan ML, Hazen SL. Emerging role of myeloperoxidase and oxidant stress markers in cardiovascular risk assessment. Curr Opin Lipidol. 2003;14:353–359. doi: 10.1097/00041433-200308000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Bae YS, Oh H, Rhee SG, Yoo YD. Regulation of reactive oxygen species generation in cell signaling. Mol Cells. 2011;32:491–509. doi: 10.1007/s10059-011-0276-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287:4434–4440. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang F, Zhang Y, Dusting GJ. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011;63:218–242. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- 11.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nature Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 12.Aguirre J, Lambeth JD. Nox enzymes from fungus to fly to fish and what they tell us about Nox function in mammals. Free Radic Biol Med. 2010;49:1342–1353. doi: 10.1016/j.freeradbiomed.2010.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imlay JA. Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem. 2008;77:755–776. doi: 10.1146/annurev.biochem.77.061606.161055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yazdanpanah B, et al. Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature. 2009;460:1159–1163. doi: 10.1038/nature08206. [DOI] [PubMed] [Google Scholar]
- 15.Mailloux RJ, Harper ME. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med. 2011;51:1106–1115. doi: 10.1016/j.freeradbiomed.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 16.Corzo CA, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182:5693–5701. doi: 10.4049/jimmunol.0900092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 18.Gonzalez-Nieto D, et al. Connexin-43 in the osteogenic BM niche regulates its cellular composition and the bidirectional traffic of hematopoietic stem cells and progenitors. Blood. 2012;119:5144–5154. doi: 10.1182/blood-2011-07-368506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holmgren A, Lu J. Thioredoxin and thioredoxin reductase: current research with special reference to human disease. Biochem Biophys Res Commun. 2010;396:120–124. doi: 10.1016/j.bbrc.2010.03.083. [DOI] [PubMed] [Google Scholar]
- 20.Weissbach H, et al. Peptide methionine sulfoxide reductase: structure, mechanism of action, and biological function. Arch Biochem Biophys. 2002;397:172–178. doi: 10.1006/abbi.2001.2664. [DOI] [PubMed] [Google Scholar]
- 21.Bryk R, Griffin P, Nathan C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- 22.Morgan B, et al. Multiple glutathione disulfide removal pathways mediate cytosolic redox homeostasis. Nature Chem Biol. 2012;9:119–125. doi: 10.1038/nchembio.1142. [DOI] [PubMed] [Google Scholar]
- 23.Anastasiou D, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Donnell-Tormey J, Nathan CF, Lanks K, DeBoer CJ, de la Harpe J. Secretion of pyruvate. An antioxidant defense of mammalian cells. J Exp Med. 1987;165:500–514. doi: 10.1084/jem.165.2.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bertini R, et al. Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells. J Exp Med. 1999;189:1783–1789. doi: 10.1084/jem.189.11.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shichita T, et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nature Med. 2012;18:911–917. doi: 10.1038/nm.2749. [DOI] [PubMed] [Google Scholar]
- 27.Seifert U, et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell. 2010;142:613–624. doi: 10.1016/j.cell.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 28.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Thorpe GW, Fong CS, Alic N, Higgins VJ, Dawes IW. Cells have distinct mechanisms to maintain protection against different reactive oxygen species: oxidative-stress-response genes. Proc Natl Acad Sci USA. 2004;101:6564–6569. doi: 10.1073/pnas.0305888101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nathan C. Specificity of a third kind: reactive oxygen and nitrogen intermediates in cell signaling. J Clin Invest. 2003;111:769–778. doi: 10.1172/JCI18174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferrer-Sueta G, et al. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem Res Toxicol. 2011;24:434–450. doi: 10.1021/tx100413v. [DOI] [PubMed] [Google Scholar]
