

Abstract
Mechanical stretch of cardiac muscle modulates action potential propagation velocity, causing potentially arrhythmogenic conduction slowing. The mechanisms by which stretch alters cardiac conduction remain unknown, but previous studies suggest that stretch can affect the conformation of caveolae in myocytes and other cell types. We tested the hypothesis that slowing of action potential conduction due to cardiac myocyte stretch is dependent on caveolae.
Cardiac action potential propagation velocities, measured by optical mapping in isolated mouse hearts and in micropatterned mouse cardiomyocyte cultures, decreased reversibly with volume loading or stretch, respectively (by 19±5% and 26±4%). Stretch-dependent conduction slowing was not altered by stretch-activated channel blockade with gadolinium or by GsMTx-4 peptide, but was inhibited when caveolae were disrupted via genetic deletion of caveolin-3 (Cav3 KO) or membrane cholesterol depletion by methyl-β-cyclodextrin. In wild-type mouse hearts, stretch coincided with recruitment of caveolae to the sarcolemma, as observed by electron microscopy. In myocytes from wild-type but not Cav3 KO mice, stretch significantly increased cell membrane capacitance (by 98±64%), electrical time constant (by 285±149%), and lipid recruitment to the bilayer (by 84±39%).
Recruitment of caveolae to the sarcolemma during physiologic cardiomyocyte stretch slows ventricular action potential propagation by increasing cell membrane capacitance.
Keywords: cardiac mechanoelectric feedback, caveolae, capacitance
1. INTRODUCTION
It is well recognized that mechanical loading can affect cardiac myocyte electrophysiology, but the cellular mechanisms of cardiac mechanoelectric feedback remain poorly understood.1 One of the least investigated consequences of brief mechanical loading on cardiac electrophysiology is the effect of stretch on action potential propagation velocity. Although there are conflicting reports, most recent studies suggest that increased stretch of intact cardiac muscle slows conduction velocity,2,3 which may be proarrhythmic under pathologic conditions such as heart failure that result in increased diastolic wall stretch. The two main mechanisms of stretch-dependent conduction slowing that have been suggested are: stretch-activated ion currents (SACs),4 which may reduce excitability by elevating resting membrane potential; and increased sarcolemmal capacitance3 that may be caused by addition of cell surface area5 during mechanical loading. Mills et al3 observed that non-specific SAC blockade by gadolinium did not prevent conduction slowing during ventricular filling in isolated perfused rabbit hearts. They found that ventricular loading greatly increased the tissue time constant, suggesting that an increase in myocyte membrane capacitance could explain observed reductions in conduction velocity due to ventricular filling.
Using electron microscopy to compare myocyte membrane morphologies in rabbit hearts fixed with and without passive ventricular filling, Kohl and colleagues5 observed myocyte membrane unfolding and incorporation of sub-sarcolemmal caveolae into the plasma membrane in loaded hearts. Recent data in a variety of other cell types suggest that caveolae may act as membrane mechanosensors. Sinha et al.6 showed that uniaxial stretch or osmotic swelling in endothelial, muscle, and caveolin-1-expressing HeLa cells caused a reversible loss of caveolae at the cell surface membrane within minutes. They proposed that stretch-induced recruitment and flattening of caveolae buffers surges in membrane tension during cell stretch. Opening or flattening of caveolae above a critical membrane tension is also consistent with the results of computational models7,8 of membrane bending mechanics. We therefore sought to test the hypothesis that caveolae are required for stretch-dependent conduction slowing in the mouse heart.
We used caveolin-3 knockout (Cav3 KO) mice that lack caveolae in myocytes and pharmacological depletion of membrane cholesterol with methyl-β-cyclodextrin (MβCD) to disrupt caveolar structure in whole mouse hearts and isolated neonatal mouse ventricular myocytes (NMVMs). We found that Cav3 is required for stretch-dependent conduction slowing both in whole hearts and isolated myocytes, and showed that recruitment of caveolae to the sarcolemma by stretch reversibly increases lipid recruitment to the bilayer and cell membrane capacitance sufficiently to explain the observed conduction slowing.
