

Abstract
Aim
Excess mitochondrial reactive oxygen species (mROS) play a vital role in cardiac ischemia reperfusion (IR) injury. P66Shc, a splice variant of the ShcA adaptor protein family, enhances mROS production by oxidizing reduced cytochrome c to yield H2O2. Ablation of p66Shc protects against IR injury, but it is unknown if and when p66Shc is activated during cardiac ischemia and/or reperfusion and if attenuating complex I electron transfer or deactivating PKCβ alters p66Shc activation during IR is associated with cardioprotection.
Methods
Isolated guinea pig hearts were perfused and subjected to increasing periods of ischemia and reperfusion with or without amobarbital, a complex I blocker, or hispidin, a PKCβ inhibitor. Phosphorylation of p66Shc at serine 36 and levels of p66Shc in mitochondria and cytosol were measured. Cardiac functional variables and redox states were monitored online before, during and after ischemia. Infarct size was assessed in some hearts after 120 min reperfusion.
Results
Phosphorylation of p66Shc and its translocation into mitochondria increased during reperfusion after 20 and 30 min ischemia, but not during ischemia only, or during 5 or 10 min ischemia followed by 20 min reperfusion. Correspondingly, cytosolic p66Shc levels decreased during these ischemia and reperfusion periods. Amobarbital or hispidin reduced phosphorylation of p66Shc and its mitochondrial translocation induced by 30 min ischemia and 20 min reperfusion. Decreased phosphorylation of p66Shc by amobarbital or hispidin led to better functional recovery and less infarction during reperfusion.
Conclusion
Our results show that IR activates p66Shc and that reversible blockade of electron transfer from complex I, or inhibition of PKCβ activation, decreases p66Shc activation and translocation and reduces IR damage. These observations support a novel potential therapeutic intervention against cardiac IR injury.
Introduction
Mitochondria are proximal effectors and determinants of cell fate during ischemia and reperfusion (IR)-mediated oxidative stress. Thus they are also potential therapeutic targets to ameliorate oxidative damage [1]. Excess mitochondrial reactive oxygen species (mROS) emission plays a key role in contributing to cardiac IR injury [2]. It is generally accepted that in mitochondria the superoxide anion (O2 −•), the precursor of most ROS, is generated within the electron transport chain (ETC) complexes (e.g. I, II and III), wherein the leak of a single electron reduces O2 to O2 −• [3]–[5].
Recent reports indicate that p66Shc, a splice variant of the ShcA adaptor protein family, also contributes to mROS production [1], [6]. Giorgio et al. [6] suggested that p66Shc utilizes reducing equivalents of the ETC by oxidizing reduced cytochrome c (cyt c) to catalyze the reduction of O2 to H2O2. Electron transfer from cyt c to p66Shc would designate it as a mitochondrial redox enzyme [6] that could play an alternative role as a signaling molecule for mitochondrial-mediated cell apoptosis [7], [8]. Indeed, p66Shc gene ablation (p66Shc−/−) has been shown to reduce hypoxia/reoxygenation-induced damage to hepatocytes [9] and to decrease necrosis and apoptosis of myofibrils after hind limb ischemia compared to the wild type [10]. Furthermore, in isolated perfused hearts, p66Shc−/− mice compared to wild type mice exhibited both reduced IR-mediated LDH release into the coronary effluent and abrogated lipoperoxidation [11].
The pathway leading to p66Shc activation and translocation into mitochondria is unclear. Excess H2O2 or ultraviolet light (UV) irradiation has been shown to activate a serine-threonine protein kinase C β (PKCβ), which led to p66Shc phosphorylation at serine 36, and to trigger mitochondrial accumulation of the protein after its recognition by the prolyl isomerase Pin1 in mouse embryonic fibroblasts (MEF) [12], [13]. Pinton et al. [13] reported that in MEF, inhibition of PKCβ with hispidin inhibited H2O2 -induced p66Shc phosphorylation; overexpression of PKCβ mediated H2O2-induced mitochondrial dysfunction in wild type MEFs, but not in p66Shc−/− MEFs. It was reported that activation of PKCβII in ventricular tissue increased after IR and that gene deletion or pharmacological blockade of PKCβII was associated with protection against ischemia [14].
Mitochondrial ETC complexes are involved in mROS production during IR. Moreover, O2 −• generated at mitochondrial complex III can be attenuated by limiting electron transfer from complex I, thereby provide protection against IR injury. We [15], and others [16], have reported that the therapeutic targeting of complex I with amobarbital provided cardioprotection, in part, by decreasing mROS production during IR. Amobarbital, a short-acting barbiturate, reversibly attenuates complex I electron transfer at the rotenone site [17], decreased IR-induced O2 −• generation and mitochondrial [Ca2+] overload [15], retarded mitochondrial permeability transition pore (mPTP) opening [16], and improved oxidative phosphorylation (OxPhos) [16]. These mitochondrial effects culminated in appreciable protection of cardiac function on reperfusion after ischemia [15], [16]. However, targeting distal complexes of the ETC, especially complex IV, is not protective against ischemic stress and may exacerbate injury. For example, blocking complex IV before ischemia increased levels of reduced cyt c, a likely substrate for p66Shc-mediated H2O2 generation [6] leading to more oxidative stress.
Our aims were to explore if p66Shc is involved in IR induced mROS generation and how ROS and p66Shc dynamically modulate each other during different periods of cardiac ischemia and reperfusion. To address these objectives, we used the perfused ex vivo guinea pig heart model and monitored: a) if and when p66Shc is activated during cardiac ischemia and/or reperfusion; b) if activation of PKCβII during IR induces p66Shc activation and mitochondrial translocation to contribute to cardiac IR injury; c) if reversible attenuation of complex I electron transfer with amobarbital during IR is associated with p66Shc activation.
Methods
Ethics Statements
Our animal protocols conformed to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health No. 85-23, Revised 1996). The Medical College of Wisconsin IACUC, with the number AUA 1647, approved all our animal studies.
Isolated heart preparation and measurements
Hearts were removed and prepared for study as described previously [18]–[22]. In brief, guinea pigs were given heparin (1000 units) and ketamine (50 mg/kg) i.p. before sacrifice. Hearts were harvested and perfused retrograde at constant pressure (55 mmHg) via the aortic root with an oxygenated Krebs-Ringer's (KR) solution of the following composition (in mM): 138 Na+, 4.5 K+, 1.2 Mg2+, 2.5 Ca2+, 134 Cl−, 15 HCO3 −, 1.2 H2PO4 −, 11.5 glucose, 2 pyruvate, 16 mannitol, 0.1 probenecid, 0.05 EDTA, and 5 U/L insulin and gassed with 3% CO2, 97% O2 (pH 7.4) at 37°C. Systolic and diastolic left ventricular pressures (LVP), coronary flow (CF) and heart rate were measured online continuously for all of perfused hearts as described previously [18]–[22]. The aortic inflow line was clamped to induce global ischemia. If ventricular fibrillation (VF) occurred on reperfusion, 250 µg of lidocaine was given via the aortic cannula to restore sinus rhythm.
