

Significance
The ubiquitin system controls a wide range of processes in cells and provides attractive drug targets for the treatment of cancer and other diseases. However, it has proved difficult to obtain inhibitors of the ligases that conjugate ubiquitin to substrates, of which there are hundreds. One class, the HECT (homologous to E6AP C terminus) domain ligases, receives ubiquitin from an E2 enzyme and transfers it to substrate. We have selected bicyclic peptides that block the E2 binding site of individual HECT ligases, as well as a small molecule, heclin (HECT ligase inhibitor), that broadly inhibits these ligases in cells. These inhibitors demonstrate that HECT domains are druggable targets and provide tools to study ubiquitination. The same approach could be used to select further HECT inhibitors.
Keywords: inhibitor, ubiquitin, HECT ligase, phage display
Abstract
The human genome encodes several hundred E3 ubiquitin ligases containing RING domains, and around 28 containing HECT domains. These enzymes catalyze the transfer of ubiquitin from E2 enzyme thioesters to a huge range of substrates and play crucial roles in many cellular functions. This makes them attractive potential therapeutic targets. However, they have proven difficult to inhibit: very few good inhibitors exist for RING domain ligases, and none have been described for HECT ligases. Here we show that bicyclic peptides isolated by phage display [Heinis C, Rutherford T, Freund S, Winter G (2009) Nat Chem Biol. 5(7):502–507] can target the E2 binding sites on the HECT domains of Smurf2, Nedd4, Mule/Huwe1, and WWP1, and thus act as specific inhibitors of these enzymes in vitro. By screening for displacement of one of these peptides from Smurf2, we were able to identify a small molecule, heclin (HECT ligase inhibitor), which inhibits several HECT ligases in tissue culture cells. In vitro, heclin does not block E2 binding but causes a conformational change that results in oxidation of the active site Cys. This demonstrates that HECT domains are potentially druggable and provides molecules that may be of experimental use. Heclin kills HEK293 cells growing in culture, consistent with an essential role for HECT ligase activity in mammalian cells.
Ubiquitin ligases are involved in almost every important function of cells, including the removal of misfolded proteins, the regulation of signaling pathways, DNA repair, the cell cycle, and apoptosis (1–4). This broad range of functions is mediated by a large number of E3 ubiquitin ligases, which recognize a similarly large range of substrates. Ubiquitination begins with the ATP-dependent formation of a ubiquitin (Ub)-E2 enzyme thioester intermediate by the E1 enzyme. The E3 enzymes then transfer ubiquitin from this intermediate to a lysine residue on the substrate (3). There are two main classes of E3: the RING domain ligases and their relatives, of which there are several hundred in humans, and the HECT domain ligases, which number around 28 (5–7). Because of their diversity and numerous functions and the fact that some ligases are frequently overexpressed in cancer cells, ubiquitin ligases have attracted much attention as possible targets for therapeutic intervention (2, 5, 6, 8).
Despite this interest, it has proven challenging to make small molecule inhibitors of ubiquitin ligases. The action of these enzymes involves primarily protein–protein interactions among them, the Ub-E2 conjugate, and the substrate. For the RING enzymes, this is simultaneous, and the Ub moiety is transferred directly from E2 to substrate; for the HECT ligases, the interaction is sequential, with a thioester-linked Ub-E3 intermediate forming transiently. Such protein–protein interactions are considered difficult to block with small molecules. Added to this difficulty has been the complexity of the ubiquitin system, with ligases having many substrates and many proteins being substrates for multiple ligases (6, 9). Despite these problems, peptide and small molecule inhibitors have been successfully obtained that specifically target the substrate binding site of Mdm2, a RING ligase that acts on p53, and consequently activate the p53 pathway (10, 11). However, only a few other inhibitors of RING ligases have been published (12, 13), and none have been published that inhibit HECT ligases in vivo.
Here we describe both peptide-based specific inhibitors of individual HECT ligases and a small molecule inhibitor with broad specificity for these enzymes. Using a previously described phage display method (14), we isolated bicyclic peptides (bicycles) that bind specifically to the HECT domains of Smurf2, Nedd4, WWP1, and Mule/Huwe1. We found that these frequently targeted the E2 binding site and inhibited ligase activity in vitro. We then used Alpha screening technology (15) to identify molecules that disrupted the bicycle–Smurf2 interaction. We identified one compound, termed heclin (HECT ligase inhibitor), that inhibits a range of HECT ligases in vitro and in tissue culture cells, with IC50s in the low micromolar range. In vitro, heclin does not prevent E2 binding but induces a conformational change that results in spontaneous oxidation of the active site cysteine. Heclin allows clear distinction between RING and HECT-mediated ubiquitination, and on prolonged exposure, heclin can kill growing cells.