- 32.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 33.Kuiper JW, Sun C, Magalhaes MA, Glogauer M. Rac regulates PtdInsP3 signaling and the chemotactic compass through a redox-mediated feedback loop. Blood. 2011;118:6164–6171. doi: 10.1182/blood-2010-09-310383. [DOI] [PubMed] [Google Scholar]
- 34.Paulsen CE, et al. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nature Chem Biol. 2012;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wani R, et al. Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc Natl Acad Sci USA. 2011;108:10550–10555. doi: 10.1073/pnas.1011665108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 37.Erickson JR, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burgoyne JR, et al. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317:1393–1397. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- 39.Kroncke KD, Klotz LO. Zinc fingers as biologic redox switches? Antioxid Redox Signal. 2009;11:1015–1027. doi: 10.1089/ARS.2008.2269. [DOI] [PubMed] [Google Scholar]
- 40.de Keizer PL, Burgering BM, Dansen TB. Forkhead box O as a sensor, mediator, and regulator of redox signaling. Antioxid Redox Signal. 2011;14:1093–1106. doi: 10.1089/ars.2010.3403. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Yang J, Yi J. Redox sensing by proteins: oxidative modifications on cysteines and the consequent events. Antioxid Redox Signal. 2012;16:649–657. doi: 10.1089/ars.2011.4313. [DOI] [PubMed] [Google Scholar]
- 42.Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid oxygenates the cysteine switch domain of promatrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J Biol Chem. 2001;276:41279–41287. doi: 10.1074/jbc.M106958200. [DOI] [PubMed] [Google Scholar]
- 43.Taggart C, et al. Oxidation of either methionine 351 or methionine 358 in α1-antitrypsin causes loss of anti-neutrophil elastase activity. J Biol Chem. 2000;275:27258–27265. doi: 10.1074/jbc.M004850200. [DOI] [PubMed] [Google Scholar]
- 44.Reddy VY, et al. Oxidative dissociation of human α2-macroglobulin tetramers into dysfunctional dimers. J Biol Chem. 1994;269:4683–4691. [PubMed] [Google Scholar]
- 45.Carp H, Janoff A. Inactivation of bronchial mucous proteinase inhibitor by cigarette smoke and phagocyte-derived oxidants. Exp Lung Res. 1980;1:225–237. doi: 10.3109/01902148009065462. [DOI] [PubMed] [Google Scholar]
- 46.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295:858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 47.Doucette CD, Schwab DJ, Wingreen NS, Rabinowitz JD. α-Ketoglutarate coordinates carbon and nitrogen utilization via enzyme I inhibition. Nature Chem Biol. 2011;7:894–901. doi: 10.1038/nchembio.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leichert LI, Jakob U. Protein thiol modifications visualized in vivo. PLoS Biol. 2004;2:e333. doi: 10.1371/journal.pbio.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.White AA, Crawford KM, Patt CS, Lad PJ. Activation of soluble guanylate cyclase from rat lung by incubation or by hydrogen peroxide. J Biol Chem. 1976;251:7304–7312. [PubMed] [Google Scholar]
- 50.Feng W, Liu G, Allen PD, Pessah IN. Transmembrane redox sensor of ryanodine receptor complex. J Biol Chem. 2000;275:35902–35907. doi: 10.1074/jbc.C000523200. [DOI] [PubMed] [Google Scholar]
- 51.Karisch R, et al. Global proteomic assessment of the classical protein-tyrosine phosphatome and “Redoxome”. Cell. 2011;146:826–840. doi: 10.1016/j.cell.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011;30:546–555. doi: 10.1038/emboj.2010.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ito K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nature Med. 2006;12:446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- 55.Okuno Y, Nakamura-Ishizu A, Otsu K, Suda T, Kubota Y. Pathological neoangiogenesis depends on oxidative stress regulation by ATM. Nature Med. 2012;18:1208–1216. doi: 10.1038/nm.2846. [DOI] [PubMed] [Google Scholar]
- 56.Storz G, Tartaglia LA, Ames BN. Transcriptional regulator of oxidative stress-inducible genes: direct activation by oxidation. Science. 1990;248:189–194. doi: 10.1126/science.2183352. [DOI] [PubMed] [Google Scholar]