Protocols
At the end of the indicated times of ischemia and/or reperfusion, some hearts were removed and immediately snap frozen in liquid N2 and stored at −80°C for later measurement of p66Shc and PKCβII phosphorylation (n = 3 hearts per group). Other hearts were removed and mitochondria were immediately isolated for assessment of cytosolic and mitochondrial changes in p66Shc levels (n = 3 hearts per group). Other hearts were removed at 120 min reperfusion to measure infarct size (n = 8 hearts per group). In the complex I blocker treated groups, hearts were perfused with 2.5 mM amobarbital (Amo) for 1 min before initiating global ischemia [15]; in the PKCβ antagonist treated groups, hearts were perfused with 40 µM hispidin (His) for 10 min before ischemia. Amobarbital and hispidin were administered up to the initiation of ischemia to ensure the presence of the drugs in the heart during the entire period of ischemia and briefly during the onset of reperfusion.
Infarct size measurement
After 120 min reperfusion, hearts were removed and the atria discarded. The ventricles were cut into 3 mm sections, and then stained with 1% 2,3,5-triphenyltetrazolium chloride (TTC) to measure infarct size. In living tissue, TTC is reduced by pyridine nucleotide-linked dehydrogenase into a red, lipid-soluble formazan that stains living tissue red. Infarcted tissue lacking this dehydrogenase remains unstained. Infarct size of the TTC stained white area was determined as a percentage of total ventricular heart weight [22], [23].
Measurement of NADH in isolated hearts
Tissue autofluorescence (arbitrary fluorescence units, afu) is an indicator of NADH at λem 460 nm (λex 350 nm); it is primarily utilized to assess the mitochondrial redox state (n = 6 isolated hearts/group) [15], [24], [25]. Motion artifact was reduced by using λem 405 nm as a reference that is less sensitive to changes in NADH [26]. Therefore, the ratio of auf at λem 460/λem 405 nm indicates mitochondrial NADH.
Detection of O2 −• in isolated hearts loaded with dihydroethidium (DHE)
The intracellular fluorescent probe DHE (Molecular Probes) was used to assess O2 −• emission continuously during IR (n = 6 isolated hearts per group) as described previously [15], [20], [24], [27]. After stabilization, hearts were loaded with 10 µM DHE dissolved in KR solution for 20 min; this was followed by washout of residual, unincorporated DHE with KR solution for 20 min [15], [20]. The fluorescence emitted (afu) after washout was adjusted to 0 afu (baseline) to normalize the fluorescence intensity for all experiments. Changes in the DHE fluorescence signal, which represents O2 −• generation, were compared to the baseline fluorescence signal values [15], [18], [27].
Preparation of cytosolic and mitochondrial fractions
Mitochondrial and cytosolic fractions were prepared using procedures described previously [27]–[32] with minor modifications. All procedures were carried out at 4°C. Hearts were minced in a chilled isolation buffer containing (in mM) 200 mannitol, 50 sucrose, 5 KH2PO4, 5 MOPS, 1 EGTA and 0.1% fatty acid free BSA at pH 7.15, then homogenized. The homogenized slurries were centrifuged at 8000 g for 10 min. The supernatant were collected and further centrifuged at 50,000 g for 30 min, and then the resulting supernatant was used as the cytosolic fraction. The pellet from the 8,000 g centrifugation was resuspended in isolation buffer containing protease inhibitors and spun at 750 g for 10 min; the supernatant was collected and again centrifuged at 8000 g. The final mitochondrial pellet following this centrifugation was resuspended in isolation buffer and then purified as described by Graham [33]. Mitochondria were layered on 30% Percoll in isolation buffer, and then centrifuged for 30 min at 95,000 g. After centrifugation, the lower part of the dense brown band containing the purified mitochondria was collected and washed two times with isolation buffer. After purification, the mitochondria were resuspended in isolation buffer containing protease inhibitors and stored at −80°C for later use. Mitochondrial and cytosolic protein concentrations were assessed using Bio-Rad protein assay with bovine serum albumin (BSA) as the standard.
Immunoprecipitation of mitochondrial proteins
Immunoprecipitation (IP) was performed as described previously [27], [34], [35] with minor changes. Frozen hearts were pulverized under liquid nitrogen and then the powder was lysed in RIPA buffer containing 50 mM Tris-Cl pH 7.4, 150 mM NaCl, 1% deoxycholate, 1% Triton X-100, 0.1% SDS and protease inhibitors on ice for 30 min. The sample was pre-cleared with 30 µl protein G Sepharose-4B beads (Invitrogen) for 1 h at 4°C with constant end-over-end shaking and then centrifuged at 3000 g for 5 min. Supernatant was collected, adjusted to 2 mg/ml protein concentration with RIPA buffer containing protease inhibitors, and subjected to IP with an anti-Shc antibody (rabbit polyclonal IgG, Millipore) at 4°C with constant end-over-end shaking overnight. The next day 30 µl of protein G Sepharose-4B beads was added and the mixture was incubated for another 2 h under the same conditions as above. The beads were collected and washed (five times) with RIPA buffer. After washing and aspirating the washing buffer completely, 50 µl Laemmli sample buffer containing 50 mM Tris-Cl, 10% glycerol, 500 mM β-mercaptoethanol, 2% SDS, 0.01% w/v bromophenol blue and protease inhibitors at pH 7.4, was added to the beads and boiled at 95°C for 5 min. The immunoprecipated proteins were separated using SDS-PAGE and then subjected to Western blot analyses.
Western blot analyses
Mitochondrial protein (50 µg) or immunoprecipitates were resolved by SDS-PAGE and transferred onto a PVDF membrane. Membranes were incubated with specific primary antibodies: anti-Shc/p66 (pSer36) (mouse monoclonal, 1∶1000, Calbiochem); anti-Shc (total pool of p66Shc, mouse monoclonal, 1∶1000, BD Transduction Laboratories); PKCβII (rabbit polyclonal, 1∶500 Santa Cruz); and P-PKCβII/δ (Ser660) (rabbit polyclonal, 1∶500, Santa Cruz). Washed membranes were incubated with the appropriate secondary antibody conjugated to HRP, then immersed in an enhanced chemiluminescence detection solution (GE Healthcare) and exposed to X-Ray film for autoradiography. To monitor the amounts of loaded proteins and purity of isolated mitochondrial and cytosolic fractions, specific antibody-blotted membranes were stripped and then probed with antibodies against VDAC (rabbit polyclonal, 1∶1000, Cell Signaling) or β-actin (rabbit polyclonal, 1∶500, Santa Cruz).
