

Abstract
Panobinostat is an investigational and potent histone deacetylase inhibitor (HDACi) that has shown promise as an antimultiple myeloma agent in the preclinical setting. In this review, we discuss the rationale for the use of panobinostat as a combination therapy for multiple myeloma and provide an overview of recent and ongoing clinical trials testing the safety and efficacy of panobinostat for the treatment of the disease.
Keywords: histone deacetylase inhibitor, multiple myeloma, panobinostat
Introduction
During the past decade, significant progress has been made in the management of multiple myeloma due to the introduction of new therapeutic agents such as proteasome inhibitors (PIs) and immunomodulatory agents (IMiDs) [Mateos et al. 2013]. Despite this, multiple myeloma remains incurable and patients eventually become refractory to their treatment regimens. Thus, there is a clear need for the development of new therapeutic options. Histone deacetylase inhibitors (HDACis) are a relatively new class of agents that have demonstrated effective anticancer activity in the preclinical setting, and two HDACis have been approved by the US Food and Drug Administration (FDA) for the treatment of specific hematological malignancies [Ververis et al. 2013]. Panobinostat is an investigational and potent HDACi that has shown activity against multiple myeloma at nanomolar concentrations in preclinical studies [Atadja, 2009; Sanchez et al. 2011]. In this review, we discuss the rationale for the use of panobinostat as a combination therapy for multiple myeloma and provide an overview of recent and ongoing clinical trials testing the safety and efficacy of panobinostat for the treatment of this disease.
Multiple myeloma
Multiple myeloma (MM), a plasma cell dyscrasia, is the most common primary malignancy of the bone marrow [Morgan, 1999; Smith and Newland, 2000]. It is estimated that 24,050 new cases of MM (13,500 in men and 10,550 in women) will be diagnosed in the United States and that 11,090 men and women will die from the disease during 2014 [Siegel et al. 2014]. MM patients treated with conventional chemotherapy have an average overall survival (OS) of 4 years as these therapies are not curative. In recent years, new and more effective drugs, including IMiDs and PIs, have become available for the treatment of MM. Such drugs have been evaluated alone and in combination with established anti-MM agents, rapidly increasing the number of therapeutic options available to MM patients. As a result, the 5-year survival rate for MM patients is currently 44% [Brenner et al. 2008; Kumar et al. 2008; Pulte et al. 2014]. Unfortunately, even with these newer agents, responses to therapy are transient, and MM remains an incurable disorder with an eventual fatal outcome. Therefore, there is an urgent need to find novel therapeutic targets and develop new therapeutic strategies that are more effective and well-tolerated, particularly in the relapsed/refractory (RR) setting.
Histone acetylases and histone deacetylases
Protein acetylation is a dynamic post-translational modification that is controlled by two groups of enzymes with opposite activities: histone acetylases (HATs) and histone deacetylases (HDACs) [Khan and La Thangue, 2012]. HATs and HDACs regulate gene transcription, cell differentiation, cell cycle progression and apoptosis by targeting both histone and nonhistone proteins [Maes et al. 2013] (Figure 1). Hyperacetylation of histone proteins results in a relaxed chromatin configuration which is compatible with gene transcription, whereas hypoacetylation of histones leads to chromatin compaction and gene silencing [Maes et al. 2013]. The activity of many chaperones and transcriptional factors, as well as that of tumor suppressor and structural proteins, depends on their acetylation status [New et al. 2012]. Therefore, alterations in HATs or HDACs can affect a myriad of cellular processes.
Figure 1.
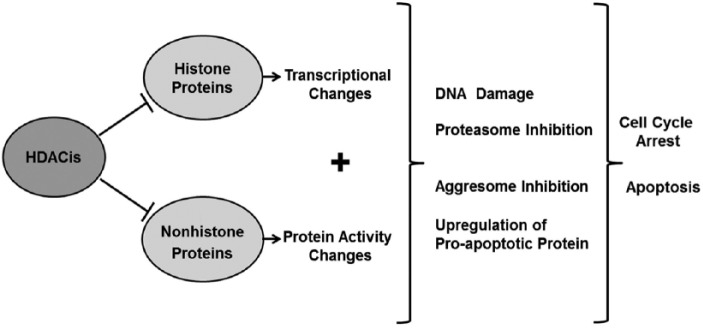
Histone deacetylase inhibitors (HDACis) block the deacetylation of both histone and nonhistone proteins, thereby causing transcriptional and protein activity changes. In multiple myeloma cells, such changes have been shown to lead to proteasome and aggresome inhibition, DNA damage and the upregulation of proapoptotic proteins, resulting in cell cycle arrest and apoptosis.
HDACs are categorized by their homology to yeast HDACs and based on their requirement for Zn2+ as a cofactor. Zn2+-dependent enzymes include: class I HDACs (1–3 and 8), which localize to the cell nucleus and are ubiquitously expressed; class II a/b HDACs (4–7, 9 and 10), which can shuttle between the cell nucleus and cytoplasm and have tissue-specific expression; and class IV HDACs, of which HDAC11 is the sole member, a predominantly nuclear HDAC with limited tissue distribution (kidney, brain, heart, skeletal muscle and testis) [Gao et al. 2002; Ropero and Esteller, 2007; Khan and La Thangue, 2012]. HDACs in class III (sirtuins, SIR 1–7) are Zn2+-independent/NAD+-dependent enzymes, for which their pattern of expression and tissue distribution remain poorly characterized [Ropero and Esteller, 2007; Khan and La Thangue, 2012].
The balance between acetylation and deacetylation is critical for normal cell function, and loss of protein acetylation has been shown to play a role in cancer initiation and progression [Ropero and Esteller, 2007; New et al. 2012]. Indeed, aberrant recruitment of HDACs to gene promoters has been shown to occur in hematological malignancies and HDAC deregulated expression has been reported in tumors of various origins including blood, colon, lung, bladder, pancreas, prostate, breast, cervix, brain, kidney, liver and stomach [Ropero and Esteller, 2007; Van Damme et al. 2012; Müller et al. 2013; Niegisch et al. 2013; Petta et al. 2013; Stenzinger et al. 2013; West and Johnstone, 2014). Because of their role in tumorigenesis, HDACs have long been considered an attractive therapeutic target.
Histone deacetylase inhibitors
HDACis are a diverse group of compounds that can be classified by chemical structure as short chain fatty acids (valproic acid, sodium butyrate and phenyl butyrate), hydroxamic acids (trichostatin A (TSA), vorinostat (SAHA), panobinostat, belinostat, dacinostat, resminostat, givinostat, suberohydroxamic acid (SBHA), rocilinostat, abexinostat, quisinostat, CHR-3996, AR-42 and pracinostat), mercaptoketones (KD5170), cyclic peptides (apicidin and romidepsin), benzamides (mocetinostat, entinostat, chidamide and tacedinaline), sirtuin inhibitors (niacinamide and sirtinol) and tubacin (Table 1) [Maes et al. 2013; West and Johnstone, 2014]. The majority of HDACis interfere with the Zn2+ ion in the catalytic site of one or more specific HDACs or multiple HDAC classes (pan-HDACi).