- 57.Brunelle JK, et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–414. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 58.Guzy RD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 59.Mansfield KD, et al. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-α activation. Cell Metab. 2005;1:393–399. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruchko MV, et al. Hypoxia-induced oxidative base modifications in the VEGF hypoxia-response element are associated with transcriptionally active nucleosomes. Free Radic Biol Med. 2009;46:352–359. doi: 10.1016/j.freeradbiomed.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Al-Mehdi AB, et al. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal. 2012;5:ra47. doi: 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ju BG, et al. A topoisomerase IIβ-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- 63.Perillo B, et al. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science. 2008;319:202–206. doi: 10.1126/science.1147674. [DOI] [PubMed] [Google Scholar]
- 64.Amente S, Lania L, Avvedimento EV, Majello B. DNA oxidation drives Myc mediated transcription. Cell Cycle. 2010;9:3002–3004. doi: 10.4161/cc.9.15.12499. [DOI] [PubMed] [Google Scholar]
- 65.Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459:996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoo SK, Starnes TW, Deng Q, Huttenlocher A. Lyn is a redox sensor that mediates leukocyte wound attraction in vivo. Nature. 2011;480:109–112. doi: 10.1038/nature10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sakai D, et al. Remodeling of actin cytoskeleton in mouse periosteal cells under mechanical loading induces periosteal cell proliferation during bone formation. PLoS ONE. 2011;6:e24847. doi: 10.1371/journal.pone.0024847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hattori H, et al. Small-molecule screen identifies reactive oxygen species as key regulators of neutrophil chemotaxis. Proc Natl Acad Sci USA. 2010;107:3546–3551. doi: 10.1073/pnas.0914351107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Henderson WR, Klebanoff SJ. Leukotriene production and inactivation by normal, chronic granulomatous disease and myeloperoxidase-deficient neutrophils. J Biol Chem. 1983;258:13522–13527. [PubMed] [Google Scholar]
- 70.Segal BH, Kuhns DB, Ding L, Gallin JI, Holland SM. Thioglycollate peritonitis in mice lacking C5, 5-lipoxygenase, or p47phox: complement, leukotrienes, and reactive oxidants in acute inflammation. J Leukoc Biol. 2002;71:410–416. [PubMed] [Google Scholar]
- 71.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nature Immunol. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.West AP, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 74.Shimada K, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dostert C, et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hogquist KA, Nett MA, Unanue ER, Chaplin DD. Interleukin 1 is processed and released during apoptosis. Proc Natl Acad Sci USA. 1991;88:8485–8489. doi: 10.1073/pnas.88.19.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shiloh MU, et al. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity. 1999;10:29–38. doi: 10.1016/s1074-7613(00)80004-7. [DOI] [PubMed] [Google Scholar]
- 78.Huang J, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci USA. 2009;106:6226–6231. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Espey MG. Role of oxygen gradients in shaping redox relationships between the human intestine and its microbiota. Free Radic Biol Med. 2013;55:130–140. doi: 10.1016/j.freeradbiomed.2012.10.554. [DOI] [PubMed] [Google Scholar]
- 80.Kumar A, et al. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J. 2007;26:4457–4466. doi: 10.1038/sj.emboj.7601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Winter SE, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. 2010;467:426–429. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maly FE, et al. The superoxide generating system of B cell lines. Structural homology with the phagocytic oxidase and triggering via surface Ig. J Immunol. 1988;140:2334–2339. [PubMed] [Google Scholar]
- 83.Maly FE, et al. Superoxide-dependent nitroblue tetrazolium reduction and expression of cytochrome b-245 components by human tonsillar B lymphocytes and B cell lines. J Immunol. 1989;142:1260–1267. [PubMed] [Google Scholar]
- 84.Singh DK, et al. The strength of receptor signaling is centrally controlled through a cooperative loop between Ca2+ and an oxidant signal. Cell. 2005;121:281–293. doi: 10.1016/j.cell.2005.02.036. [DOI] [PubMed] [Google Scholar]