Results and Discussion
Isolation of Bicycles that Bind to Smurf2 and Nedd4.
The bicycle method makes use of a phage library displaying peptides containing three Cys residues. The phages are treated under mild conditions with Tris-(bromomethyl)benzene, which reacts with all three cysteines, forming two peptide loops of six amino acids linked to the benzene ring (14, 16). We screened this library for phages that bound to His-tagged Smurf2 HECT domain immobilized on Ni-NTA beads (Methods). After three rounds of selection, using imidazole elution to release all bound phages, the population was dominated by a single bicycle sequence (bicycle A-1; Fig. 1A). Because this bicycle could bind to any part of the HECT domain, and our goal was to find inhibitors of activity, we repeated the selection, using the E2 enzyme UbcH7 to elute. The rationale was that this should preferentially elute bicycles that occupy the E2 binding site on the HECT domain, which in turn should be competitive inhibitors of activity. Surprisingly, the selected phages were again dominated by a few sequences very similar to the initial one, suggesting the E2 binding site is a preferential site for bicycles to interact (bicycles B-1 and B-2; Fig. 1A).
Fig. 1.

Isolation of bicycles that inhibit HECT ligases. (A) Sequences of bicycles selected on the Smurf2 HECT domain, either with total phage elution (A-1) or with phage eluted with UbcH7 (B-1, B-2). Affinity maturation used libraries in which the first loop was fixed as that of B-2. Similar residues are indicated; the percentage figures indicate the proportion of sequenced phage (typically 48) that had each sequence. (B) Inhibition of Smurf2 HECT auto-ubiquitination assayed by ELISA. (C) Bicycles selected on the Nedd4 HECT domain. Affinity maturation was based on A-11, which bound more tightly than A-1. (D) Inhibition of Nedd4 HECT auto-ubiquitination (using UbcH7), assayed by immunoblotting. (E) Bicycles selected on HECT domains from other ligases, with IC50s determined by ELISA as described in Methods. See Figs. S1–S3 for more data. HA, hemagglutinin.
Because the sequence convergence was strongest in the first of the bicycle loops, we sought to improve affinity by preparing a phage library in which the first loop was fixed and the second loop randomly varied. Two rounds of selection from this library showed convergence on two new sequences in the second loop (RYR and YIH; Fig. 1A and Fig. S1).
As with all members of the Nedd4 family of ligases, Smurf2 is normally inhibited by intramolecular interactions (17, 18), but the isolated HECT domain is constitutively active. We were thus able to assay chemically synthesized bicycles for their ability to inhibit auto-ubiquitination of the Smurf2 HECT domain in an ELISA (Fig. 1B). The best bicycle (Smurf2-RYR) inhibited ubiquitination with an apparent IC50 of 2 µM in the presence of 2.8 µM UbcH7 and was chosen for further study. As a direct measure of binding affinity, we used isothermal titration calorimetry, which showed a Kd of 2.5 µM, which was comparable to the IC50 value (Fig. S1).
We used a similar approach with the Nedd4 HECT domain. Again, we found convergence on a small number of bicycle sequences (Fig. 1C). Maturation of the first loop of the best binder (A-11) resulted in reselection of a very similar sequence (Nedd4-RGS), suggesting affinity could not easily be improved by this means. Assay of ubiquitination, as shown by ELISA as well as immunoblotting, gave an apparent IC50 of around 8 µM (Fig. 1D and Fig. S2). The observed IC50 increased when the concentration of E2 enzyme was increased (Fig. S2), consistent with the expected role of the bicycle as a competitive inhibitor of E2 binding.
The ability to create bicycle inhibitors appears to be a general property of HECT domains. Following the same methodology, we were able to obtain bicycles that inhibit Mule/Huwe1 and WWP1, with IC50s of 1–8 µM when assayed with 750 nM UbcH5 (Fig. 1E and Fig. S3).
Specificity of Bicycle Binding.
Testing of phage binding with different HECT domains showed that binding was highly specific. The Smurf2-RYR phage did bind the HECT domain from Smurf1, which is more than 80% identical in sequence, but did not bind the HECT domains of Nedd4, Nedd4L, or WWP1. The Nedd4-RGS phage bound only Nedd4 of these five tested HECT domains (Fig. 2A). Similarly, biotinylated synthetic versions of the Nedd4 and Smurf2 bicycles could pull down their target proteins selectively from lysates of HEK293T cells (Fig. 2B).
