

Significance
Pyrrolysyl-tRNA synthetase (PylRS) and its cognate tRNAPyl have emerged as ideal translation components for genetic code innovation. We found that a series of PylRS variants that were initially selected to be specific for the posttranslational modification Nε-acetyl-l-Lys displayed polyspecificity [i.e., activity with a broad range of noncanonical amino acid (ncAA) substrates]. Our structural and biochemical data indicate that the engineered tRNA synthetases can accommodate ncAA substrates in multiple binding modes. The data further suggest that in vivo selections do not produce optimally specific tRNA synthetases and that translation fidelity will become an increasingly dominant factor in expanding the genetic code far beyond 20 amino acids.
Keywords: aminoacyl-tRNA synthetase, genetic code, genetic selection, posttranslational modification, synthetic biology
Abstract
Pyrrolysyl-tRNA synthetase (PylRS) and its cognate tRNAPyl have emerged as ideal translation components for genetic code innovation. Variants of the enzyme facilitate the incorporation >100 noncanonical amino acids (ncAAs) into proteins. PylRS variants were previously selected to acylate Nε-acetyl-Lys (AcK) onto tRNAPyl. Here, we examine an Nε-acetyl-lysyl-tRNA synthetase (AcKRS), which is polyspecific (i.e., active with a broad range of ncAAs) and 30-fold more efficient with Phe derivatives than it is with AcK. Structural and biochemical data reveal the molecular basis of polyspecificity in AcKRS and in a PylRS variant [iodo-phenylalanyl-tRNA synthetase (IFRS)] that displays both enhanced activity and substrate promiscuity over a chemical library of 313 ncAAs. IFRS, a product of directed evolution, has distinct binding modes for different ncAAs. These data indicate that in vivo selections do not produce optimally specific tRNA synthetases and suggest that translation fidelity will become an increasingly dominant factor in expanding the genetic code far beyond 20 amino acids.
The standard genetic code table relates the 64 nucleotide triplets to three stop signals and 20 canonical amino acids. Some organisms, including humans, naturally evolved expanded genetic codes that accommodate 21 amino acids (1), or possibly 22 amino acids in rare cases (2). Engineering translation system components, including tRNAs (3, 4), aminoacyl-tRNA synthetases (AARSs) (5, 6), elongation factors (7), and the ribosome itself (8), have produced organisms with artificially expanded genetic codes. Products of genetic code engineering include bacterial, yeast, and mammalian cells and animals that are able to synthesize proteins with site-specifically inserted noncanonical amino acids (ncAAs) (9).
Genetic code expansion systems rely on an orthogonal AARS/tRNA pair (o-AARS, o-tRNA) (5, 6). The o-AARS should be specific in ligating a desired ncAA to a stop codon decoding tRNA, and both the o-tRNA and o-AARS are assumed not to cross-react with endogenous AARSs or tRNAs. Although some AARSs evolved in nature to recognize certain ncAAs (10–12), many genetic code expansion systems require a mutated AARS active site. The active site of the o-AARS is usually redesigned via directed evolution (6), including positive and negative selective rounds, to produce an enzyme that is assumed to be specific for an ncAA and not active with the 20 canonical amino acids. Genetic code expansion technology is rapidly evolving (13), and the ability to incorporate multiple ncAAs into a protein using quadruplet-codon decoding (14) or sense-codon recoding (15–19) is now becoming feasible. Protein synthesis with multiple ncAAs will require o-AARSs that are able to discriminate their ncAA substrate not only from canonical amino acids in the cell but from other ncAAs that are added to the cell.
Probing the effects of amino acid analogs on bacterial cell growth revealed, over 50 y ago, that many ncAAs were incorporated into proteins by the regular translation machinery (10). Thus, it was not surprising to see that many of the successful orthogonal Methanococcus jannaschii tyrosyl-tRNA synthetase variants (20) facilitate incorporation of multiple different ncAAs (21–23). This polyspecificity is also a property of the orthogonal pyrrolysyl-tRNA synthetase (PylRS)/tRNAPyl pair (reviewed in ref. 24).
PylRS variants that facilitate site-specific insertion of Nε-acetyl-l-Lys (AcK; 2) (Fig. 1A) into proteins were derived from directed evolution experiments (25–28). These AcK-tRNA synthetase (AcKRS) enzymes have been used to investigate the role of acetylation sites in tumor suppressor p53 (29) and histone H3 (30). Here, we present biochemical and structural studies showing that AcKRS variants are polyspecific and catalytically deficient enzymes compared with canonical AARSs. These AcKRSs selected by directed evolution to ligate AcK to tRNAPyl are actually ∼30-fold more efficient in activation with Phe derivatives. Crystallographic structures of AcKRS and PylRS variants in complex with AcK, 3-iodo-l-Phe (3-I-Phe; 4) (Fig. 1A), or 2-(5-bromothienyl)-l-Ala (3-Br-ThA; 10) (Fig. 1A) reveal the structural basis of polyspecificity in these engineered PylRS enzymes.
Fig. 1.

(A) Chemical structures of nsAAs used in the study: 1, l-pyrrolysine (Pyl); 2, AcK; 3, Nε-trifluoroacetyl-l-Lys (CF3-AcK); 4, 3-I-Phe; 5, 3-bromo-l-Phe (3-Br-Phe); 6, 3-chloro-l-Phe (3-Cl-Phe); 7, 3-trifluoromethyl-l-Phe (3-CF3-Phe); 8, 3-methyl-l-Phe (3-Me-Phe); 9, 3-methoxyl-l-Phe (3-MeO-Phe); 10, 3-Br-ThA. (B) Range of substrate specificity of AcKRS3. Translation of the sfGFP reporter (UAG codon at position 2) by the library of ncAA-tRNAPyl was measured by fluorescence intensity. A library of 94 different Lys and Phe analogs (Dataset S1) was tested. Fluorescence signals from the incorporation of ncAAs 2–5, 7, 9, and 10 are labeled in A. Well A12 is a control without added ncAA. Well A1 was a positive control experiment to detect the production signal of WT sfGFP (100%). Fluorescence data and error values (SD) are represented by bars from three independent experiments (also given in Dataset S1). Colors (0–4% in purple, 4–8% in blue, 18–24% in red, and 95–100% in brown) have been used to indicate the level of UAG read-through.