- 85.Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med. 2002;195:59–70. doi: 10.1084/jem.20010659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hara-Chikuma M, et al. Chemokine-dependent T cell migration requires aquaporin-3-mediated hydrogen peroxide uptake. J Exp Med. 2012;209:1743–1752. doi: 10.1084/jem.20112398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Los M, et al. IL-2 gene expression and NF-kappa B activation through CD28 requires reactive oxygen production by 5-lipoxygenase. EMBO J. 1995;14:3731–3740. doi: 10.1002/j.1460-2075.1995.tb00043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Savina A, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006;126:205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 89.Mishell RI, Dutton RW. Immunization of normal mouse spleen cell suspensions in vitro. Science. 1966;153:1004–1006. doi: 10.1126/science.153.3739.1004. [DOI] [PubMed] [Google Scholar]
- 90.Nathan CF, Terry WD. Differential stimulation of murine lymphoma growth in vitro by normal and BCG-activated macrophages. J Exp Med. 1975;142:887–902. doi: 10.1084/jem.142.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Angelini G, et al. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc Natl Acad Sci USA. 2002;99:1491–1496. doi: 10.1073/pnas.022630299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sido B, et al. A prominent role for mucosal cystine/ cysteine metabolism in intestinal immunoregulation. Gastroenterology. 2008;134:179–191. doi: 10.1053/j.gastro.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 93.Yan Z, Garg SK, Kipnis J, Banerjee R. Extracellular redox modulation by regulatory T cells. Nature Chem Biol. 2009;5:721–723. doi: 10.1038/nchembio.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fisher RI, Bostick-Bruton F. Depressed T cell proliferative responses in Hodgkin’s disease: role of monocyte-mediated suppression via prostaglandins and hydrogen peroxide. J Immunol. 1982;129:1770–1774. [PubMed] [Google Scholar]
- 95.Efimova O, Szankasi P, Kelley TW. Ncf1 (p47phox) is essential for direct regulatory T cell mediated suppression of CD4+ effector T cells. PLoS ONE. 2011;6:e16013. doi: 10.1371/journal.pone.0016013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gelderman KA, Hultqvist M, Holmberg J, Olofsson P, Holmdahl R. T cell surface redox levels determine T cell reactivity and arthritis susceptibility. Proc Natl Acad Sci USA. 2006;103:12831–12836. doi: 10.1073/pnas.0604571103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mougiakakos D, Johansson CC, Jitschin R, Bottcher M, Kiessling R. Increased thioredoxin-1 production in human naturally occurring regulatory T cells confers enhanced tolerance to oxidative stress. Blood. 2011;117:857–861. doi: 10.1182/blood-2010-09-307041. [DOI] [PubMed] [Google Scholar]
- 98.Colombo MP, Piconese S. Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nature Rev Cancer. 2007;7:880–887. doi: 10.1038/nrc2250. [DOI] [PubMed] [Google Scholar]
- 99.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nature Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Muhlebach TJ, et al. Treatment of patients with chronic granulomatous disease with recombinant human interferon-gamma does not improve neutrophil oxidative metabolism, cytochrome b558 content or levels of four anti-microbial proteins. Clin Exp Immunol. 1992;88:203–206. doi: 10.1111/j.1365-2249.1992.tb03062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nagaraj S, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nature Med. 2007;13:828–835. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kusmartsev S, Gabrilovich DI. Inhibition of myeloid cell differentiation in cancer: the role of reactive oxygen species. J Leuk Biol. 2003;74:186–196. doi: 10.1189/jlb.0103010. [DOI] [PubMed] [Google Scholar]