Fig. 2.

Specificity of bicycle binding. (A) ELISA of phage binding to indicated HECT domains. Smurf2* and Nedd4* contain E2 binding site mutations (see Results). (B) Pull down of full-length HECT ligases expressed in HEK293 cells with biotinylated bicycles in vitro. (C) Pull down of Nedd4 (N4) and Smurf 2 (S2) EGFP-HECT fusions by biotinylated bicycles, with E2 binding site mutations (E2m) and/or mutation of the active site cysteine (C/G) to prevent ubiquitination and degradation. The N4-E2m mutant retains some activity, and thus requires the C/G mutation for stability. (D) Substitution of the Nedd4 E2 binding site into Mule (as described in Results) switches bicycle binding specificity.
Bicycle Binding Involves the E2 Binding Site.
Crystal structures of the HECT domains of E6AP and Nedd4L bound to E2 enzyme show that the E2 binding site is a concavity on a specific subdomain whose surface is largely defined by an alpha helix, a beta strand, and a loop between them (19, 20). Point mutations in the corresponding alpha helix of Nedd4 (EY680/1AH, based on ref. 19) and Smurf2 (WI535/6CT, ref. 21) efficiently blocked binding of phages bearing the corresponding bicycles (Smurf2* and Nedd4*; Fig. 2A), as well as pull-down of the HECT domains from 293T cell lysates by biotinylated bicycles (E2m; Fig. 2C). These results suggest bicycles can inhibit the enzymes by obstructing the E2 binding site. Phage-bearing bicycles that inhibited Mule or WWP1 were similarly unable to bind to the corresponding HECT domains in which the E2 binding site was mutated (Fig. S3).
We confirmed that the bicycles recognize the E2 binding region by creating a chimera of the Mule HECT domain in which this region (residues 4,152–4,214 of the intact protein) was replaced with that of Nedd4 (residues 680–740 of Nedd4-1 isoform 4). The wild-type Mule HECT domain was pulled down by biotinylated Mule-specific bicycle M6, whereas the chimera could only be precipitated by the Nedd4-specific bicycle (Fig. 2D). Because the chimera contained only wild-type sequences, this is good evidence that the main determinant of bicycle binding is indeed the E2 binding domain.
Small Molecule Inhibitors of Bicycle Binding.
Our results show that bicycles can provide highly specific inhibitors of HECT domain activity, but we found it difficult to deliver them effectively into cells for in vivo studies. Instead, we exploited their properties in a screen for small molecule inhibitors. Specifically, we used Alpha screen technology (PerkinElmer) to search for compounds that blocked binding of a biotinylated Smurf2 bicycle to a GST-HECT fusion, reasoning that these might also block E2 binding. The bicycle and HECT were immobilized on streptavidin and anti-GST IgG coated donor and acceptor beads, respectively. In the Alpha assay, an optical signal is obtained when donor and acceptor are linked by ligands, and compounds that disrupt the binding result in a decreased signal (15). A counter screen using biotinylated GST to link the beads was used to eliminate compounds that interfere nonspecifically with the optical readout.
From some 17,500 compounds, we selected around 30 of the best hits and screened them for their ability to inhibit Smurf2 activity in vitro and in vivo. For the in vivo assay, the Smurf2 HECT domain was expressed as a GFP fusion protein in HEK293 cells; being constitutively active, this protein becomes ubiquitinated and rapidly degraded, but if it is inhibited, ubiquitination is reduced and the protein is stabilized. Around half the compounds demonstrated inhibitory activity in vitro, but only one showed consistent, albeit weak, inhibition in vivo of both Smurf2 and Nedd4 (compound I; Fig. 3A). To search for a better inhibitor, we purchased three closely related compounds (II, III, and IV) and characterized this set in more detail. The compounds showed very similar inhibitory profiles with three different HECT domains of the Nedd4 family in vitro, with the closely related compounds I and III being most potent (Table 1). Compound I was also able to inhibit the distantly related HECT ligases Ube3c and Mule.
Fig. 3.

Small molecule inhibitors. (A) Structure of compound I, from the screening library, and close relatives tested. (B) Effects of the compounds on HECT fusions expressed in HEK293 cells. Cell lysates were immunoblotted; upper bands are ubiquitinated forms of the proteins. Compounds were dissolved in DMSO, and controls without any addition (co) or with DMSO alone are shown. (C) Effects of heclin on the Smurf2 HECT fusion. Cells were cotransfected with a plasmid that expressed His-tagged ubiquitin, and the upper panel shows proteins recovered from Ni-NTA beads to enrich for ubiquitinated forms; note that the unmodified fusion protein also binds to some extent.