Table 1.
Histone deacetylase inhibitors (HDACis) currently being tested in clinical studies for hematological malignancies.
| Chemical class | HDACi | Clinical status (highest phase for hematological malignancies) | Hematological malignancy | ClinicalTrials.gov identifier |
|---|---|---|---|---|
| Short chain fatty acids | Valproic acid | II | AML, MDS, CLL, non-Hodgkin’s and Hodgkin’s lymphoma | NCT00414310; NCT00382590; NCT00339196; NCT00439673; NCT00326170; NCT01016990; NCT01356875 |
| Butyrate | In vitro | N/A | ||
| Phenylbutyrate | II | AML, MDS, non-Hodgkin’s lymphoma, MM | NCT00006019 | |
| Hydroxamic acids | Trichostatin A | In vitro | N/A | |
| Vorinostat | Approved | CTCL | NCT01554852; NCT00773747 | |
| III | MM | |||
| Panobinostat | III | MM | NCT01023308 | |
| Belinostat | II | AML, MM, MDS,, T-cell lymphomas, PTCL, B-cell non-Hodgkin’s lymphoma | Cashen et al. [2012]; NCT00131261; NCT00131261; NCT00431340*; NCT00303953; NCT00274651; NCT00865969 | |
| Dacinostat | I | N/A | ||
| Resminostat | II | Hodgkin’s lymphoma | NCT01037478 | |
| Givinostat | II | Polycythaemia vera, myeloproliferative neoplasms, MM, Hodgkin’s lymphoma | Finazzi et al. [2013]; NCT01761968; NCT00792506;* NCT00606307; NCT00496431;$ NCT00792467 | |
| Suberohydroxamic acid (SBHA) | Preclinical | N/A | ||
| Rocilinostat | I/II | MM, lymphoid malignancies | NCT01323751; NCT01997840; NCT02091063; NCT01583283 | |
| Abexinostat | I/II | Non-Hodgkin’s and Hodgkin’s lymphoma | NCT00724984 | |
| Quisinostat | II | CTCL | NCT01486277 | |
| CHR-3996 | I | N/A | NCT00697879 | |
| AR-42 | I | MM, CLL, lymphoma, AML | NCT01129193; NCT01798901 | |
| Pracinostat | II | MDS, AML, myelofibrosis | Quintás-Cardama et al. [2012]; NCT01112384; NCT01075308; NCT01873703; NCT01993641; NCT01912274 | |
| Mercaptoketones | KD5170 | Preclinical | N/A | |
| Cyclic peptides | Apicidin | Preclinical | N/A | |
| Romidepsin | Approved II |
CTCL and PCTL MM | NCT00066638; NCT00765102 | |
| Benzamides | Mocetinostat | II | MDS, CLL, lymphoma | NCT00324220; NCT00431873; NCT00359086 |
| Entinostat | II | Hodgkin’s lymphoma, AML, MDS, ALL | NCT00866333; NCT01305499; NCT00313586; NCT00462605; NCT00466115 | |
| Chidamide | II | N/A | NCT01836679 | |
| Tacedinaline | II | MM | NCT00005624 | |
| Sirtuin inhibitors | Niacinamide | I | Non-Hodgkin’s and Hodgkin’s lymphoma | NCT00691210 |
| Sirtinol | Preclinical | N/A | ||
| 1,3-dioxanes | Tubacin | Preclinical | N/A |
The study was terminated due to dose-limiting toxicities.
The study was terminated due to limited activity of the drug.
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; CTCL, cutaneous T-cell lymphoma; HDAC, histone deacetylase; HDACi, histone deacetylase inhibitor; MDS, myelodysplastic syndrome; MM, multiple myeloma; N/A, not applicable; PCTL, peripheral T-cell lymphoma.
A direct consequence of HDAC inhibition is the hyperacetylation of proteins, which results in a wide variety of responses including induction of cell cycle arrest, apoptosis, senescence and differentiation, as well as DNA damage, immunogenicity, downregulation of members of the aggresome pathway, and inhibition of angiogenesis [Maes et al. 2013; West and Johnstone, 2014] (Figure 1).
Based on their in vitro and in vivo preclinical activity, HDACis have undergone rapid clinical development. HDACis have been shown to exert effects in several types of cancers, although responses to treatment with single agent HDACis have primarily been observed in advanced hematologic malignancies and in thyroid, lung and prostate tumors [Rasheed et al. 2008; Prince et al. 2009].
Currently, vorinostat and romidepsin are the only two HDACis approved by the FDA for the treatment of cutaneous T-cell lymphoma (CTCL). Romidepsin has also been approved for the treatment of peripheral T-cell lymphoma (PTCL) [Treppendahl et al. 2014]. In in vitro studies, vorinostat has also displayed activity against MM cell lines. TSA, sodium butyrate and dacinostat (NVP-LAQ824) have been shown to inhibit proliferation and induce apoptosis in MM cell lines, patient-derived MM cells and cells resistant to various anti-MM therapies [Lavelle et al. 2001; Catley et al. 2003]. Significant decreases in tumor growth and increases in survival were also observed in response to dacinostat in a MM xenograft mouse model [Catley et al. 2003].
Other HDACis, including valproic acid, have been and continue to be evaluated in the clinical setting with mixed results [West and Johnstone, 2014]. Efforts to improve efficacy have led to both the assessment of existing compounds in combination therapies and the development of newer compounds, such as panobinostat and belinostat among others. The clinical status of the currently available HDACis is shown in Table 1.
Histone deacetylase inhibitors as therapy for MM
Malignant plasma cells produce large quantities of misfolded or unfolded immunoglobulins [Cenci, 2012] and rely heavily on their protein handling machinery, which includes both the proteasome and the aggresome, to circumvent cytotoxicity [Aronson and Davies, 2012]. Peptide degradation is also regulated by the aminopeptidase enzyme system, which catalyzes the hydrolysis of proteins and peptides from the NH2-terminus [Botbol and Scornik, 1991]. Clearly, the inhibition of any of these pathways will have a detrimental effect on cell viability.
High expression levels of proteins involved in the proteasome pathway are often observed in hematopoietic malignancies [Jankowska et al. 2013]. A s a result, malignant plasma cells are particularly sensitive to proteasome inhibition [Aronson and Davies, 2012]. Proteasome inhibition has been shown to lead to cell death in malignant cells; however, an undesirable consequence of treatment with PIs is the compensatory induction of autophagy via the aggresome pathway [Hideshima and Anderson, 2012; Kale and Moore, 2012; Mateos et al. 2013]. Degradation of misfolded proteins via the aggresome requires both the presence of intact microtubules for protein transportation and the activity of HDAC6, which targets acetylated tubulin [Simms-Waldrip et al. 2008]. Inhibitors of HDAC6 such as tubacin interfere with the activity of the aggresome pathway and cause misfolded proteins to accumulate [Simms-Waldrip et al. 2008]. Inhibition of aminopeptidases disrupts protein turnover and leads to peptide accumulation and reduced amino acid availability, which in turns causes cytotoxicity. Treatment of MM cells with the aminopeptidase inhibitor tosedostat has been shown to induce cell cycle arrest, apoptosis and autophagy, and to synergize with the PI bortezomib [Moore et al. 2009].