2. METHODS
Detailed methods are included in the online supplement.
2.1 Animal Models for Tissue Studies
All animal experiments were performed in accordance with an animal use protocol approved by the University of California, San Diego Institutional Animal Care and Use Committee. Eight- to ten- week old Cav3 KO9,10 and age-matched C57BL/6 mice were used for electron microscopy and whole heart optical mapping studies.
2.2 Pressure-Loaded Isolated Heart Studies
Hearts were isolated and Langendorff perfused as described previously.2 Mice were administered an intraperitoneal injection of 100 units of heparin, briefly anesthetized with isoflurane, and sacrificed by cervical dislocation. The heart was excised and placed in ice cold, hyperkalemic arrest solution. The aorta was rapidly cannulated on a 20 gauge stainless steel cannula. A fluid-filled length of polyethylene tubing fitted onto a 20 gauge luer adapter was inserted into the left ventricular (LV) cavity through the pulmonary vein and mitral valve and a tie was secured around the opening of the pulmonary vein. The heart was transferred to a Langendorff perfusion apparatus. The heart was perfused with a heated (35–37°C), oxygenated (95% O2, 5% CO2) modified Krebs-Henseleit solution (24.9 mM NaHCO3, 1.2 mM KH2PO4, 11.1 mM dextrose, 1.2 mM MgSO4, 4.7 mM KCl, 118 mM NaCl, and 2.5 mM CaCl2) at a constant pressure of 65–70 mmHg. Gd3 experiments were done using a HEPES buffered perfusate rather than Krebs-Henseleit to prevent precipitation. Coronary flow rate was monitored with an inline flow probe (Transonic Systems, Inc.) and was maintained at 1–2 mL/min in the unloaded and loaded states.
The tube in the LV cavity was connected to a reservoir of warmed, oxygenated perfusate, and an in-line pressure transducer was used to monitor LV pressure. The LV filling pressure (LVP) was controlled by the height of the reservoir with respect to the heart.
2.3 Isolated Heart Optical Mapping
Langendorff perfused hearts were optically mapped to investigate the effects of ventricular pressure on action potential propagation. Once the heart was cannulated, it was submerged in a bath of warmed perfusate in a water-jacketed chamber with a window for optical access. Electrodes in the bath chamber provided a volume-conducted electrocardiogram that was monitored and recorded throughout the experiment. The heart was allowed to beat freely for a 15-minute equilibration period. Myocardium was stained with a bolus injection of the potentiometric fluorophore di-4-ANEPPS (8 ml, 3.25 μM) into the perfusion line. The LV epicardium was excited with a light-emitting diode array (LEDtronics, Torrance, CA) at 470 nm. Fluorescence emission was collected by a custom-built tandem lens optical system (two 1× Plan APO macro objectives, Leica, Solms, Germany) filtered with >610 nm long-pass filter and focused onto a high-speed 14-bit CMOS camera (MiCAM Ultima L, SciMedia). Fluorescence images were acquired at 1000 frames per second and a spatial resolution of 100×100 pixels at 0.1 mm × 0.1 mm per pixel. The heart was paced at the lateral left ventricular midwall with a platinum unipolar electrode coated with Teflon except at the tip. A 2-ms rectangular constant current stimulus was applied at two times threshold and a cycle length of 200 ms, delivered by a digital stimulator (DS8000 stimulator and DLS100 isolator, World Precision Instruments, Sarasota, FL). Data were acquired in the unloaded state before loading with an LVP of 0 mmHg to measure baseline conduction velocity. The ventricular pressure was increased to 30 mmHg via the tube in the LV cavity for 1 minute and data was acquired at this loaded state. Finally, the LVP was returned to 0 mmHg for 1 minute, and data was acquired in this unloaded state after loading. No chemical or mechanical intervention was implemented to restrict motion. Images were imported into MATLAB and analyzed using custom software as described previously.2,3 Activation time was identified at each pixel at the maximum rate of change of fluorescence for each beat. Conduction velocity vector fields were calculated from the spatial gradients of activation time over the surface of the ventricle.11 Conduction velocity magnitudes were calculated from these vector fields and an apparent maximum and minimum conduction velocity (CVmax, CVmin) were defined as the 95th and 5th percentile values in the regions of fastest and slowest conduction respectively.