Statistical analysis
All results were expressed as means ±SEM and were analyzed by one-way ANOVA followed by a post-hoc analysis (Student-Newman-Keuls' test) to determine significant differences of means among groups. Isolated heart data were collected for statistical evaluation at time points 30, 45, 50, and 65 min. P<0.05 was considered significantly different (two-tailed).
Results
Activation of p66Shc occurred during reperfusion after ischemia
We first examined if and when p66Shc was activated during cardiac IR by determining the level of phosphorylation of p66Shc at serine 36 (Ser36), an indication of p66Shc activation [12], [13]. After normalizing to total immunoprecipated p66Shc (Fig. 1A, bottom panel), phosphorylation of p66Shc at Ser36 increased on reperfusion in the IR groups compared to the time control (TC) and ischemia only groups (Fig. 1A, upper panel); the increase in phosphorylation of p66Shc at Ser36 occurred as early as 10 min reperfusion after 30 min ischemia and was detectable for up to 60 min reperfusion. In the 20 and 30 min ischemia only groups, the phosphorylation of p66Shc at Ser36 did not change compared to the TC group. These results indicated that IR induced p66Shc activation and, moreover, that p66Shc was activated only during reperfusion after ischemia and not by ischemia alone.
Figure 1. Phosphorylation of p66Shc at Ser36 increased during reperfusion (R) after ischemia (I) in isolated guinea pig hearts compared to no ischemia (time controls, TC).
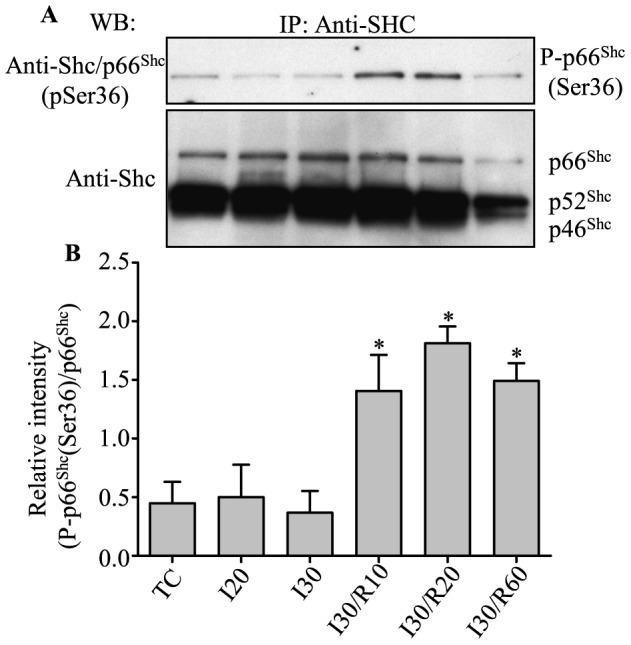
A, upper panel: phosphorylation of p66Shc at Ser36 from total protein lysate of heart tissue. A, lower panel: total p66Shc level for loading control. Summary of mean band intensities (B) derived from three independent experiments (n = 3 hearts/group); band intensities were determined by densitometry using imageJ software and the intensities of P-p66 (Ser36) were normalized to the total p66Shc in each sample. P<0.05: *IR vs. TC and I.
P66Shc localized to mitochondria during reperfusion after ischemia
It was reported that after phosphorylation at Ser36, p66Shc is recognized by prolyl isomerase Pin1, which enables the phosphorylated p66Shc to be translocated to the IMM [13]. We therefore examined the translocation of p66Shc into mitochondria during cardiac IR injury. At the end of the indicated ischemia and/or reperfusion period, hearts were removed and cytosolic and mitochondrial fractions were prepared (see Methods), and then subjected to Western blot analyses with anti-Shc antibody. The level of p66Shc in mitochondria increased by approximately 2-fold at 10 and 60 min reperfusion, and by approximately 3-fold at 20 min reperfusion after 30 min ischemia, compared to TC (Fig. 2). The level of p66Shc in the cytosolic fraction decreased in the 30 min ischemia alone group and in the 30 min ischemia followed by 10, 20 or 60 min reperfusion groups when compared to the TC and 20 min ischemia groups (Fig. 2).
Figure 2. P66Shc expression levels changed in cytosol and mitochondria during reperfusion after ischemia.

A, upper panel: p66Shc level in mitochondrial and cytosolic fractions. Anti-VDAC (A, middle panel) and anti-β–actin (A, lower panel) confirm protein loading, mitochondrial (β–actin) and cytosolic purity (VDAC). Summary of mean band intensities (B) derived from three independent experiments (n = 3 hearts/group) (see Fig. 1 for imaging). P<0.05: *IR vs. TC and I20; # IR vs I30; a I30R60 vs I30R10 or I30R20.
The incongruity in the reverse changes in p66Shc levels in cytosolic and mitochondrial fractions in the 30 min ischemia alone group indicated that p66Shc might translocate to other parts of the cell, including organelles other than mitochondria. Moreover, the cytosolic p66Shc level was lower in the 30 min ischemia plus 10 and 20 of reperfusion groups when compared to the 30 min ischemia plus 60 min of reperfusion group (Fig. 2). These results suggested greater cell injury/death after 60 min of reperfusion contributed to the lower p66Shc levels. The translocation of p66Shc into mitochondria during reperfusion after 30 min ischemia was consistent with phosphorylation of p66Shc at Ser36 during the indicated periods (Fig. 1). Insofar as ischemia alone did not lead to phosphorylation of p66Shc, correspondingly, ischemia alone (20 or 30 min) also did not show significant mitochondrial translocation of p66Shc when compared to their respective TCs.
Prolonged ischemia required to activate p66Shc during reperfusion after ischemia
Our previous reports show that the magnitude of ROS production during ischemia is dependent on the duration of ischemia [15], [20], [27]. Next, we investigated if the activation of p66Shc was associated with the duration of ischemia. Guinea pig isolated hearts were subjected to 5, 10, 20 or 30 min ischemia, followed by 20 min reperfusion. At the end of reperfusion, hearts were harvested and evaluated for phosphorylation of p66Shc at Ser36. Phosphorylation of p66Shc at Ser36 increased after 20 min reperfusion following 20 or 30 min global ischemia, but not after 5 or 10 min ischemia plus 20 min reperfusion compared to their respective TCs (Fig. 3). These results indicated that at least 20 min ischemia was required for some p66Shc activation during reperfusion after ischemia.
Figure 3. Phosphorylation of p66Shc at Ser36 during reperfusion after ischemia was dependent on the duration of ischemia.