Results
Polyspecificity in Engineered PylRS Enzymes.
We determined the substrate range of two AcKRS enzymes by screening with a chemically diverse library of 313 ncAAs (11, 32), followed by rescreening with a selected subset of Phe and Lys derivatives (Fig. 1B). For this screen, we used a superfolder GFP (sfGFP) reporter (32) bearing a TAG codon at position 2, a site known to be permissive for diverse ncAAs (32, 33). The reporter was coexpressed with plasmids encoding the tRNAPyl and AcKRS; quantification was by sfGFP fluorescence, the indication of ncAA incorporation efficiency. As a control WT sfGFP (AGC Ser codon at position 2) was monitored. Compared with this WT control, translation of UAG (codon 2) with AcK was 2% (for AcKRS1) and 4% (for AcKRS3).
AcKRS3 showed significant incorporation of six ncAAs (Fig. 1B). Even though AcK (2) is among them (Fig. 1A), four meta substituted Phe analogs (4, 5, 7, and 9) enabled more sfGFP production compared with AcK (Fig. 1B). Incorporation of 4 and 10 was analyzed by full-length protein MS and LC-tandem MS (MS/MS), which identified the expected ncAA but no other amino acids inserted at position 2 in sfGFP (Figs. S1–S4). The AcKRS1 substrate range is similar (Fig. S5) with four ncAAs (3–5 and 7) showing better incorporation than AcK (2).
The fact that both AcKRS variants showed good activity with 3-I-Phe suggests that the PylRS active site may be better suited for binding of metasubstituted Phe derivatives. This conclusion is supported by a recent report (34) showing the PylRS variant Asn346Ala/Cys348Ala to possess significant activity with 3-I-Phe yet weak activity with Phe. Based on these data, we created PylRS mutant libraries that randomized active site positions, including N346 and C348 (SI Methods). The libraries were selected for 3-I-Phe incorporation into chloramphenicol acetyl transferase with a TAG codon at position 112. Selection from this library produced a mutant [iodo-phenylalanyl-tRNA synthetase (IFRS); Asn346Ser/Cys348Ile] that showed significantly higher UAG translation (∼50% of WT sfGFP) than any known PylRS variant (Table 1). The other metasubstituted Phe analogs (5–8) displayed 30–68% incorporation efficiencies (Table 1, Fig. S6, and Dataset S1). IFRS also shows a broader substrate range than AckRS1 (Fig. S6). The electrospray ionization (ESI)-MS and MS/MS analyses (Figs. S1–S4) indicate that UAG translation led only to the desired ncAA-containing protein product, and no canonical amino acid insertion at the UAG-encoded locus was observed. These data indicate that the sfGFP fluorescence data are a quantitative measure of ncAA incorporation.
Table 1.
Apparent kinetic parameters of PylRS variants for aminoacylation
| Enzymes | Amino acids | Km, μM × 103 | kcat, s−1 × 10−2 | kcat/Km, μM−1⋅s−1 × 10−5 | Relative catalytic efficiency | UAG translation efficiency* |
| MmPylRS | Pyl (1) | 0.02 ± 0.004 | 0.83 ± 0.03 | 41.5 | 100 | 15± 1.5 |
| MbPylRS | Pyl (1) | 0.02 ± 0.002 | 3.01 ± 0.04 | 151 | 364 | —† |
| IFRS | 3-I-Phe (4) | 0.44 ± 0.04 | 0.44 ± 0.02 | 1.00 | 2.4 | 48± 1.2 |
| 3-Br-Phe (5) | 0.62 ± 0.24 | 0.48 ± 0.03 | 0.77 | 1.9 | 68 ± 5.2 | |
| 3-Cl-Phe (6) | 1.36 ± 0.13 | 0.76 ± 0.05 | 0.56 | 1.3 | 46 ± 3.2 | |
| 3-CF3-Phe (7) | 0.45 ± 0.09 | 0.28 ± 0.01 | 0.62 | 1.5 | 53 ± 2.3 | |
| 3-Me-Phe (8) | 0.95 ± 0.17 | 0.34 ± 0.05 | 0.36 | 0.9 | 30 ± 0.5 | |
| 3-MeO-Phe (9) | 1.64 ± 0.19 | 0.07 ± 0.01 | 0.04 | 0.1 | <1 | |
| 3-Br-ThA (10) | 0.37 ± 0.09 | 0.14 ± 0.01 | 0.38 | 0.9 | 7 ± 0.9 |
Apparent kinetic parameters of PylRS variants for aminoacylation were determined by quantitating amino acid ligation to radiolabeled tRNAs (36). Numbers of amino acids are shown in bold. 3-CF3-Phe, 3-trifluoromethyl-l-Phe; 3-Cl-Phe, 3-chloro-l-Phe; 3-Me-Phe, 3-methyl-l-Phe; 3-MeO-Phe, 3-methoxyl-l-Phe.
Not measured.
Enzyme Kinetics of PylRS Variants.
AARSs catalyze a two-step reaction. The amino acid substrate is first activated by adenylation in a reaction with ATP. The aminoacyl-adenylate is then a substrate for aminoacyl-tRNA formation in the second step. Both steps can be monitored biochemically; amino acid-AMP formation is monitored by the ATP-PPi exchange assay (35), whereas aminoacylation is assayed by quantitating amino acid ligation to radiolabeled tRNAs (36, 37) (SI Methods). The enzyme kinetic constants (kcat and Km) are used to compare activity for the different ncAAs. As reported in earlier studies (38, 39), the truncated version (to overcome solubility problems) of the PylRS enzyme (residues 185–454) was used to measure the ATP-PPi exchange. However, in the aminoacylation experiments, full-length PylRS variants could be used because the assay (37) required low enzyme concentrations.