- 103.Molon B, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. 2011;208:1949–1962. doi: 10.1084/jem.20101956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nathan C, Cohn Z. Role of oxygen-dependent mechanisms in antibody-induced lysis of tumor cells by activated macrophages. J Exp Med. 1980;152:198–208. doi: 10.1084/jem.152.1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nathan CF, Klebanoff SJ. Augmentation of spontaneous macrophage-mediated cytolysis by eosinophil peroxidase. J Exp Med. 1982;155:1291–1308. doi: 10.1084/jem.155.5.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nathan CF, Silverstein SC, Brukner LH, Cohn ZA. Extracellular cytolysis by activated macrophages and granulocytes. II Hydrogen peroxide as a mediator of cytotoxicity. J Exp Med. 1979;149:100–113. doi: 10.1084/jem.149.1.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nathan CF, Arrick BA, Murray HW, DeSantis NM, Cohn ZA. Tumor cell anti-oxidant defenses. Inhibition of the glutathione redox cycle enhances macrophage-mediated cytolysis. J Exp Med. 1981;153:766–782. doi: 10.1084/jem.153.4.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nathan CF, Cohn ZA. Antitumor effects of hydrogen peroxide in vivo. J Exp Med. 1981;154:1539–1553. doi: 10.1084/jem.154.5.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.O’Donnell-Tormey J, DeBoer CJ, Nathan CF. Resistance of human tumor cells in vitro to oxidative cytolysis. J Clin Invest. 1985;76:80–86. doi: 10.1172/JCI111981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 111.Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44:479–496. doi: 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ishikawa K, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–664. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 113.Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 114.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Weitzman SA, Weitberg AB, Clark EP, Stossel TP. Phagocytes as carcinogens: malignant transformation produced by human neutrophils. Science. 1985;227:1231–1233. doi: 10.1126/science.3975611. [DOI] [PubMed] [Google Scholar]
- 116.Lonkar P, Dedon PC. Reactive species and DNA damage in chronic inflammation: reconciling chemical mechanisms and biological fates. Int J Cancer. 2011;128:1999–2009. doi: 10.1002/ijc.25815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kohanski MA, DePristo MA, Collins JJ. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell. 2010;37:311–320. doi: 10.1016/j.molcel.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ishimoto T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc− and thereby promotes tumor growth. Cancer Cell. 2011;19:387–400. doi: 10.1016/j.ccr.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 119.Gilbertson RJ, Rich JN. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nature Rev Cancer. 2007;7:733–736. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- 120.Diehn M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Knoefler D, et al. Quantitative in vivo redox sensors uncover oxidative stress as an early event in life. Mol Cell. 2012;47:767–776. doi: 10.1016/j.molcel.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gomes A, Fernandes E, Lima JL. Fluorescence probes used for detection of reactive oxygen species. J Biochem Biophys Methods. 2005;65:45–80. doi: 10.1016/j.jbbm.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 123.Kim JH, et al. Single-molecule detection of H2O2 mediating angiogenic redox signaling on fluorescent single-walled carbon nanotube array. ACS Nano. 2011;5:7848–7857. doi: 10.1021/nn201904t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee D, et al. Detection of hydrogen peroxide with chemiluminescent micelles. Int J Nanomed. 2008;3:471–476. [PMC free article] [PubMed] [Google Scholar]
- 125.Belousov VV, et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nature Methods. 2006;3:281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- 126.Gutscher M, et al. Real-time imaging of the intracellular glutathione redox potential. Nature Methods. 2008;5:553–559. doi: 10.1038/nmeth.1212. [DOI] [PubMed] [Google Scholar]
- 127.Raj L, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 128.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nature Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 129.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 130.Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science. 2012;336:315–319. doi: 10.1126/science.1219192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nathan C. Fresh approaches to anti-infective therapies. Sci Transl Med. 2012;4:140sr2. doi: 10.1126/scitranslmed.3003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liby KT, Sporn MB. Synthetic oleanane triterpenoids: multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol Rev. 2012;64:972–1003. doi: 10.1124/pr.111.004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pineda-Molina E, et al. Glutathionylation of the p50 subunit of NF-κB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 134.Warburg O. Beobachtungen über die Oxydationsprozesse im Seeigelei. Z Physiol Chem. 1908;57:1–16. [Google Scholar]