2.4 Electron Microscopy
Mouse hearts were prepared for electron microscopy to investigate the effects of increased load on membrane morphology and caveolae. Hearts were fixed either at unstretched baseline, i.e. after cannulation and perfusion but without additional ventricular pressure loading; during loading to a LV pressure of 30 mmHg; or following loading to 30 mmHg for 1 min and subsequent unloading to 0 mmHg for 1 min. One minute after the final load change, the perfusate was switched to a standard Karnovsky’s fixative of 4% paraformaldehyde, 1.5% gluteraldehyde in 0.1 M cacodylate buffer while hanging until the contractions stopped. Fixed specimens were dehydrated with ethanol and embedded in LX112 (Ladd, Willston, VT #21210) with a longitudinal orientation. 70 nm sections were counterstained with lead citrate and uranyl acetate, and examined on JEOL CX100 TEM scope. 10 micrographs at 4800× magnification and 10 at 6800× magnification were taken per heart at random from sections for quantization of plasma membrane effects. Measurements were made manually for each micrograph with ImageJ (NIH open source software). Caveolae were counted in each of the 6800× magnification images and identified as sub-sarcolemmal (no visible connection to the sarcolemma in the image plane) or visibly integrated into the sarcolemma (flask-like connection to the sarcolemma observed). Total density of caveolae and their localization were quantified, with respect to the membrane length as illustrated in Supplemental Figure S2. Additionally, the ratio of membrane length to absolute length was calculated in the 4800× magnification images to quantify slack membrane.
2.5 Stretch of Micropatterned Neonatal Murine Ventricular Myocyte (NMVM) Cultures
Polydimethylsiloxane (PDMS) tissue culture substrates were molded from silicon wafers that had been patterned with microgrooves by photolithograpy, using SU-8 2005 negative photoresist (MicroChem Corp., Newton, MA) and a custom designed photomask (Advance Reproductions Corp., North Andover, MA), as described previously.12,13 The microgrooves were 10 μm wide, 10 μm apart and 5 μm deep in the silicone elastomer (Sylgard 186, Dow Corning Corp., Midland, MI). Murine laminin (Sigma-Aldrich, St. Louis, MO) was adsorbed onto the PDMS using a ten-minute treatment with ultraviolet radiation (350 nm wavelength). The substrate was rinsed twice with 1× phosphate-buffered saline (MediaTech, Manassas, VA) prior to plating cells.
The PDMS substrates were mounted onto custom elliptical stretch devices described previously.12,14 The stretchers applied homogeneous anisotropic biaxial stretch to the patterned PDMS elastomers, which were mounted in the stretch devices such that the principal axis of maximum stretch was directed parallel to the longitudinal axis of the microgrooves.
Neonatal murine ventricular myocytes (NMVMs) were isolated from P1–P2 CD-1 mouse pups (Charles River Labs) using methods adapted from standard protocols.15 Hearts were excised and enzymatically digested. Fibroblasts were removed from the cell suspension using a 90-minute pre-plating incubation and discarded. Cardiomyocytes were plated in the stretch device culture chambers in 15% serum plating media, and maintained at 37°C and 5% CO2 for 2–4 days with media changes every 1–3 days. Cultures were maintained in standard 6% serum media until 24 hours prior to optical mapping experiments, when the media was changed to antibiotic-free media. Cultured NMVMs were confluent, rod-shaped with parallel striations and aspect ratios and coupled via gap junctions (Fig. S1).