The combination of HDACis and other anti-MM therapies has also been evaluated in preclinical studies. For instance, tubacin, vorinostat, romidepsin, belinostat, rocilinostat and panobinostat (see below) have all demonstrated synergistic cytotoxicity with bortezomib in MM cell lines, and primary cells from MM patients that are sensitive or resistant to bortezomib [Pei et al. 2004; Hideshima et al. 2005; Maiso et al. 2006; Feng et al. 2007; Simms-Waldrip et al. 2008; Campbell et al. 2010; Santo et al. 2012]. Conversely, bortezomib has been shown to downregulate the expression of class I HDACs in MM cells, thereby affecting gene transcription [Kikuchi et al. 2010]. For instance, basal expression of Kruppel-like family factor 9 (KLF9), a transcription factor that regulates pro-apoptotic genes, has been shown to be higher in MM cells from patients who respond to bortezomib, and treatment of MM cell lines with this PI has shown to upregulate KLF9 [Mannava et al. 2012]. HDACis have also been shown to potentiate the anti-MM activity of IMiDs such as lenalidomide and thalidomide, chemotherapeutic agents and steroids [Sanchez et al. 2011; Hajek et al. 2014]. Together, these studies have provided support for the use of HDACi as anti-MM therapy, especially when combined with other active anti-MM agents.
Panobinostat
Panobinostat (LBH589) is a potent cinnamic hydroxamic acid analogue capable of inhibiting class I, II and IV HDACs at nanomolar concentrations [Atadja, 2009]. Panobinostat was originally formulated for both intravenous (IV) and oral administration. This HDACi has demonstrated potent antiproliferative and cytotoxic activities in a variety of cell lines derived from hematological malignancies, including CTCL, chronic myelogenous leukemia (CML), acute myeloid leukemia (AML), Hodgkin lymphoma and MM, and cell lines derived from breast, prostate, colon and pancreatic cancers, while displaying minimal toxicity on normal cells [Catley et al. 2006; Maiso et al. 2006; Atadja, 2009; Bruzzese et al. 2013].
Panobinostat and MM preclinical studies
Panobinostat causes cell cycle arrest and caspase dependent and independent apoptosis in MM cell lines [Catley et al. 2006; Maiso et al. 2006]. Panobinostat has been shown to have cytotoxic effects on MM cell lines and tumor cells derived from MM patients known to be refractory to anti-MM drugs, including the anthracycline doxorubicin, anthracenedione antineoplastic agent mitoxantrone, alkylating agent melphalan, glucocorticosteroid dexamethasone and bortezomib. [Catley et al. 2006; Maiso et al. 2006]. Recently, it has been suggested that the inhibition of class I HDACs is sufficient to induce significant MM cell death and therefore that pan-HDACis such as panobinostat are more effective as single agents than inhibitors that target only HDAC6 such as tubacin [Mithraprabhu et al. 2013]. In MM cells, panobinostat can also reactivate the expression of genes which silencing is thought to enable the proliferation of differentiated B cells, thereby inducing cell death [Kalushkova et al. 2010].
Panobinostat has been shown to increase the anti-MM activity of the bisphosphonate zoledronic acid, the insulin-like growth factor type 1 receptor tyrosine kinase inhibitor picropodiphyllin, the aminopeptidase inhibitor tosedostat, dexamethasone, bortezomib, doxorrubicin, and melphalan [Maiso et al. 2006; Moore et al. 2009; Sanchez et al. 2011; Lemaire et al. 2012; Bruzzese et al. 2013]. Similar to tubacin, panobinostat was shown to induce α-tubulin hyperacetylation, decrease the 20S chymotryptic activity of the proteasome, and reduce bortezomib-induced aggresome formation, which may help explain, at least in part, panobinostat’s activity in bortezomib-resistant cells and the synergism observed between this HDACi and bortezomib [Catley et al. 2003, 2006].
Our previous studies in various MM cell lines demonstrated induction of tubulin and histone acetylation as well as caspase-dependent apoptosis in response to treatment with panobinostat, and these effects were potentiated when the HDACi was combined with either melphalan or doxorubicin [Sanchez et al. 2011]. We also observed significant decreases in human paraprotein levels (a measurement of MM burden) and tumor size after treatment with panobinostat (once daily for 5 days) in our human MM xenograft mouse model LAGλ-1, which carries uncultured, patient-derived MM cells. Similar to our in vitro studies, the anti-MM effect was shown to be enhanced when this HDACi was combined with melphalan (once weekly) or pegylated liposomal doxorubicin (PLD; three consecutive days a week) in this in vivo model [Sanchez et al. 2011].
Synergistic effects have also been observed in triple combinations of newer anti-MM drugs. In vitro treatment with panobinostat, dexamethasone and bortezomib or lenalidomide showed more cytotoxic activity than each anti-MM agent used alone or in dual combinations [Ocio et al. 2010]. In these xenograft mouse models of disseminated and extramedullary MM, the triple combinations also conferred a significant survival advantage compared with double agent combinations or single agent treatment. A summary of HDACis with activity in MM cells is shown in Table 2.
Table 2.
Histone deacetylase inhibitors (HDACis) with activity in multiple myeloma cell lines.
| Chemical class | HDACi | References |
|---|---|---|
| Short chain fatty acids | Sodium butyrate | Lavelle et al. [2001] |
| Pei et al. [2004] | ||
| Hydroxamic acids | Trichostatin A | Lavelle et al. [2001] |
| Vorinostat | Pei et al. [2004] | |
| Matthews et al. [2013] | ||
| Panobinostat | Catley et al. [2006] | |
| Maiso et al. [2006] | ||
| Ocio et al. [2010] | ||
| Sanchez et al. [2011] | ||
| Matthews et al. [2013] | ||
| Belinostat | Feng et al. [2007] | |
| Dacinostat | Catley et al. [2003] | |
| Rocilinostat | Santo et al. [2012] | |
| Romidepsin | Matthews et al. [2013] | |
| 1,3-dioxanes | Tubacin | Hideshima et al. [2005] |
| Mithraprabhu et al. [2013] |
HDACi, Histone deacetylase inhibitor.
These findings provided support for the clinical development of panobinostat in combination with alkylating agents, IMiDs and/or PIs for the treatment of MM patients.
Panobinostat and MM clinical studies
The initial phase I studies evaluating single-agent panobinostat were carried out in solid tumors and hematological malignancies using the IV formulation [Giles et al. 2006; Sharma et al. 2013]. QTc prolongation and cardiac arrhythmias reported in these trials led to the discontinuation of the IV administration route [Khot et al. 2013; Sharma et al. 2013].