The enzymatic characterization of WT PylRS was stalled by the lack of pyrrolysine (Pyl). Because there is no commercial source of Pyl and its early chemical synthesis had proven to be challenging (40), we used a sample from an alternative synthetic route (41). We determined the kinetic parameters with Pyl for WT Methanosarcina mazei PylRS (MmPylRS) and Methanosarcina barkeri PylRS (MbPylRS). The data show a Km of ∼50 μM and a catalytic turnover of 0.1–0.3 s−1 in amino acid-AMP formation (Table 2) and values of 20 μM and 0.008–0.03 s−1 for Pyl-tRNAPyl formation (Table 1). Thus, even with its native substrate, PylRS is less efficient than many AARSs with kcat values about >1,000-fold lower than measured for many canonical AARSs (e.g., phenylalanyl-tRNA synthetase) (42) (Tables 1 and 2). This lower activity is not surprising, because in Methanosarcina, Pyl is translated by as few as ∼50 codons, whereas a “canonical” tRNA synthetase [e.g., Escherichia coli leucyl-tRNA synthetase (43)] normally needs to provide substrate to translate ∼150,000 codons.
Table 2.
Apparent kinetic parameters of M. mazei PylRS variants for amino acid activation
| Enzymes | Amino acids | Km, μM × 103 | kcat, s−1 × 10−2 | kcat/Km, μM−1⋅s−1 × 10−5 | Relative catalytic efficiency | UAG translation efficiency* |
| MmPylRS | Pyl (1) | 0.05 ± 0.008 | 29.8 ± 1.2 | 596 | 100 | 15± 1.5 |
| MbPylRS† | Pyl (1) | 0.055 ± 0.005 | 10.5 ± 1.1 | 191 | 32 | —‡ |
| AcKRS1 | 3-I-Phe (4) | 6.14 ± 0.32 | 12.6 ± 1.5 | 2.05 | 0.34 | 19 ± 2.0 |
| 3-Br-Phe (5) | 11.9 ± 0.30 | 13.6 ± 0.35 | 1.14 | 0.19 | 3 ± 0.5 | |
| 3-Cl-Phe (6) | 9.46 ± 3.85 | 8.54 ± 2.73 | 0.90 | 0.15 | <1 | |
| 3-CF3-Phe (7) | 1.09 ± 0.28 | 3.91 ± 0.48 | 3.59 | 0.60 | 3 ± 0.3 | |
| 3-Me-Phe (8) | 0.80 ± 0.19 | 1.37 ± 0.19 | 1.71 | 0.29 | <1 | |
| 3-MeO-Phe (9) | 6.27 ± 1.17 | 4.15 ± 0.35 | 0.66 | 0.11 | <1 | |
| CF3-AcK (3) | 0.61 ± 0.06 | 1.24 ± 0.02 | 2.03 | 0.34 | 3 ± 0.2 | |
| AcK§ (2) | 35.3 ± 10.9 | 3.23 ± 0.61 | 0.09 | 0.02 | 2 ± 0.1 | |
| Pyl (1) | nd | nd | ||||
| AcKRS3 | AcK (2) | 137 ± 59 | 14.6 ± 3.8 | 0.11 | 0.02 | 4 ± 0.2 |
| 3-I-Phe (4) | 16.6 ± 1.9 | 34.3 ± 2.4 | 2.07 | 0.35 | 21 ± 2.1 | |
| 3-Br-Phe (5) | 22.4 ± 2.0 | 26.3 ± 1.0 | 1.17 | 0.20 | 5 ± 0.1 | |
| 3-CF3-Phe (7) | 3.21 ± 0.97 | 4.68 ± 0.39 | 1.46 | 24 | 6 ± 0.3 | |
| IFRS | 3-I-Phe (4) | 0.82 ± 0.09 | 8.71 ± 0.33 | 10.6 | 1.8 | 48 ± 0.1 |
| 3-CF3-Phe (7) | 1.13 ± 0.10 | 7.29 ± 0.21 | 6.45 | 1.1 | 53 ± 2.3 | |
| 3-Br-ThA (10) | 1.57 ± 0.32 | 4.43 ± 0.27 | 2.82 | 0.47 | 7 ± 0.9 |
Apparent kinetic parameters of MmPylRS variants for amino acid activation were determined by ATP-PPi exchange for the amino acids indicated. Numbers of amino acids are shown in bold. CF3-AcK, Nε-trifluoroacetyl-l-Lys; nd, not detectable.
UAG translation efficiency is from data in Figs. S5 and S6. Translation of the WT sfGFP (AGC is codon 2) is considered 100%, and the percentage of sfGFP observed resulting from UAG translation with the indicated ncAA is referred to as the UAG translation efficiency.
Kinetic data (Km and kcat) for MbPylRS were reproduced from previous work (66).
Not measured.
Kinetic data were reproduced from a study by Umehara et al. (26).
AcKRS1 is most catalytically efficient with the substrate 3-trifluoromethyl-l-Phe (7) (Fig. 1). This activity is 166-fold less efficient compared with WT PylRS activity with Pyl. Surprisingly, the Km for AckRS1 with AcK is 35 mM, but its kcat is only 10-fold lower than WT PylRS for Pyl. The best substrate for AcKRS3 is 3-I-Phe (4) (Fig. 1), but the activity is 288-fold less efficient compared with WT PylRS with Pyl as a substrate.
The UAG translation efficiencies observed in vivo in the sfGFP reporter (Table 2 and Figs. S5 and S6) are in moderate agreement with the enzyme-kinetic data. However, aminoacylation is only one component that influences translation efficiency; ncAA uptake (44) and stability and elongation factor Tu (EF-Tu) compatibility (7) are known to alter the efficiency of translation. It is also possible that some ncAAs or translation of off-target UAG codons could alter the cellular proteome or metabolism in ways that may have an impact on protein synthesis and its fidelity.
The improved translation efficiency observed for IFRS is evident in biochemical assays, which show IFRS to be fivefold more efficient in 3-I-Phe–adenylate formation compared with AckRS1 or AcKRS3 (Table 2). However, IFRS is still less efficient in the aminoacylation of tRNAPyl (∼40-fold) compared with WT PylRS (Table 1). Despite the fact that PylRS is significantly more active for Pyl than are engineered AARSs for their substrates, we recorded only 15% UAG read-through with Pyl, whereas Pyl analogs support up to 40% UAG read-through (35). These data may indicate that E. coli imports Pyl less efficiently than other ncAAs or that it is less stable; however, a firm conclusion requires determination of the concentrations of these amino acids in the cell.