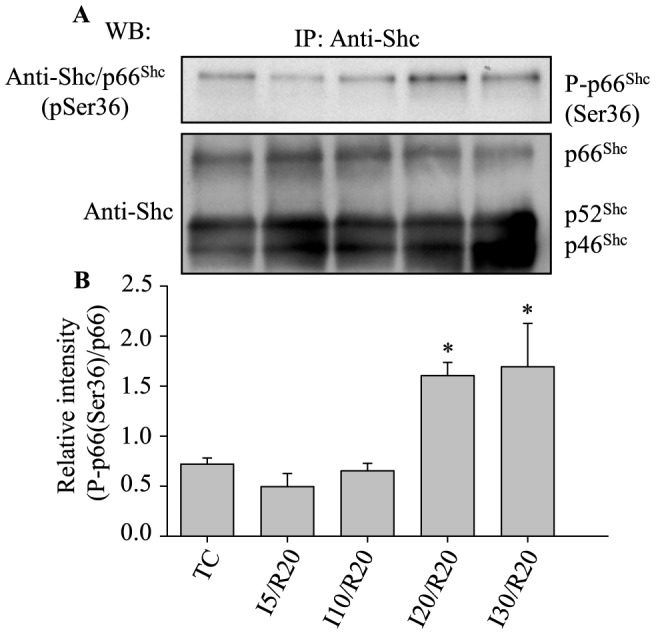
A, upper panel: phosphorylation of p66Shc at Ser36; A, lower panel: total p66Shc level for loading control. B: Summary of mean band intensities derived from three independent experiments (n = 3 hearts/group) (see Fig. 1 for imaging). P<0.05: *I20 and I30 vs. I5, I10 and TC.
To evaluate a possible correlation between p66Shc activation and translocation with the mitochondrial redox state and generation of ROS, online mitochondrial NADH and DHE (O2 −• emission) fluorescence intensities were measured during 30 min ischemia and 20 min reperfusion. During early ischemia NADH increased markedly, but then gradually declined toward baseline levels as ischemia progressed (Fig. 4A, IR); during reperfusion, NADH was lower than baseline (Fig. 4A, IR). These findings indicated a shift in mitochondrial redox towards more oxidized mitochondria during early reperfusion. The corresponding time-dependent changes in DHE fluorescence signals were characterized by a modest steady increase during early ischemia and a marked and accelerated increase during late ischemia (Fig. 4B, IR); the DHE fluorescence signal intensity remained elevated above baseline during reperfusion in the IR group.
Figure 4. NADH autofluorescence (afu) (A) was higher and DHE fluorescence intensity (O2 −• emission) (B) was lower during ischemia and reperfusion after amobarbital (Amo) treatment compared to IR alone. DHE and NADH were recorded online in isolated hearts at the LV free wall.

n = 6 hearts/group/fluorescence measure. Cardiac contractile and relaxant function: developed LVP (C), diastolic LVP (D), dLVP/dt min (E) and dLVP/dt max (F) were improved on reperfusion after amobarbital (Amo) treatment during ischemia. Inset in D shows detailed changes in diastolic LVP during late ischemia and early reperfusion. n = 9 hearts/group/functional measure. All values are means±SEM. TC, time control; IR, ischemia and reperfusion. P<0.05: *IR vs. TC; #Amo+IR vs. IR.
Amobarbital decreased p66Shc activation induced by ischemia and reperfusion
We next sought to determine if reversible blockade of complex I with amobarbital, which we showed previously reduced O2 −• emission during IR [15], would reduce p66Shc activation during reperfusion. To achieve this, hearts were treated with or without amobarbital (Amo) prior to ischemia followed by 20 or 30 min ischemia and 20 min reperfusion. At the end of reperfusion, hearts were collected and evaluated for phosphorylation of p66Shc at Ser36 (see Methods). Compared to the IR alone group, the amobarbital treated groups exhibited reduced phosphorylation of p66Shc at Ser36 by 27.5±0.1% and 14.4±0.1% at 20 min reperfusion after 20 min (Fig. 5A) and 30 min (Fig. 5B) ischemia, respectively. However, phosphorylation of p66Shc at Ser36 in the amobarbital treated groups was significantly higher than in the TC group. These data indicated, and supported our previous findings [15], that amobarbital-induced interference with mitochondrial electron transfer provides protection, at least in part, by attenuating p66Shc activation during IR.
Figure 5. IR-induced phosphorylation (P) of p66Shc at Ser36 was decreased by amobarbital (Amo) after 20 (A) or 30 (B) min ischemia (I) followed by 20 min reperfusion (R).
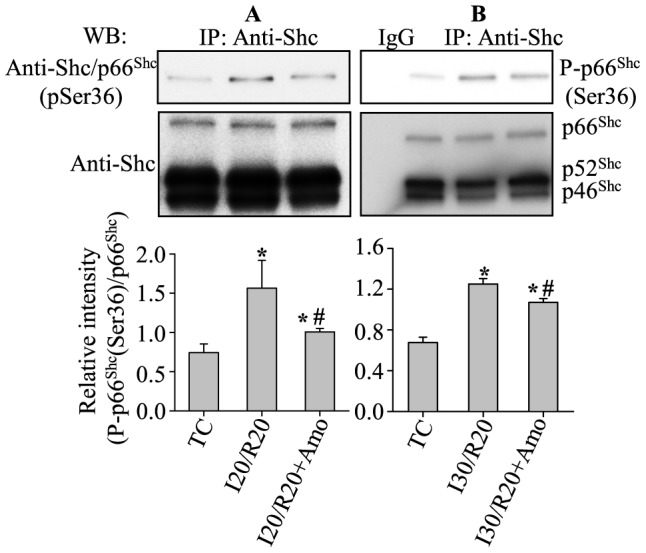
A, B, upper panel: phosphorylation of p66Shc at Ser36; equal amounts of p66Shc loading were verified with anti-ShC antibody (A, B: middle panel). Summary of mean band intensities derived from three independent experiments (n = 3 hearts/group) (A, B: lower panel). IP, immunoprecipitation; TC, time controls (no IR). P<0.05: *IR vs. TC; #Amo+IR vs. IR.
Therefore, to ascertain if attenuation of p66Shc activation by amobarbital correlated with mitochondrial redox state, ROS production, and functional recovery during IR injury, we monitored NADH, O2 −• generation and several indices of contractility and relaxation online in amobarbital treated and untreated hearts during IR. Compared to untreated hearts, amobarbital treated hearts displayed better preserved mitochondrial NADH levels during 30 min ischemia and 20 min reperfusion (Fig. 4A; Amo+IR), and a decrease in DHE fluorescence (O2 −• production) during late ischemia (Fig. 4B; Amo+IR). Moreover, amobarbital treatment resulted in a significantly lower diastolic LVP during late ischemia and early reperfusion (Fig. 4D; Amo+IR) and improved developed LVP, dLVP/dt min and dLVP/dt max during early reperfusion (Fig. 4C, E, F; Amo+IR) compared to the IR alone group. The incidence of ventricular fibrillation (VF) in the IR untreated hearts was 100%, with an average of approximately 3 VF occurrences/heart during reperfusion; amobarbital treated hearts exhibited no incidence of VF during reperfusion. Each VF that occurred was subsequently reversed to sinus rhythm with lidocaine. It is noteworthy that although the 20 min ischemia plus 20 min reperfusion group demonstrated activation of p66Shc, there was no significant compromise in cardiac function or mitochondrial bioenergetics when compared to the TC group (data not shown). This suggested that p66Shc activation is an early marker of ischemia.