The safety and efficacy of single-agent panobinostat administered orally on Monday, Wednesday and Friday (thrice weekly) of every week or every other week was evaluated in a phase Ia/II study for patients with hematological malignancies, including MM [DeAngelo et al. 2013]. The maximum tolerated dose (MTD) of panobinostat was dependent on the indication and one partial response (PR) was observed in a MM patient [DeAngelo et al. 2013]. Following this, the activity of single-agent oral panobinostat administered at 20 mg thrice weekly for 2 weeks of a 21-day cycle was investigated in heavily pretreated RRMM patients. Panobinostat demonstrated durable, albeit modest, responses in two (one PR, one minimal response) of the 38 evaluable patients [Wolf et al. 2012]. As a result, the focus of clinical studies with oral panobinostat has shifted to combination therapies.
The vast majority of the clinical studies examining the safety and efficacy of panobinostat as combination therapy for MM patients have been carried out in the RR setting (Table 3). To date, the most promising combination appears to be that of panobinostat and bortezomib. The combination of oral panobinostat and IV bortezomib was investigated in a phase Ib trial [San-Miguel et al. 2013]. Panobinostat was administered on Monday, Wednesday and Friday for 3 consecutive weeks and bortezomib was administered at 1.0 mg/m2 on days 1, 4, 8 and 11 of a 21-day cycle. In the dose escalation phase of the study, the MTD of panobinostat in combination with bortezomib was established at 20 mg [San-Miguel et al. 2013]. Thrombocytopenia was the most frequent hematological event and QTc prolongation was only observed in one patient. In the expansion phase of the trial, the schedule of panobinostat was changed and the drug was administered at the MTD thrice weekly but for only the first two weeks of a 21-day cycle to allow for platelet recovery. Dexamethasone at 20 mg administered after bortezomib was also allowed after cycle 2 because, as stated above, the triple combination of panobinostat, bortezomib and dexamethasone was shown to have greater anti-MM activity than any dual combination [Ocio et al. 2010]. The overall response rate (ORR) was 51.5% (n = 62) and the ORR of the expansion phase was 73.3% (n = 11). Responses (26.3%) were also observed in bortezomib-refractory patients [San-Miguel et al. 2013].
Table 3.
Panobinostat clinical trials for relapsed/refractory multiple myeloma.
| Combination with | Phase | Number of patients | Efficacy | Adverse events (G3/G4) |
|---|---|---|---|---|
| BTZ | I | 11 | ORR = 36.4% | Leukopenia = 54.0% |
| Neutropenia = 45.0% | ||||
| Thrombocytopenia = 36.0% | ||||
| LEN/DEX | Ib | 46 (30 evaluated for response) | ORR = 56.66% | Thrombocytopenia = 44.0% |
| Neutropenia = 37.0% | ||||
| BTZ | Ib | 47 (dose escalation) | ORR = 52.9% (dose escalation) | Dose escalation: |
| Thrombocytopenia = 85.5% | ||||
| 15 (expansion) | Neutropenia = 63.8% | |||
| ORR = 73.3% (expansion) | Asthenia = 29.8% | |||
| Expansion | ||||
| Thrombocytopenia = 66.7% | ||||
| Neutropenia = 46.7% | ||||
| Fatigue = 20.0% | ||||
| CFZ | I/Ib | 17 | ORR = 35.0% | Thrombocytopenia = 59% |
| Fatigue = 59.0% | ||||
| Anemia = 41.0% | ||||
| Neutropenia = 35.2% | ||||
| Pneumonia-23.5% | ||||
| CFZ | I/II | 10 enrolled, 9 evaluated | ORR = 64% | Thrombocytopenia = 30.0% |
| Neutropenia = 20.0% | ||||
| Fatigue = 11.0% | ||||
| MEL | I/II | 40 | ORR = 7.5% | Thrombocytopenia = 30.8% |
| Neutropenia = 23.1% | ||||
| MEL/PRED/THAL | I/II | 24 | ORR = 38.5% | Neutropenia = 71.0% |
| Thrombocytopenia = 35.5% | ||||
| BTZ/DEX (PANORAMA 2) | II | 55 | ORR = 38.5%CBR = 49.0% | Neutropenia = 53.0% |
| PN = 2.0% | ||||
| LEN/DEX | II | 5 | ORR = 40.0% | Neutropenia = 60.0% |
| CBR = 60.0% | Thrombocytopenia = 60.0% | |||
| Febrile neutropenia = 20.0% | ||||
| Pulmonary embolism = 20.0% | ||||
| BTZ/DEX (PANORAMA 1) | III | 536 enrolled525 evaluated | Thrombocytopenia = 36.2% | |
| Anemia = 13.0% | ||||
| Diarrhea = 14.5% | ||||
| Fatigue = 12.2% | ||||
| Neutropenia = 11.4% | ||||
| Peripheral neuropathy = 5.3% |
BTZ, bortezomib; CBR, clinical benefit rate; CFZ, carfilzomib; DEX, dexamethasone; LEN, lenalidomide; MEL, melphalan; ORR, overall response rate; PRED, prednisone; THAL, thalidomide.
On the basis of these early studies, PANORAMA 2 (PANobinostat ORAl in Multiple MyelomA), a phase II, single arm, two-stage trial, evaluated the triple combination of panobinostat, bortezomib and dexamethasone for bortezomib-refractory MM patients [Richardson et al. 2012]. In the stage 1 of the trial, panobinostat was administered at 20 mg three times a week, on weeks 1 and 2 of a 21-day cycle for a total of 8 cycles. Bortezomib was given IV at 1.3 mg/m2 on days 1, 4, 8 and 11, and oral dexamethasone was given at 20 mg on the day of, and the day after each bortezomib administration. Patients showing clinical benefit were eligible to continue therapy as part of the stage 2 of the trial. In stage 2, panobinostat was given three times a week on weeks 1, 2, 4 and 5 of a 6-week cycle, whereas bortezomib was administered once a week on weeks 1, 2, 4 and 5, and dexamethasone was given the day of, and the day after bortezomib administration. Responses were observed in 19 out of 55 evaluable patients, including one near complete response (CR) and 18 PRs; the ORR was 34.5% [Richardson et al. 2012]. Median progression-free survival (PFS) was 5.4 months and the median OS was 17.5 months [Schlossman et al. 2013]. The triple combination displayed manageable toxicities, with thrombocytopenia being the most common grade 3/4 hematological adverse event. Treatment emergent peripheral neuropathy was mild and observed in 27.3% of patients, with only one grade 3/4 event reported [Richardson et al. 2013]. A phase III randomized trial, PANORAMA 1, is comparing the efficacy of bortezomib and dexamethasone versus panobinostat, bortezomib and dexamethasone for MM patients who have previously received but were not refractory to bortezomib. Preliminary safety data from the first 525 evaluable patients enrolled in the trial have been reported and suggest that the safety profile of the triple combination is similar to that shown in the PANORAMA 2 trial [San-Miguel et al. 2012].