Table 1.
IC50 (µM) for HECT ligase inhibition by compounds I–IV
| Compound | HECT domain | ||||
| Smurf2 | Nedd4 | WWP1 | Ube3c | Mule | |
| I | 7.4 | 7.1 | 8.7 | 8.0 | 23 |
| II | 112 | 55 | 88 | nd* | nd |
| III/heclin | 6.8 | 6.3 | 6.9 | nd | nd |
| IV | 19 | 24 | 28 | nd | nd |
Although they had similar activities in vitro, compound III was strikingly more effective than compound I at inhibiting auto-ubiquitination of the Smurf2 HECT domain in vivo (Fig. 3B). Ubiquitinated Smurf2 disappeared after about 30 min of treatment, whereas unmodified protein steadily accumulated (Fig. 3C). A dose–response showed loss of ubiquitinated protein with an apparent IC50, based on densitometry of the ubiquitinated bands of 9 µM, which is comparable to the in vitro value (Fig. S4). It may be that the weaker activity of compound I in cells is a result of reduced availability, perhaps because of interaction with a serum or cellular component. Given its activity, we named compound III heclin.
In vivo, heclin remained active for at least 4 h, and its action on Smurf2 was reversible even in the presence of cycloheximide, implying reactivation of existing ligase (Fig. S4). We next tested it in an assay for the ubiquitination and degradation of the Dishevelled protein (Dvl). Dvl contains a PY motif and is a substrate for several members of the Nedd4 family (22–25), which bind PY motifs via their WW domains. This was readily demonstrated by coexpression of Dvl2 with Smurf2 and other ligases. Full-length Smurf2, Nedd4, and WWP2 all showed inhibition by heclin in this assay (Fig. 4A). Smad1 ubiquitination by Smurf2 was also inhibited (Fig. S4). We also tested a variety of enzymes with Ndfip2, a membrane protein that activates, and is a substrate for, Nedd4 family ligases (17). WWP1, WWP2, Nedd4, and Nedd4L all promoted ubiquitination and degradation of Ndfip2, and this was inhibited by heclin (Fig. 4B). Lower levels of heclin-sensitive Ndfip2 ubiquitination were detectable without coexpression of a ligase, presumably resulting from the activity of endogenous Nedd4-like ligases.
Fig. 4.
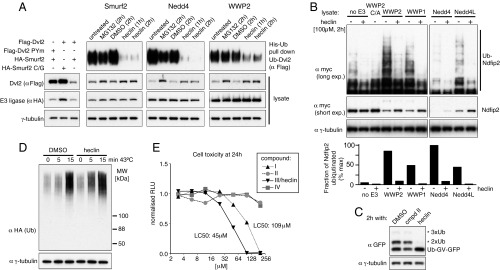
Effects of heclin on ubiquitination and cell viability. (A) Tagged full-length Smurf2 and Dvl2 were coexpressed with His-Ub, and ubiquitination of Dvl2 detected by Ni-NTA enrichment and immunoblotting. The first three lanes show Dvl2 destabilization prevented by mutation of the Smurf2 active site (C/G) or the PY motif in Dvl2 (PYm). Other lanes contain wild-type proteins and are from cells treated with heclin or the proteasome inhibitor MG132 (20 µM), as indicated. WWP2 activity on Dvl2 is considerably greater than that of the other ligases, and the His-Ub pull down shown is a lighter exposure. (B) Cells were cotransfected with myc-tagged Ndfip2 and the indicated full-length HECT ligases (C/A indicates active site mutation). The histogram shows the amount of ubiquitination normalized for the amount of Ndfip2 present, estimated by densitometry of the blots. (C) Cells expressing noncleavable Ub-GFP were treated as indicated, and their lysates were immunoblotted. (D) Cells expressing hemagglutinin (HA)-Ub were treated with heclin and harvested after incubation at 43 °C for the times indicated. (E) HEK293 cell toxicity measured after 24 h exposure to the indicated compounds. The CellTiter-Glo assay (Promega) measures total ATP in the samples. In A–D, heclin (in DMSO) was added at 100 µM, and controls contained the same final concentration of DMSO (1%).