Anatomy of an Engineered Active Site.
Seven distinct AckRS variants have been reported previously (25–28, 30). Three variants were derived from the MbPylRS (25, 30), whereas the others originated from the MmPylRS (26–28). Biochemical studies revealed that the AcKRS enzymes were far less catalytically efficient than natural tRNA synthetases (26). This result led us to characterize AcKRS complexed with AcK structurally. Because we had previously optimized crystallization conditions for MmPylRS, we constructed AcKRS3, an MmPylRS variant that carried the active site residues of an M. barkeri AcKRS (Table S1). Cocrystals of AcKRS3 complexed to AcK and the ATP analog 5′-adenylyl β,γ-imidodiphosphate (ADPNP) were obtained following previously published protocols (38), with specific modifications (SI Methods). The final structure was refined at a resolution of 2.3 Å with an R factor of 18.6% and an Rfree value of 21.4% (Table S2).
AckRS3 crystals contain one protein molecule in the asymmetrical unit. The anticipated AcKRS3 dimer is created by crystallographic symmetry between neighboring enzymes in the crystal lattice. The Fourier difference (Fo-Fc) electron density maps show clear density for the ADPNP contoured at 4σ. Density for AcK appears at 4σ, with the strongest density around the α- and β-carbons of the R group. There is no density for the methyl group in the acetyl moiety (Fig. S7). High β-factors associated with the terminus of the AcK R group indicate that the amino acid substrate is highly mobile in the active site. In addition, the AcK shows ∼70% occupancy even at a 75 mM concentration in the cryogen solution. The remaining molecules in the crystal contain water molecules with well-defined hydrogen bonding partners in the active site. In comparison to the WT PylRS structure, one of the most noticeable changes in the amino acid binding pocket is the movement of Asn346 (Fig. 2). The density for Tyr384 is not resolved, and the aromatic ring appears to be disordered (45).
Fig. 2.
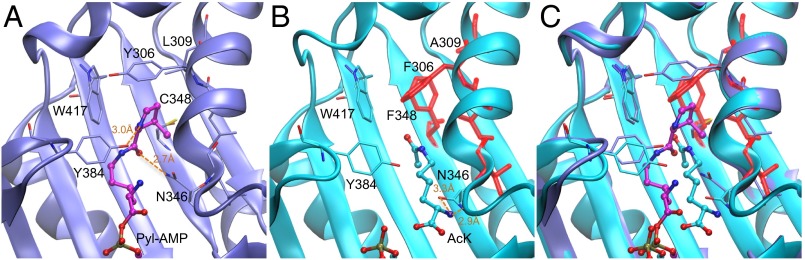
Comparison of PylRS and AcKRS3 substrate complexes. (A) View of the active site of PylRS [purple, Protein Data Bank (PDB) ID code 2ZIM (38)] in complex with Pyl-adenylate (Pyl-AMP; magenta). Hydrogen bonds to the substrate amino acid are shown (dashed lines), with distances between the heavy atoms indicated. (B) View of the active site of AcKRS3 (cyan, PDB ID code 4Q6G) complex with AcK and ATP analog ADPNP with mutated residues shown in red. (C) Active site view of a structural overlap of PylRS and AcKRS3. The substrate (Pyl-AMP) and key recognition residues from PylRS are shown (purple, sticks) overlaid with residues in the AcKRS3 active site (cyan or red, sticks).
Comparison of the MmPylRS and AcKRS3 show that the structures are nearly identical, except in the active site (Fig. 2). All AcKRS variants differ from their parent PylRS at four to five positions in the active site. With the exception of AcKRS2, all other AcKRSs have the mutations Cys348Phe and Leu309Ala (Table S1). The Tyr at position 306 is either a Leu or Phe residue. The AcKRS3 structure indicates that the Leu309Ala substitution creates space for a new cluster of hydrophobic and bulky residues at positions 348 and 306. These mutations lead to an AcKRS active site that is smaller and more hydrophobic than that of the parent PylRS, which demonstrates why AcKRS is no longer active with the larger substrate, Pyl (26) (Table 2) and why the parent PylRS enzyme is not active in inserting AcK into proteins in vivo (26). Interestingly, one PylRS variant (Tyr306Ala/Tyr384Phe) was shown to accommodate furan-containing ncAAs that are substantially larger than Pyl (46). In a crystal structure, the Ala substitution at position 306 was implicated in extending the size of the active site pocket to accommodate larger ncAAs.
A number of structural studies have shown significant plasticity in the active site of the WT PylRS (38, 47, 48), which explains earlier biochemical studies demonstrating that PylRS is able to accommodate several different Pyl and Lys derivatives (35, 49). Structural analyses have also revealed how mutant PylRS enzymes can accommodate ncAAs that are chemically distinct (50) and larger than Pyl (47), including furan-containing Lys (46) and norbornene-containing Pyl analogs (51). Larger ncAAs are accepted by these PylRS variants by creating larger amino acid recognition pockets with mutations to smaller active site residues, particularly at position 306.
The structure suggests that hydrophobic shape complementarity is the principal means by which AcKRS recognizes AcK. In the PylRS/Pyl complex, Tyr384 and Asn346 make specific hydrogen bonding interactions with the amide group and nitrogen atom on the pyrroline ring (Fig. 2B). In the AcKRS structure, the Nε-amide group has no hydrogen bonding partners. In fact, Asn346 forms a novel interaction with the α-amino group of the AcK substrate (Fig. 2B). This interaction shifts the AcK substrate away from a catalytically competent conformation, which is exemplified by the Pyl substrate in the PylRS complex (Fig. 2C).
Structural Basis of Polyspecificity.