The promising results achieved with the combination of an HDACi and a PI have provided rationale for a phase I/Ib study testing panobinostat in combination with carfilzomib, a second generation PI that is very active in MM and has an improved safety profile compared with bortezomib [Shah et al. 2012]. Panobinostat was administered 3 times a week for the first 2 weeks of every 28-day cycle and carfilzomib was given as an infusion over 30 minutes on days 1, 2, 8, 9, 15 and 16. Dose levels started with panobinostat at 15 mg and carfilzomib at 20/27 mg/m2 and escalated to 20 mg or 30 mg (panobinostat) and 20/36 or 20/45 mg/m2 (carfilzomib) using a classic 3+3 schema based on dose-limiting toxicities. For the 17 evaluable patients, the ORR was 35%, including 2 very good partial responses (VGPRs) and 1 PR. Grade 3/4 toxicities included thrombocytopenia, fatigue, anemia, neutropenia and pneumonia [Shah et al. 2012]. An ongoing phase I/II study is also evaluating this combination [Berdeja et al. 2012]. Panobinostat was administered thrice weekly on weeks 1 and 3 of a 28-day cycle and carfilzomib was given as an infusion over 30 minutes on days 1, 2, 8, 9, 15 and 16 of weeks 1–3 of a 28-day cycle. Four dose levels were evaluated: panobinostat 20 mg and carfilzomib 20 (first cycle)/27 (subsequent cycles) mg/m2; panobinostat 20 mg and carfilzomib 20/36 mg/m2; panobinostat 20 mg and carfilzomib 20/45 mg/m2; and panobinostat 30 mg and carfilzomib 20/45 mg/m2 [Berdeja et al. 2013]. No dose limiting toxicities were observed in the dose-escalating phase of the study; and, therefore, the expansion phase opened at the maximum administered dose (MAD). In 9 evaluable patients, the ORR of this drug combination was 64% with responses observed among both bortezomib and IMiD refractory patients. A total of 61% of patients experienced ≥ grade 3 hematological toxicities, whereas 34% of patients experienced nonhematological toxicities. Peripheral neuropathy was infrequent (5% of patients) and no grade 3/4 cases were reported. A total of 59% of patients who received the MAD required panobinostat dose reductions [Berdeja et al. 2013]. Because of the observed toxicity, two additional dose levels are currently being evaluated.
Panobinostat in combination with lenalidomide and dexamethasone was also tested based on promising preclinical studies [Ocio et al. 2010]. A phase Ib clinical trial evaluated the MTD of the triple combination. Oral panobinostat was administered at 5, 10, 20 and 25 mg thrice weekly for 3 weeks, lenalidomide was given by mouth (PO) at 25 mg daily on days 1–21 and dexamethasone was administered PO 40 mg daily on days 1–4, 9–12 and 17–20 of a 21-day cycle [Mateos et al. 2010]. The MTD of panobinostat was 20 mg in this combination. Out of 30 evaluable patients, 17 showed responses, including 1 stringent CR, 1 CR, 7 VGPRs and 8 PRs. A subsequent phase II study evaluated panobinostat administered at the previously determined MTD (20 mg) [Mateos et al. 2010] on a thrice weekly schedule but on only weeks 1 and 3 of a 21-day cycle, and lenalidomide administered at the same dose and schedule used in the phase Ib trial [Biran et al. 2013]. Dexamethasone was administered at 40 mg once a week and a reduced dose of dexamethasone (20 mg) was administered to older patients (≥75 years old). At the time of the report, only five lenalidomide-refractory patients were enrolled in the study. The regimen showed hematological toxicities and produced durable responses in 3 patients (1 VGPR, 1 PR and 1 minor response (MR), including lenalidomide-refractory patients [Biran et al. 2013].
Finally, our group has evaluated panobinostat in combination with the alkylating agent melphalan in RRMM patients. Based on preclinical results from our severe combined immune deficient human (SCID-hu) MM model [Sanchez et al. 2011], we evaluated the safety and efficacy of melphalan and panobinostat in a phase I/II trial [Berenson et al. 2014]. Due to tolerability issues, including grade 4 thrombocytopenia (n = 2), grade 3 fatigue (n = 1) and grade 4 neutropenia (n = 1), the drug dosing and schedule was changed three times during the trial, resulting in four different schedules. The MTD was established at 20 mg of panobinostat and only 0.05 mg/kg of oral melphalan, both administered only during the first week (on days 1, 3 and 5) of a 28-day cycle. Using this schedule, ≥ grade 3 neutropenia and thrombocytopenia were observed in 25 and 10% of patients, respectively. Both efficacy and toxicity appeared to have a direct correlation with the cumulative panobinostat exposure per cycle. Despite its tolerability, 20 mg of panobinostat and 0.05 mg/kg of melphalan, administered during the first week of each cycle produced no responses. Overall, responses were observed in only 3 (2 VGPRs, 1 PR) of the 45 patients evaluated for efficacy receiving panobinostat and melphalan, and this combination was associated with significant hematological and nonhematological toxicities including neutropenia (75%), thrombocytopenia (72.5%), anemia (52.5%), fatigue (58%) and nausea (55%) [Berenson et al. 2014]. Similar tolerability issues were observed when panobinostat was used in combination with melphalan, thalidomide and prednisone [Offidani et al. 2012]. In that phase II trial, oral melphalan was administered at 0.18 mg/kg on days 1–4, oral prednisone at 1.5 mg/kg on days 1–4, thalidomide at 50 mg/day continuously, and panobinostat at doses ranging from 10 to 20 mg three times a week for 3 weeks of each 28-day cycle. The ORR was 38.5%; however, the MTD of the drug combination could not be established due to the number of dose-limiting toxicities (grade 3 atrial fibrillation (n = 1), fatigue (n = 1), gastrointestinal toxicity (n = 2), and febrile neutropenia (n = 2) as well as grade 4 neutropenia (n = 10) and thrombocytopenia (n = 2)) observed in patients receiving 10 mg or 15 mg of panobinostat [Offidani et al. 2012]. Overall, the panobinostat–melphalan combination appears to be both too ineffective and toxic.
Other drug combinations are currently being tested for the treatment of MM. For instance, a phase I/II trial is evaluating panobinostat in combination with dexamethasone and the mammalian target of rapamycin (mTOR) inhibitor everolimus [ClinicalTrials.gov identifier: NCT00918333]. In addition, a phase I trial is assessing panobinostat in combination with the oral PI ixazomib and dexamethasone [ClinicalTrials.gov identifier: NCT02057640].
Side effects associated with panobinostat
The most common side effects observed after treatment with panobinostat include thrombocytopenia, neutropenia, anemia, diarrhea, and fatigue, which have been observed in all clinical studies and across a variety of diseases [Rasheed et al. 2008; Khot et al. 2013]. Platelet count nadir occurs during the second week of therapy and is self-limited.[Rasheed et al. 2008; DeAngelo et al. 2013]. Electrolyte and biochemical disturbances including hypokalemia and hypocalcemia have also been reported [Rasheed et al. 2008]. Cardiac effects include prolonged QT interval on day 3 of treatment and nonspecific ST-T electrocardiogram (ECG) changes have been reported in patients receiving intravenous panobinostat; however, the incidence of QT prolongation is substantially reduced among subjects receiving the oral formulation [Rasheed et al. 2008; Khot et al. 2013]. The toxicity profile of panobinostat shows similarities with that of the FDA-approved HDACis vorinostat and romidepsin. Common adverse events reported for vorinostat used as a single agent were fatigue, anorexia, dehydration, nausea and diarrhea, whereas QT interval prolongation, fatigue and hematological toxicities (thrombocytopenia, anemia and neutropenia) were observed with the vorinostat-bortezomib combination treatment [Orlowski, 2013]. Thrombocytopenia, nausea, fatigue and reversible QT prolongation were also observed with single agent romidepsin [Niesvizky et al. 2011]. Thrombocytopenia and fatigue were also common in MM patients treated with romidepsin, bortezomib and dexamethasone [Harrison et al. 2011].