Activation of PKCβII induced by cardiac ischemia and reperfusion required for p66Shc activation
Next we examined if PKCβII is activated during cardiac IR, and if activation of p66Shc during reperfusion after ischemia is mediated through PKCβII activated pathways as previously reported in MEFs [13], [36]. Western blot was used to determine phosphorylation of PKCβII at Ser660 in hearts subjected to 20 or 30 min ischemia plus 20 min reperfusion. Figure 6A shows that 20 or 30 min ischemia plus 20 min reperfusion increased phosphorylation of PKCβII at Ser660, suggesting IR activated PKCβII. Next we determined if PKCβII activation is required for p66Shc activation during reperfusion. For this, hearts were treated with or without hispidin (His) for 10 min before the start of 30 min ischemia plus 20 min reperfusion. At the end of reperfusion, heart tissue was evaluated for phosphorylation of p66Shc at Ser36; alternatively, mitochondria were isolated immediately after reperfusion to evaluate mitochondrial p66Shc translocation. Figures 6B and C show that when hearts were perfused with hispidin before ischemia compared to IR only (control), both p66Shc phosphorylation at Ser36 and p66Shc mitochondrial translocation were decreased. These observations support the signaling role of PKCβ in activating the oxidoreductase, p66Shc, during oxidative stress in IR.
Figure 6. IR-induced activation of p66Shc occurred via the PKCβII signaling pathway.

A: Representative WB of 3 independent experiments of phosphorylation of PKCβII. B: Phosphorylation of p66Shc at Ser36 during IR with or without hispidin (His) treatment. C: p66Shc accumulation in mitochondria during IR with or without hispidin (His) treatment. B, C Lower panels show summary of mean band intensities derived from three independent experiments (n = 3 hearts/group) (see Fig. 1 for imaging). P<0.05: *IR vs. TC; #IR+His vs. IR.
We correlated attenuation of p66Shc activation by hispidin with functional recovery and ventricular infarct size. Figure 7A shows that administration of hispidin before ischemia significantly reduced diastolic contracture, i.e. diastolic LVP, when compared to the IR only hearts. The incidence of VF after ischemia was significantly abated in hearts treated with hispidin before ischemia when compared to IR only hearts (data not shown). Figure 7B also shows that hispidin attenuated infarct size to 33±3% compared to the untreated IR (control) group in which infarct size was 45±3%. These results showed that ischemia followed by reperfusion induces p66Shc activation through the PKCβII signaling pathway to contribute to IR injury.
Figure 7. Inhibiting PKCβ activation reduced cardiac damage after IR.
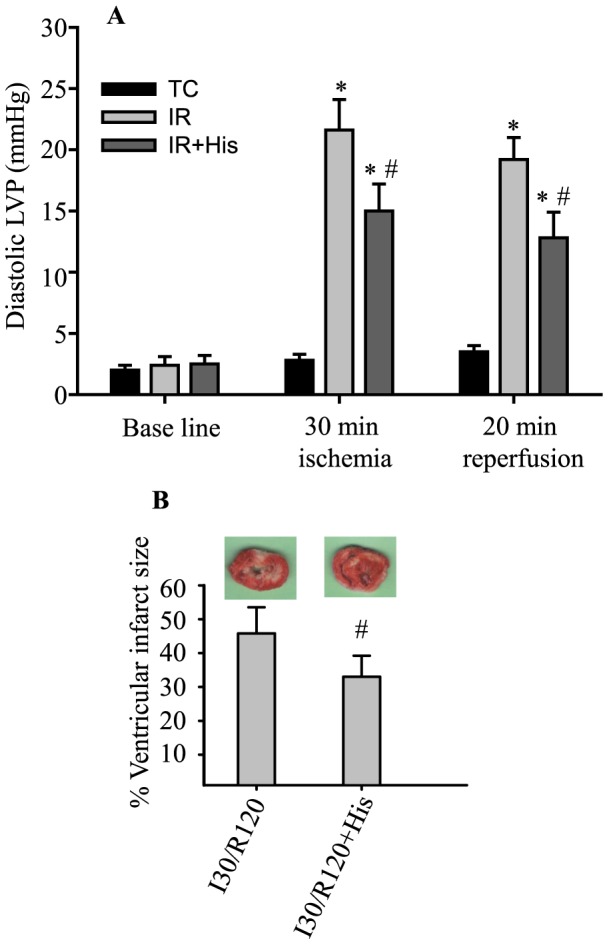
A: Diastolic LVP without IR (TC; n = 8 hearts/group) and before ischemia, at 30 min of ischemia and at 20 min of reperfusion (IR; n = 12 hearts/group) with or without hispidin (His; n = 10 hearts/group) treatment. B: Infarct size after 30 min of ischemia and 120 min of reperfusion (n = 8 hearts/group) with or without hispidin (His; n = 8 hearts/group) treatment. P<0.05: #IR+His vs. IR.
Discussion
We have demonstrated that p66Shc is activated and translocated into mitochondria during global no-flow IR in the ex vivo perfused heart model. Activation of p66Shc and its subsequent translocation into mitochondria are dependent both on the duration of ischemia and on the occurrence and timing of reperfusion after ischemia. Reversible attenuation of respiration via complex I by amobarbital reduced both ROS emission and activation of p66Shc and improved function on reperfusion. Inhibiting PKCβ during IR with hispidin reduced activation/translocation of p66Shc, improved cardiac function, and decreased cell injury. Thus, p66Shc is both a marker and a mitochondrial effector of oxidative stress-mediated mitochondrial dysfunction and ROS production. The effects of amobarbital on reducing mROS emission, improving redox state, and attenuating p66Shc activation during IR injury, suggest an important link between oxidative stress and activation of p66Shc.
P66Shc is activated during reperfusion after global cardiac ischemia
IR injury can occur under a number of situations including cardiac arrest and resuscitation, hypoxia and reoxygenation, and coronary artery occlusion and reperfusion. Although ischemia itself can induce cardiac injury, much of the injury occurs during early reperfusion [37]. The causative and interactive factors of myocardial reperfusion injury include excess ROS, Ca2+ overload, and induction of apoptosis [38]. It is well known that the major source of ROS emission during reperfusion in cardiomyocytes is the ETC, which is progressively damaged during ischemia and contributes to further ROS generation during reperfusion [2], [20], [39]–[41].