Following baseline recordings before stretch, biaxial stretch of 14% stretch parallel to the myocyte longitudinal axis and 3.6% perpendicular to it was applied for 5 minutes prior to stretched measurements, and subsequently reversed with 5 minutes of rest prior to unstretched measurements after stretch.
2.6 Optical Mapping of Micropatterned Monolayers
The stretch devices were mounted into an optical assembly for voltage mapping experiments. This method utilizes high temporal and spatial resolution imaging of a transmembrane voltage-sensitive fluorescent dye and avoids the electrical disturbances of electrode measurements.16,17 The device is configured such that the culture substrate is maintained in the same position relative to the camera during loading and unloading. The imaging chamber was maintained at 37°C and cells were electrically stimulated at 300 ms intervals for the duration of the experiment.
Changes in voltage were detected using voltage-sensitive transmembrane fluorescent probe di-8-ANEPPS (Life Technologies, Grand Island, NY), loaded at 30 μM in a solution of antibiotic-free media and 0.1% Pluronic F-127 (Life Technologies, Grand Island, NY). The dye solution was replaced with serum- and antibiotic-free imaging media prior to imaging. Antioxidants were added to the dye and imaging solutions at physiological levels to reduce generation of reactive oxygen species by the fluorescent probe.18 100 μM ascorbic acid (Fisher Scientific, Waltham, MA) and 450 μM uric acid (Sigma-Aldrich, St. Louis, MO) were added to each solution.
A 470-nm wavelength LED was used to excite the di-8-ANEPPS for electrophysiological measurement. Excitation light was directed to the cell culture substrate with a dichroic mirror, and emitted light passed through a 610 nm long-pass filter and was detected at 500 frames per second by a MiCAM Ultima-L CMOS camera. Depolarization was assessed according to time of maximum rate of fluorescence change at each pixel. The velocity of propagation of 5–6 consecutive, stimulated, and smoothly propagating action potentials per condition was measured along the longitudinal and transverse axes of the cell culture. Conduction along the same region relative to the pace site was compared for baseline, stretched and unstretched measurements.
2.7 Myocyte Patch Clamp
Individual NMVMs were patch-clamped on stretch-devices that were sparsely seeded so as to prevent cell coupling within 3 days of isolation. The measurements between stretched and unstretched cells were unpaired. Measurements were recorded within 60–90 minutes from loading condition change. Borosilicate pipettes were 2–7 MΩ when filled with (in mM) 120 K-aspartate, 10 KCl, 10 NaCl, 5 Mg-ATP, 1 MgCl2, and 0.3 Li-GTP (pH 7.2, KOH), and immersed in the bath solution (mM: 140 NaCl, 4 KCl, 1 CaCl2, 1 MgCl2, 10 D-glucose, and 10 HEPES; pH: 7.4, NaOH). An Axopatch 200B amplifier was used to apply 7 voltage steps of 200 ms between −120 and −60 mV (at 10 mV increments) from each of 5 holding potentials between −100 and −60 mV (again in 10 mV increments). For all steps, cell membrane capacitance (Cm) was calculated by standard approximation: Cm = (Q + i∞•τ)/ΔV, where Q is the integral of the measured current above the steady state value (i∞), τ is the time constant of current decay, and ΔV is the size of the voltage step. This approximation19 accounts for the exponential resistive component of the current response. Importantly, reported capacitances were taken from transients during −60 to −70 mV voltage steps (i.e. from the negative going phases of the −70 mV step at −60mV holding potential, and the −60 mV step from −70 mV holding potential). This was done to avoid contamination by INa, which was induced by the depolarizing phase of the steps in many NMVMs, and to further minimize error in the capacitive estimate due to IK1-mediated changes in membrane resistance (Rm), which are exaggerated at more negative potentials.20 Reported Rm values were taken from the negative-going steps between −80 and −90 mV, which is similar to the range of resting or maximum diastolic potential in these preparations.21 This range of potentials also allowed more reliable Rm measurement, because at more positive potentials i∞ was small enough (high Rm) to limit resolution of the signal (7 pA near resting potential versus 3 pA at more positive potentials). Cell membrane electrical time constants (Tm) were calculated from the product of Cm and Rm.