Challenges and future directions
Despite advances in the development of new anti-MM agents during the past decade, MM remains an incurable disease. Therefore, there is a constant search for newer and better therapies. HDACs regulate a plethora of cellular functions and HDACis have shown potent anticancer activity in preclinical studies [Neri et al. 2012], and thus their use as multitarget therapeutic agents is appealing.
Studies demonstrating the dynamic interplay between protein acetylation status, cell cycle progression and apoptosis in MM cell lines have provided a strong rationale for the use of panobinostat as a therapeutic option for MM. Despite the promising preclinical data, the clinical responses achieved after treatment of MM patients with single-agent panobinostat have been disappointing. Panobinostat is a nonselective HDACi and its wide spectrum of inhibition is associated with significant toxicities including thrombocytopenia, fatigue and gastrointestinal symptoms [Rasheed et al. 2008; Khot et al. 2013], which can limit exposure. The use of suboptimal doses and schedules, such as those used in the melphalan–panobinostat trials [Offidani et al. 2012; Berenson et al. 2013], has helped minimize untoward side effects but, unfortunately, it has also compromised efficacy. Thus far, the panobinostat–bortezomib–dexamethasone triple combination appears to be the most effective, with predictable and manageable toxicities [Richardson et al. 2013; San-Miguel et al. 2013]. The eagerly awaited results from the PANORAMA 1 trial may shed some light on whether or not this particular panobinostat combination produces clinical benefit.
Results from ongoing clinical studies evaluating panobinostat in combination with other anti-MM agents such as lenalidomide, carfilzomib and ixazomib [Mateos et al. 2010; Biran et al. 2013] may demonstrate broader therapeutic windows than those observed with melphalan and bortezomib. However, it is likely that further trials will be required to fine tune the best dose and schedule for each particular drug combination. A better understanding of the molecular pathways targeted by panobinostat in MM cells may provide a better rationale for the selection of new drug combinations with synergistic potential.
The FDA approval of vorinostat and romidepsin has propelled the use of currently available HDACis and the development of new ones. However, a number of issues remain unresolved. For instance, there is a need for good response and prognostic biomarkers to both assess HDAC inhibition and to help physicians make informed decisions about the therapeutic value of HDACis for their patient population [Hajek et al. 2014; Treppendahl et al. 2014]. Current biomarkers for HDACi activity, such as histone acetylation and gene expression changes, show correlation between dose and histone hyperacetylation; however, they have no prognostic value and/or are tissue- and, likely, HDACi-specific [Prince et al. 2009; Treppendahl et al. 2014].
In the context of MM, the relative contribution of each HDAC to the disease is still unknown and elucidating it will allow the use of specific HDACis which, in turn, may improve tolerability. Newer, HDAC-specific or class-specific inhibitors are being developed [West and Johnstone, 2014], and these compounds may prove to be more effective and to have better toxicity profiles than panobinostat.
In conclusion, panobinostat is a potent new HDACi with a potential role for the treatment of MM. Current clinical data suggest that the panobinostat–bortezomib–dexamethasone combination is the most promising in the RRMM setting. Cumulative toxicity is still a main concern and it remains to be seen whether other panobinostat combinations are effective with acceptable tolerability profiles.
Footnotes
Conflict of interest statement: J.R.B. is a member of the speaker’s bureau for Norvatis and acknowledges consulting and research funds from Novartis.
Funding: This work was supported with funds from Novartis.
Contributor Information
Claudia V. Andreu-Vieyra, Oncotherapeutics, West Hollywood, CA, USA
James R. Berenson, Institute for Myeloma and Bone Cancer Research, 9201 W. Sunset Blvd., Suite 300, West Hollywood, CA 90069, USA Oncotherapeutics, West Hollywood, CA, USA.
References
- Aronson L., Davies F. (2012) DangER: protein ovERload. Targeting protein degradation to treat myeloma. Haematologica 97: 1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atadja P. (2009) Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett 280: 233–241. [DOI] [PubMed] [Google Scholar]
- Berdeja J., Mace J., Lamar R., Gian V., Murphy P., Patel M., et al. (2012) A single-arm, open-label, multicenter phase I/II study of the combination of panobinostat (pan) and carfilzomib (cfz) in patients (pts) with relapsed/refractory multiple myeloma (RR MM). J Clin Oncol 30: TPS8115. [Google Scholar]
- Berdeja J., Savona M., Mace J., Hart L., Essell J., Owera R., et al. (2013) A single-arm, open-label, multi-center phase I/II study of the combination of panobinostat and carfilzomib in patients (pts) with relapsed or relapse/refractory multiple myeloma (MM). Blood 122: 1937. [Google Scholar]
- Berenson J., Hilger J., Yellin O., Boccia R., Matous J., Dressler K., et al. (2014) A phase 1/2 study of oral panobinostat combined with melphalan for patients with relapsed or refractory multiple myeloma. Ann Hematol 93: 88–98. [DOI] [PubMed] [Google Scholar]
- Biran N., Shahnaz S., Jagannath S., Cho H., Osman K., Parekh S., et al. (2013) A phase II, single-center, open-label study of oral panobinostat in combination with lenalidomide and weekly dexamethasone in patients with multiple myeloma. Blood 122: 5392. [Google Scholar]
- Botbol V., Scornik O. (1991) Measurement of instant rates of protein degradation in the livers of intact mice by the accumulation of bestatin-induced peptides. J Biol Chem 266: 2151–2157. [PubMed] [Google Scholar]
- Brenner H., Gondos A., Pulte D. (2008) Recent major improvement in long-term survival of younger patients with multiple myeloma. Blood 111: 2521–2526. [DOI] [PubMed] [Google Scholar]
- Bruzzese F., Pucci B., Milone M., Ciardiello C., Franco R., Chianese M., et al. (2013) Panobinostat synergizes with zoledronic acid in prostate cancer and multiple myeloma models by increasing ROS and modulating mevalonate and p38-MAPK pathways. Cell Death Dis 4: e878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell R., Sanchez E., Steinberg J., Shalitin D., Li Z., Chen H., et al. (2010) Vorinostat enhances the antimyeloma effects of melphalan and bortezomib. Eur J Haematol 84: 201–211. [DOI] [PubMed] [Google Scholar]