To understand how IFRS accommodates chemically diverse ncAAs, we solved two of the crystal structures of IFRS in complex with 3-I-Phe and 3-Br-ThA. Crystals of 3-I-Phe or 3-Br-ThA complexes diffracted at 2.1 or 2.7 Å, respectively (Table S2). Overall, the structures are highly similar, and electron densities corresponding to the ncAA substrates were clearly assigned in the active site (Fig. S8). In the 3-I-Phe complex structure, we found evidence that the ncAA substrate is present in at least two confirmations in the active site. The residual Fourier difference (mFo-dFc) electron density map at 4σ suggests two locations for the iodo atom (Fig. S8A).
Most strikingly, 3-I-Phe and 3-Br-ThA occupy distinct binding pockets within the active site of IFRS (Fig. 3). The twisted five-membered ring of 3-Br-ThA allows this ncAA to bind deeper into the active site cleft, which positions the carbonyl further away from the site of adenylation catalysis compared with 3-I-Phe. The mFo-dFc map at 4σ suggests a single clearly defined location for the Br atom (Fig. S8B).
Fig. 3.

Structural comparison of WT PylRS (A) and PylRS (B–D) variants in complex with ncAA substrates. PylRS variants and ncAAs are indicated in each panel. Mutations from the parent PylRS are indicated in bold, and the mutated side chains are shown in red. In C, the IFRS/3-I-Phe complex shows two confirmations of 3-I-Phe (magenta, pink) observed in the crystal structure. The gray surfaces show the solvent-accessible volume within the active site pockets. These volumes were computed in the absence of the ncAA substrate. The loop, including Tyr384, is disordered in the IFRS complex structures in C and D and is not visible. (E) Structural overlay of the IFRS complex structures with 3-I-Phe in two confirmations (magenta, pink) and 3-Br-ThA (cyan) shows distinct binding modes of the ncAA substrates.
One of the 3-I-Phe conformations is more similar to 3-Br-ThA, and these structures may represent nonproductive complexes. The enhanced activity IFRS shows toward 3-I-Phe compared with 3-Br-ThA may be due to the fact that 3-I-Phe is able to bind in a more productive conformation for catalysis to proceed. Interestingly, in one conformation, the iodo atom of 3-I-Phe binds at exactly the same position as the S atom in 3-Br-ThA, and in the second 3-I-Phe conformation, the iodo and bromo groups are colocalized (Fig. 3E). A number of PylRS structures complexed with Lys derivatives have led to the conclusion that the large size of the PylRS active site pocket is principally responsible for its activity with multiple ncAAs (52). Although this conclusion is likely correct, the fact that IFRS has (at least) two binding modes for different ncAAs is clearly related to the polyspecificity that this enzyme displays.
Discussion
Genetic selection underlies evolution in nature as well as directed evolution in the laboratory. The powerful nature of genetic selection leads one to feel that these procedures should reveal from a library of genetic variants the best enzyme, in terms of activity and specificity. However, more than half a century ago, many ncAAs were shown to be incorporated into proteins by the natural protein synthesis machinery (10). These ncAAs are close structural analogs of the natural amino acids, and the endogenous tRNA synthetases cannot effectively discriminate against these unnatural substrates (11). It is perhaps self-evident that natural selection cannot optimize enzymes to reject unnatural substrates that the cell has never encountered. The same is true for directed evolution experiments. It is normally assumed that off-target activities are weak or residual compared with natural substrates (53). Therefore, it should not be surprising that directed evolution produces polyspecific tRNA synthetases. Genetic code expansion technologies are moving toward genetically encoding multiple diverse ncAAs. Cross-reactivity between engineered tRNA synthetases and diverse ncAA substrates will become a limiting factor in creating high-fidelity orthogonal translation systems with multiple varied ncAAs (16, 54, 55).
The extent of protein engineering is limited by the fitness of the organism. Another reason why directed evolution experiments may fail to produce enzymes with the desired level of activity or specificity is that certain variants may be toxic to the cell. It is well known that protein synthesis quality control (56) ensures cellular fitness by preventing the accumulation of mistranslated proteins and the associated unfolded protein response mechanisms that underlie diverse human diseases (57). For genetic codes expanded in the laboratory, protein quality control is also an important issue. A system developed to reassign UAG codons to O-phosphoserine (Sep) leads to significant growth defects when expressed in WT E. coli cells (33). Translational read-through of endogenous UAG codons with Sep lengthened native proteins beyond their normal termination point and reduced cell viability. This and related work motivated an extensive genome editing effort using multiplex automated genome engineering (MAGE) that produced an E. coli strain in which all genomic TAG codons were mutated to TAA, thus providing a clean genetic background for reassignment of UAG to any desired ncAA (58).
Engineering other components of the translation machinery may also be required for optimal protein synthesis with ncAAs (59). In redesigning EF-Tu to accommodate site-specific selenocysteine incorporation, certain EF-Tu variants that specifically recognized selenocysteinyl-tRNA in vitro were toxic to the cell when expressed in vivo (60). Similarly, EF-Tu mutants designed to carry a bulky fluorescent ncAA-tRNA did not release the ncAA-tRNA fast enough to support efficient translation (61).
The best enzyme may not be the same as the enzyme most “orthogonal” to the natural amino acids. Traditionally, o-AARS/o-tRNA pairs were derived from successive positive and negative rounds of selection (5, 6). In the positive rounds, UAG translation of an antibiotic marker leads to cell survival. This selection reveals tRNA synthetase variants that translate UAG with ncAAs and/or canonical amino acids. The ncAA is omitted from negative selection, and translation of a toxic protein containing an in-frame UAG codon eliminates variants that promote incorporation of canonical amino acids. This stringent negative selection may eliminate some highly active synthetase variants. Indeed, it was recently demonstrated that relaxing selection conditions and screening variants for high-yield ncAA incorporation in vivo produced a second-generation 3-nitro-tyrosyl-tRNA synthetase that is an order of magnitude more efficient than the first-generation enzyme (62). Because the mutant synthetase showed significant activity with the natural Tyr, relaxing selection stringency also has an impact on translation fidelity with ncAAs.