To test activity on other endogenous HECT ligases, we used a construct in which ubiquitin is fused to GFP, with the final Ub glycine residue mutated to valine, which prevents its removal. Such fusions are subject to additional ubiquitination and degradation, with the ubiquitination being ascribed to multiple HECT domain ligases including TRIP12, Mule/Huwe1, and possibly HectD1 (26, 27). Fig. 4C shows that addition of extra Ub molecules was prevented by heclin, but not by the inactive compound II. In contrast, heclin did not reduce steady-state levels of ubiquitinated total protein (Fig. S4), nor prevent the increase in ubiquitination immediately after a brief heat shock (Fig. 4D); in fact, both were slightly enhanced. This ubiquitination is presumably mediated predominantly by RING domain ligases, and as a further control, we also observed no effect of heclin on the auto-ubiquitination of the RING domain ligase Mdm2 in vitro, using the same E1 and E2 enzymes as for the HECT ligases (Fig. S4). We conclude that heclin is active against a range of HECT ligases but does not perturb the ubiquitin system more generally.
Cell Killing by Heclin.
Although the above experiments show that cells can tolerate heclin treatment for several hours, exposure for 24 h led to the death of growing HEK293 cells. Given the involvement of HECT ligases in many important cellular processes, this is likely to be the direct result of their inhibition. In support of this, cell killing by the related compounds I–IV correlated well with their inhibitory abilities in vivo (Fig. 4E; cf. Fig. 3B), with compound I being less potent than heclin and compounds II and IV being nontoxic even at 200 µM. The median lethal concentration for heclin on growing cells was 45 µM, which is about five times higher than the IC50 for Smurf2. However, precisely which HECT ligase or ligases are critical for life, and whether heclin has any additional effects, remain unknown.
Mechanism of Heclin Action.
In vitro, heclin inhibited ubiquitination activity within a few minutes, did not cause significant aggregation of the E3 ligase, and did not inhibit the activity of the E1 enzyme in the assay (Fig. S5). However, unlike the bicycles, it did not appear to be a competitive inhibitor of E2 binding, as the IC50 was little affected by an eightfold increase in E2 concentration. Moreover, a fluorescence polarization assay that directly measures HECT binding to E2 (28) also showed no effect of heclin (Fig. S6).
To gain further insight into the mechanism of inhibition, we performed hydrogen/deuterium exchange studies with the Smurf2 HECT domain in the presence or absence of heclin or bicycle. In this assay, the protein is exposed to D2O, which allows hydrogens in the amino group of the peptide bonds to be exchanged for deuterium. Exchange is stopped at various times by reducing the pH, the protein digested with pepsin, and the deuterium content of peptides determined by mass spectrometry (29). Because the rate of exchange depends strongly on tertiary structure and solvent accessibility, such studies can give information about binding or protein conformation, which are revealed by changes in the kinetics of deuterium incorporation into specific peptides (30, 31).
Fig. 5 (Lower left) shows a plot of the difference in percentage deuteration of each Smurf2 peptide induced by the bicycle, averaged over all replicates and time points (see Dataset S1 for the full data). Two regions, each covered by multiple peptides, showed significant reduction in exchange. Both of these are located in the E2 binding region, as defined by reference to the crystal structures of Nedd4L and E6AP HECT/E2 complexes (19, 20) (Fig. 5). This suggests that, as expected, the bicycle binds in this region, thereby reducing solvent accessibility. Other minor differences were also observed, but these are closer to the background variation seen when different datasets for the HECT domain alone are compared (SI Methods and Fig. S7), and thus are harder to interpret.
Fig. 5.

H/D exchange assays. (Lower) Plots of the difference in deuteration of each peptide induced by bicycle or heclin, with the position of the peptide plotted along the protein sequence axis. Note many regions contain multiple overlapping peptides. See SI Methods and Dataset S1 for detailed experimental and analytic methods and a full dataset. (Upper) Locations of the peptides corresponding to peaks of ligand-induced protection significantly above background on the crystal structure of Smurf2 (Protein Data Bank ID code 1ZVD).
The effects of heclin were quite different. Increased protection was observed for a loop on the underside of the C-lobe (676–697) close to the active site cysteine, and more weakly for the adjacent hinge region and the tip of a helix on the N-lobe that abuts the loop in the Smurf2 crystal structure (Fig. 5). Conversely, decreased protection was seen for the FDEKE sequence at position 617 and two additional regions that are adjacent to FDEKE in the structure (see plot in Fig. 5). The HECT C-lobe is very flexible in position, and its function requires oscillation between different orientations (32). One places the active site cysteine close to the E2 site, where it can receive ubiquitin (as exemplified by the crystal structure of WWP1) (33), and another places it far away, in a conformation that allows transfer of ubiquitin to a substrate lysine (as seen in the Smurf2 crystal structure) (21). The changes induced by heclin are unlikely to reflect direct contact with this small ligand but are, instead, consistent with movement of the C-lobe from a conformation similar to that in the WWP1 crystal to one more similar to that in the Smurf2 crystal (Fig. S7). Such movement could hinder transfer of Ub thioester from the E2 to the HECT domain.