2.8 Lipophilic Dye
NMVMs were plated on micropatterned substrates for stretch, and incubated with 5 μM Vybrant DiO lipophilic dye (Molecular Probes, Life Technologies, Grand Island, NY) for 3 days from plating. Fluorescence was excited using a Lambda DG-4 fluorescent light source and captured with Photometrics Cascade 512F camera using MetaMorph v6.1 acquisition software. Fluorescent intensity per unit cell area was measured in the same cells before and immediately after 14% longitudinal by 3.6% transverse stretch. Each observation consisted of 6–20 cell measurements.
2.9 Statistics
Calculations were performed in MATLAB, Excel, and SigmaPlot. Measurements are reported as mean ± SEM. 1- and 2-way analyses of variance were conducted as regular or repeated measures where appropriate. If the data did not satisfy a Kolmogorov-Smirnov test of normality or a Levene Median test for equal variance, a generalized linear model nonparametric test was used. All analyses of variance were followed by Tukey pairwise multiple comparison procedures. Values of P<0.05 were accepted as statistically significant.
3. RESULTS
Extended data are presented in the Online Supplement (Table S1 and S2).
3.1 Mouse Ventricular Action Potential Propagation is Slowed by Increased Ventricular Filling Pressure Independently of SAC Blockade
Epicardial activation isochrones (Fig. 1A) showed that fastest conduction (CVmax) in the epicardially-paced wild-type (WT) mouse LV was approximately parallel to the epicardial fiber direction, with slowest conduction (CVmin) along the perpendicular axis. At zero left ventricular filling pressure (LVP), mean CVmax was 1.8- to 2.7-fold faster (653±15 mm.s−1) than mean CVmin (311±26 mm.s−1) as seen in Fig. 1A. The mean anisotropy ratio of conduction (CVmax/CVmin) did not vary significantly from before loading (2.2±0.2) to loaded (2.1±0.2, P=N.S.), as such, maximum epicardial conduction velocity (CVmax) is described in the remaining results except where stated otherwise. When LVP was increased from zero to 30 mmHg in the isolated heart, epicardial conduction slowed significantly within one minute by 19±5% (P<0.01) (Fig. 1B). After LVP was returned to 0 mmHg, conduction velocity returned to 104±2% of baseline (P=N.S. vs. baseline, P<0.01 vs. loaded). Conduction velocities at each loading condition are plotted per heart in Fig. 1C.