In addition to specificity, enzymatic activity of engineered tRNA synthetases may be far lower (by up to ∼1,000-fold) than what is observed in many natural tRNA synthetases (42, 43, 63). Despite this fact, even these lower activity tRNA synthetases can facilitate almost “normal” levels of protein synthesis as judged by sfGFP production. ESI-MS and LC-MS/MS analysis confirmed that the sfGFP produced resulted only from insertion of the desired ncAA in response to the UAG codon (Figs. S1–S4). We also found that modest increases in catalytic efficiency (kcat/Km) can increase translational read-through of UAG to a level of 68% compared with normal sense codon decoding (Table 2). In fact, these and other findings challenge the widely held idea that tRNA synthetases display “typical” Km or kcat values. Apart from the well-known AARS variants, tRNA synthetases and functionally distinct paralogs of these enzymes show a wide range of enzymatic activity in nature. PoxA, a paralog of lysyl-tRNA synthetase (LysRS) that uses (R)-β-Lys to modify elongation factor P posttranslationally (64), is 400-fold less catalytically efficient than LysRS, yet the enzyme is perfectly acceptable to the cell, which would otherwise have evolved it to greater efficiency.
Recent success in synthesizing proteins with 21 (58) or 22 (54) amino acids at efficiencies nearing that of normal protein synthesis are promising for the field. New technologies, including MAGE (58) and phage-assisted continuous evolution (65), are expected to play important roles in further expanding the genetic code by allowing sampling of larger libraries of enzyme variants and enabling improvement of moderately active orthogonal translation components. Even with these technological advances, it is clear that genetic code engineers face complex issues in protein engineering and design related to tRNA synthetase activity, specificity, and translation fidelity in their attempts to evolve cells to make proteins with more than 22 amino acids.
Methods
Enantiomerically pure l-Pyl was synthesized exactly as described previously (41). For crystallographic studies, overexpression and purification of the C-terminal domain (CTD; residues 188–454) of AckRS1 were performed in the same manner as for the CTD of PylRS described previously (38). The CTD of MmPylRS and IFRS was used for assaying ATP-PPi exchange. Aminoacylation was measured with full-length MbPylRS, MmPylRS, and IFRS. The enzyme substrate range was determined by the sfGFP assay using 313 ncAAs at a 1 mM concentration (SI Methods).
Supplementary Material
Acknowledgments
We thank Markus Englert, Chenguang Fan, Ilka Heinemann, Jae-hyeong Ko, Jiqiang Ling, Yuchen Liu, Laure Prat, and Don Spratt for insightful discussions. This work was supported by NIH Grants GM055984 (to L.L.K.), GM22854 (to D.S.), and P01 GM022778 (to T.A.S.); Natural Sciences and Engineering Research Council of Canada Grant RGPIN 04282-2014 (to P.O.); and Defense Advanced Research Projects Agency Contract N66001-12-C-4211 (to D.S.).
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequences reported in this paper have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4Q6G, 4TQD, and 4TQF).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1419737111/-/DCSupplemental.
References
- 1.Yuan J, et al. Distinct genetic code expansion strategies for selenocysteine and pyrrolysine are reflected in different aminoacyl-tRNA formation systems. FEBS Lett. 2010;584(2):342–349. doi: 10.1016/j.febslet.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ambrogelly A, Palioura S, Söll D. Natural expansion of the genetic code. Nat Chem Biol. 2007;3(1):29–35. doi: 10.1038/nchembio847. [DOI] [PubMed] [Google Scholar]
- 3.Liu DR, Magliery TJ, Pastrnak M, Schultz PG. Engineering a tRNA and aminoacyl-tRNA synthetase for the site-specific incorporation of unnatural amino acids into proteins in vivo. Proc Natl Acad Sci USA. 1997;94(19):10092–10097. doi: 10.1073/pnas.94.19.10092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aldag C, et al. Rewiring translation for elongation factor Tu-dependent selenocysteine incorporation. Angew Chem Int Ed Engl. 2013;52(5):1441–1445. doi: 10.1002/anie.201207567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma N, Furter R, Kast P, Tirrell DA. Efficient introduction of aryl bromide functionality into proteins in vivo. FEBS Lett. 2000;467(1):37–40. doi: 10.1016/s0014-5793(00)01120-0. [DOI] [PubMed] [Google Scholar]
- 6.Liu CC, Schultz PG. Adding new chemistries to the genetic code. Annu Rev Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 7.Park HS, et al. Expanding the genetic code of Escherichia coli with phosphoserine. Science. 2011;333(6046):1151–1154. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang K, Neumann H, Peak-Chew SY, Chin JW. Evolved orthogonal ribosomes enhance the efficiency of synthetic genetic code expansion. Nat Biotechnol. 2007;25(7):770–777. doi: 10.1038/nbt1314. [DOI] [PubMed] [Google Scholar]
- 9.Chin JW. Expanding and reprogramming the genetic code of cells and animals. Annu Rev Biochem. 2014;83:379–408. doi: 10.1146/annurev-biochem-060713-035737. [DOI] [PubMed] [Google Scholar]
- 10.Richmond MH. The effect of amino acid analogues on growth and protein synthesis in microorganisms. Bacteriol Rev. 1962;26:398–420. doi: 10.1128/br.26.4.398-420.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan C, Ho JM, Chirathivat N, Söll D, Wang YS. Exploring the substrate range of wild-type aminoacyl-tRNA synthetases. ChemBioChem. 2014;15(12):1805–1809. doi: 10.1002/cbic.201402083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merkel L, Budisa N. Organic fluorine as a polypeptide building element: In vivo expression of fluorinated peptides, proteins and proteomes. Org Biomol Chem. 2012;10(36):7241–7261. doi: 10.1039/c2ob06922a. [DOI] [PubMed] [Google Scholar]
- 13.Lemke EA. The exploding genetic code. ChemBioChem. 2014;15(12):1691–1694. doi: 10.1002/cbic.201402362. [DOI] [PubMed] [Google Scholar]