Indeed, analysis of reactions on nonreducing gels, which preserve thioesters, showed no E3-linked Ub when heclin was present (Fig. 6A). The E3-Ub thioester is normally a transient intermediate, but certain point mutations in the yeast HECT ligase Rsp5 have been shown to prevent transfer of Ub to substrate lysines, thus allowing the thioester to accumulate (32). An equivalent mutation in Smurf2 (D433A, corresponding to Rsp5 D495A) allowed detection of the thioester and confirmation that heclin prevented its appearance (Fig. 6B).
Fig. 6.

Heclin blocks formation of a Ub-E3 thioester. In vitro assays were analyzed after varying times by blotting for ubiquitin under conditions that preserve (nonreducing) or destroy (reducing) thioesters. (A) Wild-type Smurf2. (B) Smurf2 D433A, which is impaired in transfer of ubiquitin from the active site cysteine to lysine residues. ME, mercaptoethanol.
In our in vitro assays, heclin was typically preincubated with the GST-HECT fusion protein before addition of the rest of the reaction mix. Surprisingly, we found that inhibition was greatly reduced if DTT was added to the preincubation, but not if it were added later (Fig. 7A; for more comprehensive data, see Fig. S8A). This suggests that the full effect of heclin requires the ligase to be oxidized. Analysis of GST-Smurf2 on nonreducing SDS gels showed that heclin induced both dimeric and heterogeneously migrating monomeric forms of the protein, and the concentration of heclin required for this effect matched that required for inhibition of activity (Fig. 7B). This effect was rapid and could be prevented by prior addition of DTT, but was not easily reversed if was added later. It occurred also with GST-Nedd4 but was abolished by mutation of the active site cysteine; heating the samples in SDS and mercaptoethanol largely restored the normal monomeric forms (Fig. 7 C and D and Fig. S8B).
Fig. 7.

Heclin induces DTT-sensitive modifications. (A) DTT prevents heclin inhibition if present during preincubation with the HECT ligase. (B) Heclin was preincubated with GST-Smurf2 for 15 min and half analyzed by immunoblotting on a nonreducing gel (Lower); the other half was assayed for activity, detected by immunoblotting for Ub conjugates (Upper). (C) Rapid heclin-induced changes to GST-Smurf2 visualized by Coomassie staining. Where indicated, DTT (1 mM) was added before or 5 min after heclin. (D) Heclin also induces dimers with GST-Nedd4, but not if the active site cysteine is mutated to serine (C/S). For further data relating to A and C, see Fig. S8.
These results suggest that in the presence of heclin, the active site Cys is reversibly oxidized, presumably to the highly reactive sulfenic acid form (34). This is then likely to form a disulfide link, which would be reduced more slowly. GST fusions are dimeric in solution (Fig. S5D), and some of the links are evidently to the other subunit of this dimer. Incubation of heclin-treated enzyme in glutathione-coated plates, which provide both a reducing agent and a binding site for GST, restored activity to the bound protein, showing that inhibition is reversible (Fig. S8C).
We used tryptic digestion and mass spectrometry to characterize the cross-links in GST-Smurf2. Free peptide containing the active site Cys was absent after heclin treatment, but it could be found disulfide-linked to a Cys close to the C terminus (Fig. S8 D–F). Because these two residues are not close enough in the crystal structure to allow easy intrachain linkage, this explains the tendency to form dimers. The heterogeneity of the oxidized protein suggests multiple linkages may occur, which potentially could include sulfenamide formation with lysine residues (34), but no other linkages were detected in this experiment. These results are distinct from those of the deuterium exchange experiments, which were performed with the isolated monomeric HECT domain under conditions that preserve the active site Cys in thiol form, even after heclin treatment (as shown by the recovery of peptides covering this site; see Dataset S1).
Together, our results are consistent with a model in which heclin binds to the HECT domain and perturbs the natural orientation of the C lobe (as shown by hydrogen/deuterium exchange), and in so doing, not only makes the active site Cys more prone to oxidation but also allows it to reach side chains with which it can react. The relatively slow subsequent reduction of the reaction products means inhibition of the enzyme is greatly enhanced.