- Cashen A., Juckett M., Jumonville A., Litzow M., Flynn P., Eckardt J., et al. (2012) Phase II study of the histone deacetylase inhibitor belinostat (PXD101) for the treatment of myelodysplastic syndrome (MDS). Ann Hematol 91: 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catley L., Weisberg E., Kiziltepe T., Tai Y., Hideshima T., Neri P., et al. (2006) Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 108: 3441–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catley L., Weisberg E., Tai Y., Atadja P., Remiszewski S., Hideshima T., et al. (2003) NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood 102: 2615–2622. [DOI] [PubMed] [Google Scholar]
- Cenci S. (2012) The proteasome in terminal plasma cell differentiation. Semin Hematol 49: 215–222. [DOI] [PubMed] [Google Scholar]
- DeAngelo D., Spencer A., Bhalla K., Prince H., Fischer T., Kindler T., et al. (2013) Phase Ia/II, two-arm, open-label, dose-escalation study of oral panobinostat administered via two dosing schedules in patients with advanced hematologic malignancies. Leukemia 27: 1628–1636. [DOI] [PubMed] [Google Scholar]
- Feng R., Oton A., Mapara M., Anderson G., Belani C., Lentzsch S. (2007) The histone deacetylase inhibitor, PXD101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br J Haematol 139: 385–397. [DOI] [PubMed] [Google Scholar]
- Finazzi G., Vannucchi A., Martinelli V., Ruggeri M., Nobile F., Specchia G., et al. (2013) A phase II study of Givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br J Haematol 161: 688–694. [DOI] [PubMed] [Google Scholar]
- Gao L., Cueto M., Asselbergs F., Atadja P. (2002) Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem 277: 25748–25755. [DOI] [PubMed] [Google Scholar]
- Giles F., Fischer T., Cortes J., Garcia-Manero G., Beck J., Ravandi F., et al. (2006) A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res 12: 4628–4635. [DOI] [PubMed] [Google Scholar]
- Hajek R., Siegel D., Orlowski R., Ludwig H., Palumbo A., Dimopoulos M. (2014) The role of HDAC inhibitors in patients with relapsed/refractory multiple myeloma. Leuk Lymphoma 55: 11–18. [DOI] [PubMed] [Google Scholar]
- Harrison S., Quach H., Link E., Seymour J., Ritchie D., Ruell S., et al. (2011) A high rate of durable responses with romidepsin, bortezomib, and dexamethasone in relapsed or refractory multiple myeloma. Blood 118: 6274–6283. [DOI] [PubMed] [Google Scholar]
- Hideshima T., Anderson K. (2012) Biologic impact of proteasome inhibition in multiple myeloma cells–from the aspects of preclinical studies. Semin Hematol 49: 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideshima T., Bradner J., Wong J., Chauhan D., Richardson P., Schreiber S., et al. (2005) Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A 102: 8567–8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowska E., Stoj J., Karpowicz P., Osmulski P., Gaczynska M. (2013) The proteasome in health and disease. Curr Pharm Des 19: 1010–1028. [PubMed] [Google Scholar]
- Kale A., Moore B. (2012) Molecular mechanisms of acquired proteasome inhibitor resistance. J Med Chem 55: 10317–10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalushkova A., Fryknäs M., Lemaire M., Fristedt C., Agarwal P., Eriksson M., et al. (2010) Polycomb target genes are silenced in multiple myeloma. PLoS One 5: e11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan O., La Thangue N. (2012) HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol 90: 85–94. [DOI] [PubMed] [Google Scholar]
- Khot A., Dickinson M., Prince H. (2013) Panobinostat in lymphoid and myeloid malignancies. Expert Opin Investig Drugs 22: 1211–1223. [DOI] [PubMed] [Google Scholar]
- Kikuchi J., Wada T., Shimizu R., Izumi T., Akutsu M., Mitsunaga K., et al. (2010) Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood 116: 406–417. [DOI] [PubMed] [Google Scholar]
- Kumar S., Rajkumar S., Dispenzieri A., Lacy M., Hayman S., Buadi F., et al. (2008) Improved survival in multiple myeloma and the impact of novel therapies. Blood 111: 2516–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavelle D., Chen Y., Hankewych M., DeSimone J. (2001) Histone deacetylase inhibitors increase p21(WAF1) and induce apoptosis of human myeloma cell lines independent of decreased IL-6 receptor expression. Am J Hematol 68: 170–178. [DOI] [PubMed] [Google Scholar]
- Lemaire M., Fristedt C., Agarwal P., Menu E., Van Valckenborgh E., De Bruyne E., et al. (2012) The HDAC inhibitor LBH589 enhances the antimyeloma effects of the IGF-1RTK inhibitor picropodophyllin. Clin Cancer Res 18: 2230–2239. [DOI] [PubMed] [Google Scholar]
- Maes K., Menu E., Van Valckenborgh E., Van Riet I., Vanderkerken K., De Bruyne E. (2013) Epigenetic modulating agents as a new therapeutic approach in multiple myeloma. Cancers 5: 430–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiso P., Carvajal-Vergara X., Ocio E., López-Pérez R., Mateo G., Gutiérrez N., et al. (2006) The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res 66: 5781–5789. [DOI] [PubMed] [Google Scholar]
- Mannava S., Zhuang D., Nair J., Bansal R., Wawrzyniak J., Zucker S., et al. (2012) KLF9 is a novel transcriptional regulator of bortezomib- and LBH589-induced apoptosis in multiple myeloma cells. Blood 119: 1450–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos M., Ocio E., San Miguel J. (2013) Novel generation of agents with proven clinical activity in multiple myeloma. Semin Oncol 40: 618–633. [DOI] [PubMed] [Google Scholar]
- Mateos M., Spencer A., Taylor K., Lonial S., De La, Rubia J., Facon T., et al. (2010) Phase Ib study of oral panobinostat (LBH589) plus lenalidomide (LEN) plus dexamethasone (DEX) in patients (Pts) with relapsed (Rel) or Rel and refractory (Ref) multiple myeloma (MM). J Clin Oncol 28: 8030. [Google Scholar]
- Matthews G., Lefebure M., Doyle M., Short J., Ellul J., Chesi M., et al. (2013) Preclinical screening of histone deacetylase inhibitors combined with ABT-737. rhTRAIL/MD5-1 or 5-azacytidine using syngeneic Vk*MYC multiple myeloma. Cell Death Dis 4: e798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithraprabhu S., Khong T., Jones S., Spencer A. (2013) Histone deacetylase (HDAC) inhibitors as single agents induce multiple myeloma cell death principally through the inhibition of class I HDAC. Br J Haematol 162: 559–562. [DOI] [PubMed] [Google Scholar]
- Moore H., Davenport E., Smith E., Muralikrishnan S., Dunlop A., Walker B., et al. (2009) Aminopeptidase inhibition as a targeted treatment strategy in myeloma. Mol Cancer Ther 8: 762–770. [DOI] [PubMed] [Google Scholar]
- Morgan G. (1999) Advances in the biology and treatment of myeloma. Br J Haematol 105: 4–6. [PubMed] [Google Scholar]
- Müller B., Jana L., Kasajima A., Lehmann A., Prinzler J., Budczies J., et al. (2013) Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer–overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 13: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri P., Bahlis N., Lonial S. (2012) Panobinostat for the treatment of multiple myeloma. Expert Opin Investig Drugs 21: 733–747. [DOI] [PubMed] [Google Scholar]