Figure 1.
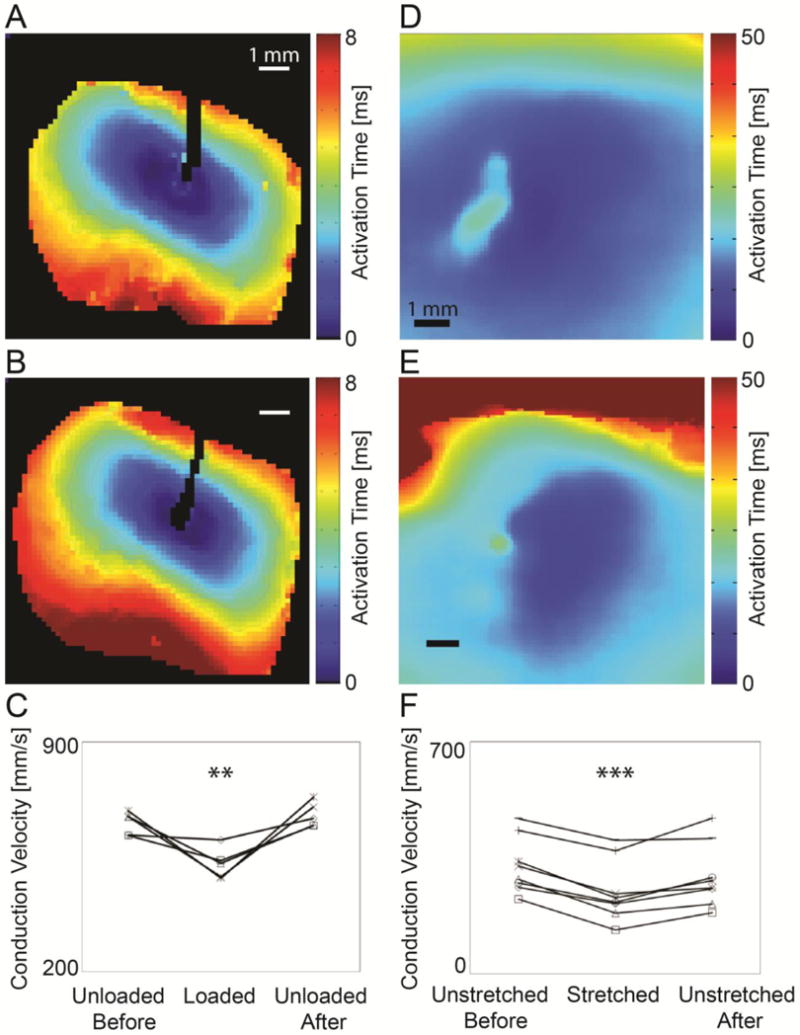
Effects of stretch on conduction velocity measured by fluorescence optical mapping in isolated hearts (A–C) and in micropatterned cardiomyocytes (D–F). Isochronal maps from the epicardium of an isolated WT mouse heart before (A) and after (B) ventricular pressure loading and from micropatterned neonatal ventricular myocyte cultures before (D) and after (E) anisotropic stretch show prolongation of activation times indicating conduction slowing during mechanical loading (scale 1 mm). Maximal epicardial conduction velocity (CVmax) in isolated mouse hearts (C) decreased significantly and reversibly during loading (N=5, **P<0.01). Maximal conduction velocity (CVmax) in cultured myocytes (F) decreased significantly and reversibly with stretch (N=8, ***P<0.001).
Conduction in unloaded hearts perfused with the non-selective stretch activated channel (SAC) blocker gadolinium (Gd3+, 50 μM) was, at 458±45 mm.s−1, 30% slower on average than in untreated hearts (N=5, P<0.01 vs. untreated). However, SAC blockade with Gd3+ did not attenuate conduction slowing (to 340±55 mm.s−1) due to LV pressure loading (P<0.01) (Fig. 2).
Figure 2.
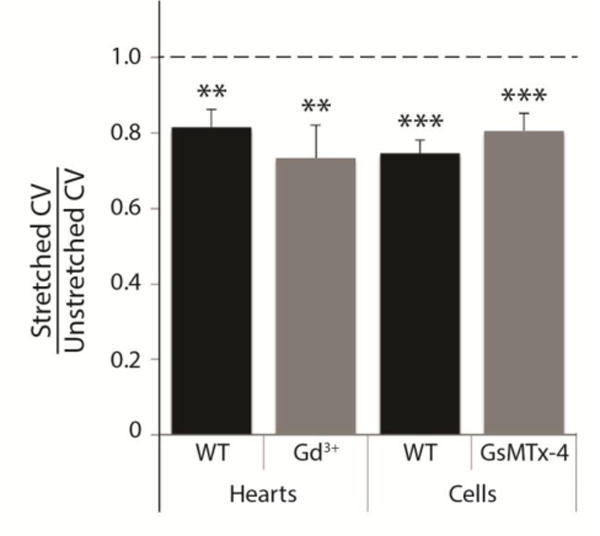
Conduction slowing with stretch is not affected by stretch-activated channel blockade in isolated hearts or myocytes. Mean conduction velocity in mechanically loaded hearts and cells was 70–80% of unstretched values, and significantly different from the original value (1.0), in the following four conditions: untreated hearts (N=5, P<0.01), hearts with stretch-activated channel blockade by Gd3+ (N=5, P<0.01), untreated cells (N=8, P<0.001), and cells with stretch-activated channel blockade by peptide GsMTx-4 (N=3, P<0.001 cells). There was no significant difference in the CV ratio between control (WT) groups and treated groups for hearts or cells.