An implication of this model is that there is a resting state for the HECT C lobe that protects the Cys from the effects of oxidation but allows it to accept Ub on binding of the E2. Once charged with Ub, the active site Cys has to be free to reach substrate lysine residues; by favoring this free conformation, heclin allows exposure of an uncharged, oxidation-prone form of the Cys. The potency of heclin in cells strongly suggests HECT inhibition is similarly enhanced by oxidation in vivo. This would not be unprecedented: Despite the generally reducing nature of the cytoplasm, active site cysteines with low pKa are readily oxidized (34), and sulfenic acid forms of many proteins, including the E1 ubiquitin activating enzyme, can be detected in living cells under normal conditions (35).
The precise molecular requirements for heclin activity, and the fact it does not inhibit other cysteine-containing enzymes such as the E1 or E2 proteins, suggest HECT ligases contain a well-defined common binding site. We have not so far been able to identify this site, although the cleft between the E2 binding domain and the rest of the C lobe is one candidate. Variation between HECT domains should allow isolation of improved inhibitors, with higher affinity and specificity. However, in some circumstances, the broad specificity of heclin may be desirable, as it can block the modification of substrates that are targets for multiple HECT ligases.
Methods
Protein Expression, Cell Transfections, and in Vitro and in Vivo Ubiquitination Assays.
These assays were performed essentially as described previously (17). For details, see SI Methods.
Phage Library Screening and Bicycle Isolation.
These were similar to reported methods (14, 16). For details, see SI Methods.
Alpha Screen for Small Molecule Inhibitors.
This was optimized and performed according to the manufacturer’s recommendations (PerkinElmer), with the assistance of MRC Technology. For details, see SI Methods.
Biophysical Methods.
Fluorescence polarization assays were performed as described in ref. 28. Hydrogen–deuterium exchange was essentially as described in ref. 30. For details of these and other biophysical methods, see SI Methods.
Supplementary Material
Acknowledgments
We are very grateful to Barbara Saxty, Keith Ansell, and David Whalley of MRC Technology for access to compound libraries and help with the Alpha screens. We also thank Greg Winter and Lutz Riechmann for bicycle phage libraries and advice; Chris Johnson and Stephen McLaughlin for help with the isothermal titration calorimetry (ITC), fluorescence polarization, and light-scattering measurements; Mark Skehel for help and advice with hydrogen-deuterium exchange and mass spectrometry; and Trevor Rutherford, John Sutherland, and Marc Fiedler for additional help and discussions. This work was supported by the Medical Research Council (MRC; file reference number MC_U105178788).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. R.D. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1412152111/-/DCSupplemental.
References
- 1.Teixeira LK, Reed SI. Ubiquitin ligases and cell cycle control. Annu Rev Biochem. 2013;82:387–414. doi: 10.1146/annurev-biochem-060410-105307. [DOI] [PubMed] [Google Scholar]
- 2.Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov. 2011;10(1):29–46. doi: 10.1038/nrd3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 4.Clague MJ, Liu H, Urbé S. Governance of endocytic trafficking and signaling by reversible ubiquitylation. Dev Cell. 2012;23(3):457–467. doi: 10.1016/j.devcel.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Bernassola F, Karin M, Ciechanover A, Melino G. The HECT family of E3 ubiquitin ligases: Multiple players in cancer development. Cancer Cell. 2008;14(1):10–21. doi: 10.1016/j.ccr.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Chen C, Matesic LE. The Nedd4-like family of E3 ubiquitin ligases and cancer. Cancer Metastasis Rev. 2007;26(3-4):587–604. doi: 10.1007/s10555-007-9091-x. [DOI] [PubMed] [Google Scholar]
- 7.Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10(6):398–409. doi: 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- 8.Micel LN, Tentler JJ, Smith PG, Eckhardt GS. Role of ubiquitin ligases and the proteasome in oncogenesis: Novel targets for anticancer therapies. J Clin Oncol. 2013;31(9):1231–1238. doi: 10.1200/JCO.2012.44.0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marín I. Animal HECT ubiquitin ligases: Evolution and functional implications. BMC Evol Biol. 2010;10:56. doi: 10.1186/1471-2148-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang YS, et al. Stapled α-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci USA. 2013;110(36):E3445–E3454. doi: 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vassilev LT, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 12.Guédat P, Colland F. Patented small molecule inhibitors in the ubiquitin proteasome system. BMC Biochem. 