- 135.Bentley R. In: The Enzymes. 2. Boyer PD, Lardy H, Myrbäck K, editors. Vol. 27. Academic; 1963. pp. 567–586. Ch. 24. [Google Scholar]
- 136.Wilson R, Turner APF. Glucose oxidase: an ideal enzyme. Biosensors & Bioelectronics. 1992;7:165–185. [Google Scholar]
- 137.Sbarra AJ, Karnovsky ML. The biochemical basis of phagocytosis. I Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J Biol Chem. 1959;234:1355–1362. [PubMed] [Google Scholar]
- 138.Iyer GYN, Islam MF, Quastel JH. Biochemical aspects of phagocytosis. Nature. 1961;192:535–541. [Google Scholar]
- 139.McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 140.Babior BM, Kipnes RS, Curnutte JT. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest. 1973;52:741–744. doi: 10.1172/JCI107236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Curnutte JT, Whitten DM, Babior BM. Defective superoxide production by granulocytes from patients with chronic granulomatous disease. N Engl J Med. 1974;290:593–597. doi: 10.1056/NEJM197403142901104. [DOI] [PubMed] [Google Scholar]
- 142.Nathan CF, Root RK. Hydrogen peroxide release from mouse peritoneal macrophages: dependence on sequential activation and triggering. J Exp Med. 1977;146:1648–1662. doi: 10.1084/jem.146.6.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Foerder CA, Klebanoff SJ, Shapiro BM. Hydrogen peroxide production, chemiluminescence, and the respiratory burst of fertilization: interrelated events in early sea urchin development. Proc Natl Acad Sci USA. 1978;75:3183–3187. doi: 10.1073/pnas.75.7.3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Segal AW, Jones OT. Novel cytochrome b system in phagocytic vacuoles of human granulocytes. Nature. 1978;276:515–517. doi: 10.1038/276515a0. [DOI] [PubMed] [Google Scholar]
- 145.Klebanoff SJ. Oxygen metabolism and the toxic properties of phagocytes. Ann Intern Med. 1980;93:480–489. doi: 10.7326/0003-4819-93-3-480. [DOI] [PubMed] [Google Scholar]
- 146.Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med. 1983;158:670–689. doi: 10.1084/jem.158.3.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nathan CF, et al. Local and systemic effects of intradermal recombinant interferon-γ in patients with lepromatous leprosy. N Engl J Med. 1986;315:6–15. doi: 10.1056/NEJM198607033150102. [DOI] [PubMed] [Google Scholar]
- 148.Royer-Pokora B, et al. Cloning the gene for an inherited human disorder — chronic granulomatous disease — on the basis of its chromosomal location. Nature. 1986;322:32–38. doi: 10.1038/322032a0. [DOI] [PubMed] [Google Scholar]
- 149.Ezekowitz RA, Dinauer MC, Jaffe HS, Orkin SH, Newburger PE. Partial correction of the phagocyte defect in patients with X-linked chronic granulomatous disease by subcutaneous interferon gamma. N Engl J Med. 1988;319:146–151. doi: 10.1056/NEJM198807213190305. [DOI] [PubMed] [Google Scholar]
- 150.Suh YA, et al. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 151.Grant SS, Kaufmann BB, Chand NS, Haseley N, Hung DT. Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals. Proc Natl Acad Sci USA. 2012;109:12147–12152. doi: 10.1073/pnas.1203735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Doroshow JH, Davies KJ. Comparative cardiac oxygen radical metabolism by anthracycline antibiotics, mitoxantrone, bisantrene, 4′-(9-acridinylamino)-methanesulfon-m-anisidide, and neocarzinostatin. Biochem Pharmacol. 1983;32:2935–2939. doi: 10.1016/0006-2952(83)90399-4. [DOI] [PubMed] [Google Scholar]
- 153.Dorr RT. Bleomycin pharmacology: mechanism of action and resistance, and clinical pharmacokinetics. Semin Oncol. 1992;19:3–8. [PubMed] [Google Scholar]
- 154.Liu Y, Imlay JA. Cell death from antibiotics without the involvement of reactive oxygen species. Science. 2013;339:1210. doi: 10.1126/science.1232751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Keren I, Wu Y, Inocencio U, Mulcahy LR, Lewis K. Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science. 2013;339:1213. doi: 10.1126/science.1232688. [DOI] [PubMed] [Google Scholar]