However recent studies showed that p66Shc may also act as an oxidoreductase to generate H2O2 in mitochondria after activation following redox stimulation [6]. Furthermore, in the mouse isolated heart model of IR injury, p66Shc−/− mice showed less susceptibility to IR injury than wild type mice [11]. In the present study, we showed that p66Shc only becomes activated and translocated to mitochondria during reperfusion after 20 or 30 min ischemia (Figs. 1, 2 and 3). This suggests that p66Shc could also contribute to mROS generation during IR injury especially during reperfusion after ischemia. Why p66Shc is activated only during reperfusion is unclear, but it is possible that the preceding events during ischemia (e.g. excess ROS emission) followed by restoration of flow, O2 and substrate supply, and by the rapid re-establishment of a reduced redox state and OxPhos, may collectively contribute to p66Shc activation.
Ischemia, when prolonged, irreversibly disrupts electron transfer during reperfusion, yet ETC complexes I–III may not be the only sources of ROS during reperfusion [41]. It is possible that during the reperfusion phase of IR injury, p66Shc induces additional ROS generation via a putative direct effect on oxidoreductase activity, which depends on electron leak from upstream complexes (e.g. I and III) and then becomes an additional source of ROS production [6], [13] that may exacerbate the ROS generated from damaged complexes. This could initiate a vicious cycle of excess ROS emission by mitochondrial ROS-induced-ROS release (RIRR) during reperfusion.
Long duration of ischemia with reperfusion is necessary for p66Shc activation
We found that 5 and 10 min ischemia plus 20 min reperfusion did not increase activation of p66Shc, whereas 20 and 30 min ischemia with reperfusion increased p66Shc activation markedly. This indicated that a longer duration of ischemia plus reperfusion was required to activate p66Shc. The duration of ischemia is generally linked to the extent of injury to ETC complexes [41], [42]. In rats, complex I activity decreased early in ischemia, whereas complex III damage occurred only after a longer ischemia time [41], [43]. Consistent with the time-dependent ischemic damage to ETC complexes, ROS emission also displays an ischemia duration dependent pattern. In the guinea pig heart, we showed a correlation between NADH, O2 −• levels and duration of ischemia (Fig. 4A, B) [15], [18], [24]. Our present (Fig. 4B) and past [15], [27] results in guinea pig isolated hearts showed small amounts of O2 −• were generated during early (1–20 min) global ischemia followed by a marked surge in O2 −• generation during late (25–30 min) global ischemia. Therefore, the longer durations of ischemia (20 and 30 min), when followed by at least 10 min reperfusion, are likely to have induced ETC complex damage leading to enhance ROS emission. Excess ROS (possibly H2O2), beyond a certain threshold, may act as deleterious redox-signaling molecules that lead to activation of p66Shc, which in turn enhances mROS generation to induce further oxidative damage to ETC complexes leading to RIRR.
Unlike 30 min ischemia, 20 min ischemia (both followed by 20 min reperfusion) induced p66Shc activation (Fig. 3), but had no significant effect on cardiac function on reperfusion (data not shown). This suggests that p66Shc is more sensitive to redox modulation than the measured cardiac function during IR injury. These observations show overall that p66Shc participates in emitting ROS involved in cardiac dysfunction during longer ischemia times as with reperfusion.
Amobarbital decreases activation of p66Shc during cardiac IR injury
Our current and previous studies [15] show that amobarbital, when present during ischemia and early reperfusion, preserved NADH (Fig. 4A), minimized O2 −• emission (Fig. 4B), and reduced mitochondrial Ca2+ overload [15], which concomitantly improved functional recovery (Fig. 4 C–F) and reduced infarction when compared to IR only (control) [15]. Furthermore, in this study we also found that compared to IR alone, amobarbital reduced activation of p66Shc induced by ischemia and reperfusion (Fig. 5). Since amobarbital reversibly binds at the rotenone site of complex I to attenuate electron transfer [17], [44], [45], this suggests that amobarbital, when present in the tissue during ischemia, attenuates activation of p66Shc by blunting electron transfer to reduce O2 −• generation. Our observations further suggest that amobarbital better protected mitochondria and reduced myocardial injury during reperfusion, possibly due to attenuated p66Shc activation and its concomitant translocation into mitochondria to modulate ROS emission.
When p66Shc is activated and translocated into mitochondria, it becomes a pro-apoptotic protein that induces apoptosis by altering the redox state [6]; however, the detailed mechanism for this remains unclear [46]. Amobarbital-induced attenuation of p66Shc activation during IR demonstrates that p66Shc is affected by electron transfer related events that modulate the ETC. Thus, insofar as amobarbital was presumed present in the myocardium during ischemia, it may have reduced activation of p66Shc during reperfusion by decreasing ischemic ROS emission. Hence, the amobarbital-induced decrease in ROS production during ischemia appears to decrease ROS emission on reperfusion, attenuate activation of cytosolic PKCβII, reduce activation and translocation of p66Shc into mitochondria, and ultimately, decrease mitochondrial and cellular damage.
PKCβ induces p66Shc activation and its translocation into mitochondria during IR
The signaling pathways that lead to p66Shc phosphorylation at Ser36 during cardiac ischemia and/or reperfusion have not been reported before. In this study, we show that inhibiting PKCβ with hispidin reduced both p66Shc phosphorylation at Ser36 and mitochondrial translocation during IR (Fig. 6 B and C). These results indicate that p66Shc activation and its eventual translocation into mitochondria during IR likely occur through PKCβ-mediated signaling pathways; this is consistent with previous studies in which different stress models were used [13], [36], [47]. Several previous reports show common p66Shc phosphorylation signaling pathways, indicating an independence of the stimulus and cell type. For example, incubation of human endothelial cell with oxidized LDL led to phosphorylation [36] of p66Shc; in MEFs, treatment with H2O2 and UV induced phosphorylation [13] of p66Shc; and A549 and RAW 264.7 cells treated with Taxol, an antitumor drug, induced phosphorylation [48] of p66Shc. Among these stimuli and cell types, the signaling pathways that induce p66Shc phosphorylation first involve activating PKCβ [13], [36], followed by activation of other kinases [36], [47], [48] that directly induce phosphorylation of p66Shc at Ser36 [36].
The role of PKCβII in modulating cardiac function also has been reported. Kong et al. [14] observed significant increases in PKCβII cell membrane translocation and phosphorylation after 30 min of LAD occlusion followed by 30 min reperfusion. Moreover, PKCβ−/−, or its blockade with ruboxistaurin, reduced infarct size by 2.6 fold after 30 min regional ischemia and 48 h reperfusion in mice. Consistent with these findings, we found PKCβII was activated (specifically by its phosphorylation at Ser660) during IR (Fig. 6A). Furthermore, hispidin, when given before ischemia, reduced diastolic contracture (Fig. 7A) and infarct size (Fig. 7B). Although other studies also show that inhibition of PKCβ activation is protective against cardiac IR injury, our study provides evidence that PKCβII modulates cardiac IR injury by triggering the activation and translocation of p66Shc into mitochondria (Fig. 6B, C).