2007;8(Suppl 1):S14. doi: 10.1186/1471-2091-8-S1-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang W, Sidhu SS. Development of inhibitors in the ubiquitination cascade. FEBS Lett. 2014;588(2):356–367. doi: 10.1016/j.febslet.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heinis C, Rutherford T, Freund S, Winter G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat Chem Biol. 2009;5(7):502–507. doi: 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- 15.Eglen RM, et al. The use of AlphaScreen technology in HTS: Current status. Curr Chem Genomics. 2008;1:2–10. doi: 10.2174/1875397300801010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rentero Rebollo I, Heinis C. Phage selection of bicyclic peptides. Methods. 2013;60(1):46–54. doi: 10.1016/j.ymeth.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 17.Mund T, Pelham HR. Control of the activity of WW-HECT domain E3 ubiquitin ligases by NDFIP proteins. EMBO Rep. 2009;10(5):501–507. doi: 10.1038/embor.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiesner S, et al. Autoinhibition of the HECT-type ubiquitin ligase Smurf2 through its C2 domain. Cell. 2007;130(4):651–662. doi: 10.1016/j.cell.2007.06.050. [DOI] [PubMed] [Google Scholar]
- 19.Huang L, et al. Structure of an E6AP-UbcH7 complex: Insights into ubiquitination by the E2-E3 enzyme cascade. Science. 1999;286(5443):1321–1326. doi: 10.1126/science.286.5443.1321. [DOI] [PubMed] [Google Scholar]
- 20.Kamadurai HB, et al. Insights into ubiquitin transfer cascades from a structure of a UbcH5B approximately ubiquitin-HECT(NEDD4L) complex. Mol Cell. 2009;36(6):1095–1102. doi: 10.1016/j.molcel.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogunjimi AA, et al. Regulation of Smurf2 ubiquitin ligase activity by anchoring the E2 to the HECT domain. Mol Cell. 2005;19(3):297–308. doi: 10.1016/j.molcel.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 22.Ding Y, Zhang Y, Xu C, Tao QH, Chen YG. HECT domain-containing E3 ubiquitin ligase NEDD4L negatively regulates Wnt signaling by targeting dishevelled for proteasomal degradation. J Biol Chem. 2013;288(12):8289–8298. doi: 10.1074/jbc.M112.433185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei W, Li M, Wang J, Nie F, Li L. The E3 ubiquitin ligase ITCH negatively regulates canonical Wnt signaling by targeting dishevelled protein. Mol Cell Biol. 2012;32(19):3903–3912. doi: 10.1128/MCB.00251-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narimatsu M, et al. Regulation of planar cell polarity by Smurf ubiquitin ligases. Cell. 2009;137(2):295–307. doi: 10.1016/j.cell.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 25.Nethe M, et al. Rac1 acts in conjunction with Nedd4 and dishevelled-1 to promote maturation of cell-cell contacts. J Cell Sci. 2012;125(Pt 14):3430–3442. doi: 10.1242/jcs.100925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park Y, Yoon SK, Yoon JB. The HECT domain of TRIP12 ubiquitinates substrates of the ubiquitin fusion degradation pathway. J Biol Chem. 2009;284(3):1540–1549. doi: 10.1074/jbc.M807554200. [DOI] [PubMed] [Google Scholar]
- 27.Poulsen EG, et al. HUWE1 and TRIP12 collaborate in degradation of ubiquitin-fusion proteins and misframed ubiquitin. PLoS ONE. 2012;7(11):e50548. doi: 10.1371/journal.pone.0050548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eletr ZM, Kuhlman B. Sequence determinants of E2-E6AP binding affinity and specificity. J Mol Biol. 2007;369(2):419–428. doi: 10.1016/j.jmb.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev. 2006;25(1):158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 30.Vadas O, et al. Molecular determinants of PI3Kγ-mediated activation downstream of G-protein-coupled receptors (GPCRs) Proc Natl Acad Sci USA. 2013;110(47):18862–18867. doi: 10.1073/pnas.1304801110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung KY, et al. Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature. 2011;477(7366):611–615. doi: 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamadurai HB, et al. Mechanism of ubiquitin ligation and lysine prioritization by a HECT E3. eLife. 2013;2:e00828. doi: 10.7554/eLife.00828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verdecia MA, et al. Conformational flexibility underlies ubiquitin ligation mediated by the WWP1 HECT domain E3 ligase. Mol Cell. 2003;11(1):249–259. doi: 10.1016/s1097-2765(02)00774-8. [DOI] [PubMed] [Google Scholar]
- 34.Gupta V, Carroll KS. Sulfenic acid chemistry, detection and cellular lifetime. Biochim Biophys Acta. 2014;1840(2):847–875. doi: 10.1016/j.bbagen.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leonard SE, Reddie KG, Carroll KS. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem Biol. 2009;4(9):783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.