- New M., Olzscha H., La Thangue N. (2012) HDAC inhibitor-based therapies: can we interpret the code? Mol Oncol 6: 637–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niegisch G., Knievel J., Koch A., Hader C., Fischer U., Albers P., et al. (2013) Changes in histone deacetylase (HDAC) expression patterns and activity of HDAC inhibitors in urothelial cancers. Urol Oncol 31: 1770–1779. [DOI] [PubMed] [Google Scholar]
- Niesvizky R., Ely S., Mark T., Aggarwal S., Gabrilove J., Wright J., et al. (2011) Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer 117: 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocio E., Vilanova D., Atadja P., Maiso P., Crusoe E., Fernández-Lázaro D., et al. (2010) In vitro and in vivo rationale for the triple combination of panobinostat (LBH589) and dexamethasone with either bortezomib or lenalidomide in multiple myeloma. Haematologica 95: 794–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offidani M., Polloni C., Cavallo F., Liberati A., Ballanti S., Pulini S., et al. (2012) Phase II study of melphalan, thalidomide and prednisone combined with oral panobinostat in patients with relapsed/refractory multiple myeloma. Leuk Lymphoma 53: 1722–1727. [DOI] [PubMed] [Google Scholar]
- Orlowski R. (2013) Novel agents for multiple myeloma to overcome resistance in phase III clinical trials. Semin Oncol 40: 634–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei X., Dai Y., Grant S. (2004) Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res 10: 3839–3852. [DOI] [PubMed] [Google Scholar]
- Petta V., Gkiozos I., Strimpakos A., Syrigos K. (2013) Histones and lung cancer: are the histone deacetylases a promising therapeutic target? Cancer Chemother Pharmacol 72: 935–952. [DOI] [PubMed] [Google Scholar]
- Prince H., Bishton M., Harrison S. (2009) Clinical studies of histone deacetylase inhibitors. Clin Cancer Res 15: 3958–3969. [DOI] [PubMed] [Google Scholar]
- Pulte D., Redaniel M., Brenner H., Jansen L., Jeffreys M. (2014) Recent improvement in survival of patients with multiple myeloma: variation by ethnicity. Leuk Lymphoma 55: 1083–1089. [DOI] [PubMed] [Google Scholar]
- Quintás-Cardama A., Kantarjian H., Estrov Z., Borthakur G., Cortes J., Verstovsek S. (2012) Therapy with the histone deacetylase inhibitor pracinostat for patients with myelofibrosis. Leuk Res 36: 1124–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasheed W., Bishton M., Johnstone R., Prince H. (2008) Histone deacetylase inhibitors in lymphoma and solid malignancies. Expert Rev Anticancer Ther 8: 413–432. [DOI] [PubMed] [Google Scholar]
- Richardson P., Alsina M., Weber D., Coutre S., Lonial S., Gasparetto C., et al. (2012) PANORAMA 2: panobinostat combined with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory multiple myeloma. Blood 120: 1852. [DOI] [PubMed] [Google Scholar]
- Richardson P., Schlossman R., Alsina M., Weber D., Coutre S., Gasparetto C., et al. (2013) PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 122: 2331–2337. [DOI] [PubMed] [Google Scholar]
- Ropero S., Esteller M. (2007) The role of histone deacetylases (HDACs) in human cancer. Mol Oncol 1: 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez E., Shen J., Steinberg J., Li M., Wang C., Bonavida B., et al. (2011) The histone deacetylase inhibitor LBH589 enhances the anti-myeloma effects of chemotherapy in vitro and in vivo. Leuk Res 35: 373–379. [DOI] [PubMed] [Google Scholar]
- San-Miguel J., Moreau P., Yoon S., Dimopoulos M., de Moraes Hungria V., Jedrzejczak W., et al. (2012) Phase III study of panobinostat with bortezomib and dexamethasone in patients with relapsed multiple myeloma (PANORAMA 1). J Clin Oncol 30: e18572. [Google Scholar]
- San-Miguel J., Richardson P., Günther A., Sezer O., Siegel D., Bladé J., et al. (2013) Phase Ib study of panobinostat and bortezomib in relapsed or relapsed and refractory multiple myeloma. J Clin Oncol 31: 3696–3703. [DOI] [PubMed] [Google Scholar]
- Santo L., Hideshima T., Kung A.L., Tseng J., Tamang D., Yang M., et al. (2012) Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 119: 2579–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlossman R., Alsina M., Weber D., Coutre S., Gasparetto C., Mukhopadhyay S., et al. (2013) Time to event analyses in PANORAMA 2: a phase 2 study of panobinostat, bortezomib, and dexamethasone in patients with relapsed and bortezomib-refractory multiple myeloma. Blood 122: 1970. [DOI] [PubMed] [Google Scholar]
- Shah J., Thomas S., Weber D., Wang M., Alexanian R., Qazilbash M., et al. (2012) Phase 1/1b study of the efficacy and safety of the combination of panobinostat + carfilzomib in patients with relapsed and/or refractory multiple myeloma. Blood 120: 4081. [Google Scholar]
- Sharma S., Beck J., Mita M., Paul S., Woo M., Squier M., et al. (2013) A phase I dose-escalation study of intravenous panobinostat in patients with lymphoma and solid tumors. Invest New Drugs 31: 974–985. [DOI] [PubMed] [Google Scholar]
- Siegel R., Ma J., Zou Z., Jemal A. (2014) Cancer statistics, 2014. CA Cancer J Clin 64: 9–29. [DOI] [PubMed] [Google Scholar]
- Simms-Waldrip T., Rodriguez-Gonzalez A., Lin T., Ikeda A., Fu C., Sakamoto K. (2008) The aggresome pathway as a target for therapy in hematologic malignancies. Mol Genet Metab 94: 283–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M., Newland A. (1999) Treatment of myeloma. QJM 92: 11–14. [DOI] [PubMed] [Google Scholar]
- Stenzinger A., Endris V., Klauschen F., Sinn B., Lorenz K., Warth A., et al. (2013) High SIRT1 expression is a negative prognosticator in pancreatic ductal adenocarcinoma. BMC Cancer 13: 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treppendahl M., Kristensen L., Grønbæk K. (2014) Predicting response to epigenetic therapy. J Clin Invest 124: 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme M., Crompot E., Meuleman N., Mineur P., Bron D., Lagneaux L., et al. (2012) HDAC isoenzyme expression is deregulated in chronic lymphocytic leukemia B-cells and has a complex prognostic significance. Epigenetics 7: 1403–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ververis K., Hiong A., Karagiannis T., Licciardi P. (2013) Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics 7: 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A., Johnstone R. (2014) New and emerging HDAC inhibitors for cancer treatment. J Clin Invest 124: 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf J., Siegel D., Goldschmidt H., Hazell K., Bourquelot P., Bengoudifa B., et al. (2012) Phase II trial of the pan-deacetylase inhibitor panobinostat as a single agent in advanced relapsed/refractory multiple myeloma. Leuk Lymphoma 53: 1820–1823. [DOI] [PubMed] [Google Scholar]