- 14.Neumann H, Wang K, Davis L, Garcia-Alai M, Chin JW. Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature. 2010;464(7287):441–444. doi: 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]
- 15.Kwon I, Kirshenbaum K, Tirrell DA. Breaking the degeneracy of the genetic code. J Am Chem Soc. 2003;125(25):7512–7513. doi: 10.1021/ja0350076. [DOI] [PubMed] [Google Scholar]
- 16.Bröcker MJ, Ho JM, Church GM, Söll D, O’Donoghue P. Recoding the genetic code with selenocysteine. Angew Chem Int Ed Engl. 2014;53(1):319–323. doi: 10.1002/anie.201308584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bohlke N, Budisa N. Sense codon emancipation for proteome-wide incorporation of noncanonical amino acids: Rare isoleucine codon AUA as a target for genetic code expansion. FEMS Microbiol Lett. 2014;351(2):133–144. doi: 10.1111/1574-6968.12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng Y, Wang W, Liu WR. Towards reassigning the rare AGG codon in Escherichia coli. ChemBioChem. 2014;15(12):1750–1754. doi: 10.1002/cbic.201400075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishnakumar R, Ling J. Experimental challenges of sense codon reassignment: An innovative approach to genetic code expansion. FEBS Lett. 2014;588(3):383–388. doi: 10.1016/j.febslet.2013.11.039. [DOI] [PubMed] [Google Scholar]
- 20.Wang L, Schultz PG. A general approach for the generation of orthogonal tRNAs. Chem Biol. 2001;8(9):883–890. doi: 10.1016/s1074-5521(01)00063-1. [DOI] [PubMed] [Google Scholar]
- 21.Stokes AL, et al. Enhancing the utility of unnatural amino acid synthetases by manipulating broad substrate specificity. Mol Biosyst. 2009;5(9):1032–1038. doi: 10.1039/b904032c. [DOI] [PubMed] [Google Scholar]
- 22.Young DD, et al. An evolved aminoacyl-tRNA synthetase with atypical polysubstrate specificity. Biochemistry. 2011;50(11):1894–1900. doi: 10.1021/bi101929e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cooley RB, Karplus PA, Mehl RA. Gleaning unexpected fruits from hard-won synthetases: Probing principles of permissivity in non-canonical amino acid-tRNA synthetases. ChemBioChem. 2014;15(12):1810–1819. doi: 10.1002/cbic.201402180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wan W, Tharp JM, Liu WR. Pyrrolysyl-tRNA synthetase: An ordinary enzyme but an outstanding genetic code expansion tool. Biochim Biophys Acta. 2014;1844(6):1059–1070. doi: 10.1016/j.bbapap.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neumann H, Peak-Chew SY, Chin JW. Genetically encoding N(ε)-acetyllysine in recombinant proteins. Nat Chem Biol. 2008;4(4):232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]
- 26.Umehara T, et al. N-acetyl lysyl-tRNA synthetases evolved by a CcdB-based selection possess N-acetyl lysine specificity in vitro and in vivo. FEBS Lett. 2012;586(6):729–733. doi: 10.1016/j.febslet.2012.01.029. [DOI] [PubMed] [Google Scholar]
- 27.Mukai T, et al. Adding l-lysine derivatives to the genetic code of mammalian cells with engineered pyrrolysyl-tRNA synthetases. Biochem Biophys Res Commun. 2008;371(4):818–822. doi: 10.1016/j.bbrc.2008.04.164. [DOI] [PubMed] [Google Scholar]
- 28.Mukai T, et al. Genetic-code evolution for protein synthesis with non-natural amino acids. Biochem Biophys Res Commun. 2011;411(4):757–761. doi: 10.1016/j.bbrc.2011.07.020. [DOI] [PubMed] [Google Scholar]
- 29.Arbely E, et al. Acetylation of lysine 120 of p53 endows DNA-binding specificity at effective physiological salt concentration. Proc Natl Acad Sci USA. 2011;108(20):8251–8256. doi: 10.1073/pnas.1105028108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neumann H, et al. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol Cell. 2009;36(1):153–163. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaelin WG, Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153(1):56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ko JH, et al. Pyrrolysyl-tRNA synthetase variants reveal ancestral aminoacylation function. FEBS Lett. 2013;587(19):3243–3248. doi: 10.1016/j.febslet.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heinemann IU, et al. Enhanced phosphoserine insertion during Escherichia coli protein synthesis via partial UAG codon reassignment and release factor 1 deletion. FEBS Lett. 2012;586(20):3716–3722. doi: 10.1016/j.febslet.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang YS, Fang X, Wallace AL, Wu B, Liu WR. A rationally designed pyrrolysyl-tRNA synthetase mutant with a broad substrate spectrum. J Am Chem Soc. 2012;134(6):2950–2953. doi: 10.1021/ja211972x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polycarpo CR, et al. Pyrrolysine analogues as substrates for pyrrolysyl-tRNA synthetase. FEBS Lett. 2006;580(28-29):6695–6700. doi: 10.1016/j.febslet.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ambrogelly A, et al. Pyrrolysine is not hardwired for cotranslational insertion at UAG codons. Proc Natl Acad Sci USA. 2007;104(9):3141–3146. doi: 10.1073/pnas.0611634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolfson AD, Pleiss JA, Uhlenbeck OC. A new assay for tRNA aminoacylation kinetics. RNA. 1998;4(8):1019–1023. doi: 10.1017/s1355838298980700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kavran JM, et al. Structure of pyrrolysyl-tRNA synthetase, an archaeal enzyme for genetic code innovation. Proc Natl Acad Sci USA. 2007;104(27):11268–11273. doi: 10.1073/pnas.0704769104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yanagisawa T, et al. Crystallographic studies on multiple conformational states of active-site loops in pyrrolysyl-tRNA synthetase. J Mol Biol. 2008;378(3):634–652. doi: 10.1016/j.jmb.2008.02.045. [DOI] [PubMed] [Google Scholar]