Summary, Conclusions and Limitations
We provide evidence that p66Shc is activated and translocated from the cytosol into mitochondria in cardiac IR injury. Activation of p66Shc occurred only during reperfusion via PKCβII activation. The magnitude of p66Shc activation correlated with the degree of cell damage, so that the greatest activation/translocation of p66Shc had the worst functional recovery, a higher incidence of VF, and a greater infarct size. Reversible blockade of complex I reduced the ROS level necessary to activate p66Shc. These observations imply that excess ROS and altered redox state resulting from IR injury are single or combined factors that initiate p66Shc activation and mitochondrial translocation.
Persistent activation of p66Shc may lead to a vicious cycle of mitochondrial RIRR as a result of redox-sensitive activation of PKCβII. Therefore, preventing p66Shc activation may reduce the feed-forward cycle of RIRR and lessen reperfusion injury. The findings that p66Shc mediates only detrimental ROS production during reperfusion provides a novel opportunity for pharmacological interventions that would target this redox-signaling cascade leading to p66Shc activation and translocation into mitochondria after the onset of cardiac IR injury. Overall, this study provides additional valuable insights into the possible mechanisms of how modulating electron transfer along the ETC can minimize deleterious ROS emission during cardiac IR injury as a means to improve cardiac function. A potential limitation is that attenuation of p66Shc translocation into mitochondria by modulating electron transfer is less pronounced than the effect on functional recovery, which indicates that other factors in addition to the effects of p66Shc on translocation exert a role on ROS emission during the progression of ischemia into reperfusion.
Acknowledgments
The authors acknowledge Bhawana Agarwal and Mohammed Aldakkak for their technical assistance in the study and for reading the manuscript. We thank Anita Tredeau for her administrative assistance.
Footnote:
This work was funded in part by the National Institutes of Health (R01-HL095122 and R01-HL089514) and published in part in abstract form: Yang M, Stowe DF, Heisner JS, Camara AKS. Attenuating complex I activity decreases p66shc phosphorylation and translocation to mitochondria during cardiac ischemia reperfusion injury. FASEB J 105:1144.2, 2013.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.
Funding Statement
This work was supported by funding from the National Heart Lung and Blood Institute of the National Institutes of Health R01 HL 095122 NIH (awarded to Dr. Camara) and R01 HL 089514 NIH (awarded to Dr. Stowe). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Camara AK, Lesnefsky EJ, Stowe DF (2010) Potential therapeutic benefits of strategies directed to mitochondria. Antioxid Redox Signal 13:279–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stowe DF, Camara AK (2009) Mitochondria reactive oxygen species production in excitable cells: Modulators of mitochondrial and cell function. Antioxid Redox Signal 11:1373–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ (2003) Production of reactive oxygen species by mitochondria: Central role of complex III. J Biol Chem 278:36027–36031. [DOI] [PubMed] [Google Scholar]
- 4. St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD (2002) Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 277:44784–44790. [DOI] [PubMed] [Google Scholar]
- 5. Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, et al. (2012) Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem 287:27255–27264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, et al. (2005) Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122:221–233. [DOI] [PubMed] [Google Scholar]
- 7. Camara AK, Stowe DF (2014) ROS and cardiac ischemia and reperfusion injury. System Biology of Free Radicals and Anti-Oxidants Chapter 75:1. [Google Scholar]
- 8. Camara AK, Bienengraeber M, Stowe DF (2011) Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front Physiol 2:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haga S, Terui K, Fukai M, Oikawa Y, Irani K, et al. (2008) Preventing hypoxia/reoxygenation damage to hepatocytes by p66Shc ablation: Up-regulation of anti-oxidant and anti-apoptotic proteins. J Hepatol 48:422–432. [DOI] [PubMed] [Google Scholar]
- 10. Zaccagnini G, Martelli F, Fasanaro P, Magenta A, Gaetano C, et al. (2004) p66ShcA modulates tissue response to hindlimb ischemia. Circulation 109:2917–2923. [DOI] [PubMed] [Google Scholar]
- 11. Carpi A, Menabo R, Kaludercic N, Pelicci P, Di Lisa F, et al. (2009) The cardioprotective effects elicited by p66Shc ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim Biophys Acta 1787:774–780. [DOI] [PubMed] [Google Scholar]
- 12. Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, et al. (1999) The p66Shc adaptor protein controls oxidative stress response and life span in mammals. Nature 402:309–313. [DOI] [PubMed] [Google Scholar]
- 13. Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, et al. (2007) Protein kinase C β and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 315:659–663. [DOI] [PubMed] [Google Scholar]
- 14. Kong L, Andrassy M, Chang JS, Huang C, Asai T, et al. (2008) PKCβ modulates ischemia-reperfusion injury in the heart. Am J Physiol Heart Circ Physiol 294:H1862–70. [DOI] [PubMed] [Google Scholar]
- 15. Aldakkak M, Stowe DF, Chen Q, Lesnefsky EJ, Camara AK (2008) Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+ overload and ROS release. Cardiovasc Res 77:406–415. [DOI] [PubMed] [Google Scholar]
- 16. Chen Q, Lesnefsky EJ (2011) Blockade of electron transport during ischemia preserves bcl-2 and inhibits opening of the mitochondrial permeability transition pore. FEBS Lett 585:921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chance B, Williams GR, Hollunger G (1963) Inhibition of electron and energy transfer in mitochondria. I. effects of amytal, thiopental, rotenone, progesterone, and methylene glycol. J Biol Chem 238:418–431. [PubMed] [Google Scholar]
- 18. Aldakkak M, Stowe DF, Heisner JS, Riess ML, Camara AK (2011) Adding ROS quenchers to cold K+ Cardioplegia reduces superoxide emission during 2-hour global cold cardiac ischemia. J Cardiovasc Pharmacol Ther 17:93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Camara AK, Riess ML, Kevin LG, Novalija E, Stowe DF (2004) Hypothermia augments reactive oxygen species detected in the guinea pig isolated perfused heart. Am J Physiol Heart Circ Physiol 286:H1289–99. [DOI] [PubMed] [Google Scholar]
- 20. Kevin LG, Camara AK, Riess ML, Novalija E, Stowe DF (2003) Ischemic preconditioning alters real-time measure of O2 radicals in intact hearts with ischemia and reperfusion. Am J Physiol Heart Circ Physiol 284:H566–74. [DOI] [PubMed] [Google Scholar]
- 21. Novalija E, Varadarajan SG, Camara AK, An J, Chen Q, et al. (2002) Anesthetic preconditioning: Triggering role of reactive oxygen and nitrogen species in isolated hearts. Am J Physiol Heart Circ Physiol 283:H44–52. [DOI] [PubMed] [Google Scholar]
- 22. Stowe DF, Gadicherla AK, Zhou Y, Aldakkak M, Cheng Q, et al. (2013) Protection against cardiac injury by small Ca2+-sensitive K+channels identified in guinea pig cardiac inner mitochondrial membrane. Biochim Biophys Acta 1828:427–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stowe DF, Aldakkak M, Camara AK, Riess ML, Heinen A, et al. (2006) Cardiac mitochondrial preconditioning by big Ca2+-sensitive K+ channel opening requires superoxide radical generation. Am J Physiol Heart Circ Physiol 290:H434–40. [DOI] [PubMed] [Google Scholar]
- 24. Camara AK, Aldakkak M, Heisner JS, Rhodes SS, Riess ML, et al. (2007) ROS scavenging before 27 degrees C ischemia protects hearts and reduces mitochondrial ROS, Ca2+ overload, and changes in redox state. Am J Physiol Cell Physiol 292:C2021–31. [DOI] [PubMed] [Google Scholar]
- 25. Aldakkak M, Stowe DF, Lesnefsky EJ, Heisner JS, Chen Q, et al. (2009) Modulation of mitochondrial bioenergetics in the isolated guinea pig beating heart by potassium and lidocaine cardioplegia: Implications for cardioprotection. J Cardiovasc Pharmacol 54:298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Riess ML, Camara AK, Chen Q, Novalija E, Rhodes SS, et al. (2002) Altered NADH and improved function by anesthetic and ischemic preconditioning in guinea pig intact hearts. Am J Physiol Heart Circ Physiol 283:H53–60. [DOI] [PubMed] [Google Scholar]
- 27. Yang M, Camara AK, Wakim BT, Zhou Y, Gadicherla AK, et al. (2012) Tyrosine nitration of voltage-dependent anion channels in cardiac ischemia-reperfusion: Reduction by peroxynitrite scavenging. Biochim Biophys Acta 1817:2049–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Haumann J, Dash RK, Stowe DF, Boelens AD, Beard DA, et al. (2010) Mitochondrial free [Ca2+] increases during ATP/ADP antiport and ADP phosphorylation: Exploration of mechanisms. Biophys J 99:997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heinen A, Aldakkak M, Stowe DF, Rhodes SS, Riess ML, et al. (2007) Reverse electron flow-induced ROS production is attenuated by activation of mitochondrial Ca2+-sensitive K+ channels. Am J Physiol Heart Circ Physiol 293:H1400–7. [DOI] [PubMed] [Google Scholar]
- 30. Aldakkak M, Stowe DF, Cheng Q, Kwok WM, Camara AK (2010) Mitochondrial matrix K+ flux independent of large-conductance Ca2+-activated K+ channel opening. Am J Physiol Cell Physiol 298:C530–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gadicherla AK, Stowe DF, Antholine WE, Yang M, Camara AK (2012) Damage to mitochondrial complex I during cardiac ischemia reperfusion injury is reduced indirectly by anti-anginal drug ranolazine. Biochim Biophys Acta 1817:419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hiona A, Lee AS, Nagendran J, Xie X, Connolly AJ, et al. (2011) Pretreatment with angiotensin-converting enzyme inhibitor improves doxorubicin-induced cardiomyopathy via preservation of mitochondrial function. J Thorac Cardiovasc Surg 142:396–403.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Graham JM (2001) Purification of a crude mitochondrial fraction by density-gradient centrifugation. Curr Protoc Cell Biol Chapter 3:Unit 3.4. [DOI] [PubMed] [Google Scholar]
- 34. Kanski J, Behring A, Pelling J, Schoneich C (2005) Proteomic identification of 3-nitrotyrosine-containing rat cardiac proteins: Effects of biological aging. Am J Physiol Heart Circ Physiol 288:H371–81. [DOI] [PubMed] [Google Scholar]
- 35. Liu B, Tewari AK, Zhang L, Green-Church KB, Zweier JL, et al. (2009) Proteomic analysis of protein tyrosine nitration after ischemia reperfusion injury: Mitochondria as the major target. Biochim Biophys Acta 1794:476–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shi Y, Cosentino F, Camici GG, Akhmedov A, Vanhoutte PM, et al. (2011) Oxidized low-density lipoprotein activates p66Shc via lectin-like oxidized low-density lipoprotein receptor-1, protein kinase C-β, and c-jun N-terminal kinase kinase in human endothelial cells. Arterioscler Thromb Vasc Biol 31:2090–2097. [DOI] [PubMed] [Google Scholar]
- 37. Gorenkova N, Robinson E, Grieve DJ, Galkin A (2013) Conformational change of mitochondrial complex I increases ROS sensitivity during ischemia. Antioxid Redox Signal [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pagliaro P, Moro F, Tullio F, Perrelli MG, Penna C (2011) Cardioprotective pathways during reperfusion: Focus on redox signaling and other modalities of cell signaling. Antioxid Redox Signal 14:833–850. [DOI] [PubMed] [Google Scholar]
- 39. Piantadosi CA, Zhang J (1996) Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke 27:327–31 discussion 332. [DOI] [PubMed] [Google Scholar]
- 40. Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ (2008) Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol 294:C460–6. [DOI] [PubMed] [Google Scholar]
- 41. Sack MN (2006) Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovasc Res 72:210–219. [DOI] [PubMed] [Google Scholar]
- 42. Veitch K, Hombroeckx A, Caucheteux D, Pouleur H, Hue L (1992) Global ischaemia induces a biphasic response of the mitochondrial respiratory chain. anoxic pre-perfusion protects against ischaemic damage. Biochem J 281 (Pt 3):709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL (2001) Mitochondrial dysfunction in cardiac disease: Ischemia–reperfusion, aging, and heart failure. J Mol Cell Cardiol 33:1065–1089. [DOI] [PubMed] [Google Scholar]
- 44. Horgan DJ, Singer TP, Casida JE (1968) Studies on the respiratory chain-linked reduced nicotinamide adenine dinucleotide dehydrogenase. 13. binding sites of rotenone, piericidin A, and amytal in the respiratory chain. J Biol Chem 243:834–843. [PubMed] [Google Scholar]
- 45. Spiegel HE, Wainio WW (1969) Some features of barbiturate interaction and inhibition of NADH-cytochrome C oxidoreductase in respiring systems. J Pharmacol Exp Ther 165:23–29. [PubMed] [Google Scholar]
- 46. Galimov ER (2010) The role of p66Shc in oxidative stress and apoptosis. Acta Naturae 2:44–51. [PMC free article] [PubMed] [Google Scholar]
- 47. Chahdi A, Sorokin A (2010) Endothelin-1 induces p66Shc activation through EGF receptor transactivation: Role of β1Pix/Gαi3 interaction. Cell Signal 22:325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang CP, Horwitz SB (2002) Distinct mechanisms of taxol-induced serine phosphorylation of the 66-kDa shc isoform in A549 and RAW 264.7 cells. Biochim Biophys Acta 1590:76–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.