- 40.Hao B, et al. Reactivity and chemical synthesis of L-pyrrolysine- the 22(nd) genetically encoded amino acid. Chem Biol. 2004;11(9):1317–1324. doi: 10.1016/j.chembiol.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 41.Wong ML, Guzei IA, Kiessling LL. An asymmetric synthesis of L-pyrrolysine. Org Lett. 2012;14(6):1378–1381. doi: 10.1021/ol300045c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ibba M, Kast P, Hennecke H. Substrate specificity is determined by amino acid binding pocket size in Escherichia coli phenylalanyl-tRNA synthetase. Biochemistry. 1994;33(23):7107–7112. doi: 10.1021/bi00189a013. [DOI] [PubMed] [Google Scholar]
- 43.Chen JF, Guo NN, Li T, Wang ED, Wang YL. CP1 domain in Escherichia coli leucyl-tRNA synthetase is crucial for its editing function. Biochemistry. 2000;39(22):6726–6731. doi: 10.1021/bi000108r. [DOI] [PubMed] [Google Scholar]
- 44.Steinfeld JB, Aerni HR, Rogulina S, Liu Y, Rinehart J. Expanded cellular amino acid pools containing phosphoserine, phosphothreonine, and phosphotyrosine. ACS Chem Biol. 2014;9(5):1104–1112. doi: 10.1021/cb5000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yanagisawa T, Sumida T, Ishii R, Yokoyama S. A novel crystal form of pyrrolysyl-tRNA synthetase reveals the pre- and post-aminoacyl-tRNA synthesis conformational states of the adenylate and aminoacyl moieties and an asparagine residue in the catalytic site. Acta Crystallogr D Biol Crystallogr. 2013;69(Pt 1):5–15. doi: 10.1107/S0907444912039881. [DOI] [PubMed] [Google Scholar]
- 46.Schmidt MJ, Weber A, Pott M, Welte W, Summerer D. Structural basis of furan-amino acid recognition by a polyspecific aminoacyl-tRNA-synthetase and its genetic encoding in human cells. ChemBioChem. 2014;15(12):1755–1760. doi: 10.1002/cbic.201402006. [DOI] [PubMed] [Google Scholar]
- 47.Yanagisawa T, et al. Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode N(ε)-(o-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem Biol. 2008;15(11):1187–1197. doi: 10.1016/j.chembiol.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 48.Flügel V, Vrabel M, Schneider S. Structural basis for the site-specific incorporation of lysine derivatives into proteins. PLoS ONE. 2014;9(4):e96198. doi: 10.1371/journal.pone.0096198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kobayashi T, Yanagisawa T, Sakamoto K, Yokoyama S. Recognition of non-α-amino substrates by pyrrolysyl-tRNA synthetase. J Mol Biol. 2009;385(5):1352–1360. doi: 10.1016/j.jmb.2008.11.059. [DOI] [PubMed] [Google Scholar]
- 50.Takimoto JK, Dellas N, Noel JP, Wang L. Stereochemical basis for engineered pyrrolysyl-tRNA synthetase and the efficient in vivo incorporation of structurally divergent non-native amino acids. ACS Chem Biol. 2011;6(7):733–743. doi: 10.1021/cb200057a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schneider S, et al. Structural insights into incorporation of norbornene amino acids for click modification of proteins. ChemBioChem. 2013;14(16):2114–2118. doi: 10.1002/cbic.201300435. [DOI] [PubMed] [Google Scholar]
- 52.Yanagisawa T, Umehara T, Sakamoto K, Yokoyama S. Expanded genetic code technologies for incorporating modified lysine at multiple sites. ChemBioChem. 2014;15(15):2181–2187. doi: 10.1002/cbic.201402266. [DOI] [PubMed] [Google Scholar]
- 53.Tracewell CA, Arnold FH. Directed enzyme evolution: Climbing fitness peaks one amino acid at a time. Curr Opin Chem Biol. 2009;13(1):3–9. doi: 10.1016/j.cbpa.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang K, et al. Optimized orthogonal translation of unnatural amino acids enables spontaneous protein double-labelling and FRET. Nat Chem. 2014;6(5):393–403. doi: 10.1038/nchem.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lepthien S, Merkel L, Budisa N. In vivo double and triple labeling of proteins using synthetic amino acids. Angew Chem Int Ed Engl. 2010;49(32):5446–5450. doi: 10.1002/anie.201000439. [DOI] [PubMed] [Google Scholar]
- 56.Reynolds NM, Lazazzera BA, Ibba M. Cellular mechanisms that control mistranslation. Nat Rev Microbiol. 2010;8(12):849–856. doi: 10.1038/nrmicro2472. [DOI] [PubMed] [Google Scholar]
- 57.Drummond DA, Wilke CO. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet. 2009;10(10):715–724. doi: 10.1038/nrg2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lajoie MJ, et al. Genomically recoded organisms expand biological functions. Science. 2013;342(6156):357–360. doi: 10.1126/science.1241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pavlov MY, et al. Slow peptide bond formation by proline and other N-alkylamino acids in translation. Proc Natl Acad Sci USA. 2009;106(1):50–54. doi: 10.1073/pnas.0809211106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haruna K, Alkazemi MH, Liu Y, Söll D, Englert M. Engineering the elongation factor Tu for efficient selenoprotein synthesis. Nucleic Acids Res. 2014;42(15):9976–9983. doi: 10.1093/nar/gku691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mittelstaet J, Konevega AL, Rodnina MV. A kinetic safety gate controlling the delivery of unnatural amino acids to the ribosome. J Am Chem Soc. 2013;135(45):17031–17038. doi: 10.1021/ja407511q. [DOI] [PubMed] [Google Scholar]
- 62.Cooley RB, et al. Structural basis of improved second-generation 3-nitro-tyrosine tRNA synthetases. Biochemistry. 2014;53(12):1916–1924. doi: 10.1021/bi5001239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Donoghue P, Ling J, Wang YS, Söll D. Upgrading protein synthesis for synthetic biology. Nat Chem Biol. 2013;9(10):594–598. doi: 10.1038/nchembio.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roy H, et al. The tRNA synthetase paralog PoxA modifies elongation factor-P with (R)-β-lysine. Nat Chem Biol. 2011;7(10):667–669. doi: 10.1038/nchembio.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carlson JC, Badran AH, Guggiana-Nilo DA, Liu DR. Negative selection and stringency modulation in phage-assisted continuous evolution. Nat Chem Biol. 2014;10(3):216–222. doi: 10.1038/nchembio.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li WT, et al. Specificity of pyrrolysyl-tRNA synthetase for pyrrolysine and pyrrolysine analogs. J Mol Biol. 2009;385(4):1156–1164. doi: 10.1016/j.jmb.2008.11.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.