3.2 Conduction Slows Reversibly and Independently of SAC Blockade with Brief Stretch in Micropatterned Neonatal Mouse Ventricular Myocyte Monolayers
Velocity of action potential conduction averaged from 5–6 consecutive paced action potentials in unstretched WT cultures was 1.2- to 7.2-fold (mean ratio 3.2±0.7) faster along the longitudinal axis of patterned myocytes (325±30 mm.s−1) than in the transverse direction (124±22 mm.s−1) (Fig. 1D). The mean anisotropy ratio of conduction did not vary significantly from before stretch (3.2±0.7) to stretched (2.9±0.6, P=N.S.). Maximal (longitudinal) CV is described in remaining results except where noted. As seen in Fig. 1E, conduction velocities in micropatterned neonatal murine cardiomyocytes cultures slowed during stretch (26±4%, P<0.001). Five minutes after reversal of stretch, CVmax recovered to 90±5% of initial values (P<0.05 vs. baseline, P<0.01 from stretched) (Fig. 1F).
30-minute pre-incubation with the specific SAC blocking peptide GsMTx-4 (3 μM, kindly supplied by Dr. Frederick Sachs, SUNY Buffalo, NY) did not significantly alter the effects of stretch on CV, as seen in Figure 2. Though GxMTx-4 increased CV in unstretched cultures (1017±343 mm.s−1, N=3, P<0.01 vs. untreated), stretch still significantly decreased CV in GsMTx-4-treated cultures (P<0.001).
3.3 Caveolae are Required for Stretch-Induced Conduction Slowing in the Isolated Heart and Micropatterned Myocyte Cultures
Depletion of caveolae attenuated conduction slowing during LV pressure loading. WT hearts perfused for 15 minutes with methyl-β-cyclodextrin (MβCD, 1 mM) (Sigma-Aldrich, St. Louis, MO), which depletes caveolae by sequestering cholesterol,22 did not have significantly altered unloaded CV (692±16 m.s−1) compared with untreated hearts, as shown in Figure 3A, (N=5, P=N.S.). Conduction velocities observed in Cav3 KO hearts in the unloaded state (591±21 mm.s−1) were not significantly different from those in unloaded WT hearts (N=5, P=N.S.) (Fig. 3A). There was no significant change in CV with load in Cav3 KO or MβCD-treated hearts (P=N.S.).
Figure 3.
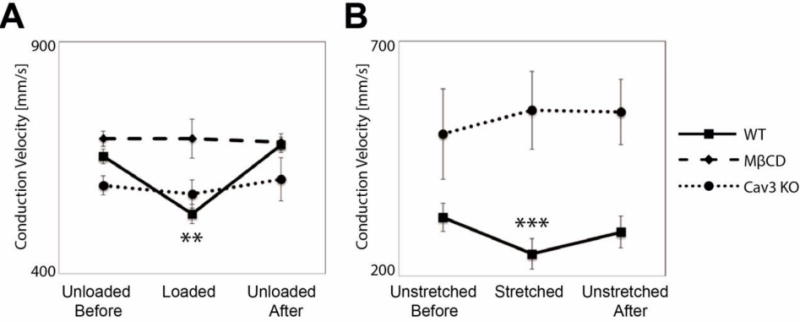
Stretch-dependent conduction slowing requires caveolae in isolated mouse hearts (A) and cultured myoctes (B). (A) Conduction slowing observed with loading in WT hearts is not seen when caveolae are depleted via either treatment with MβCD or genetic deletion of caveolin-3 (**WT hearts N=5, P<0.01; MβCD-treated and Cav3 KO hearts N=5, P=N.S.). (B) Conduction slowing observed in WT cells is prevented when caveolae are depleted via genetic deletion of caveolin-3 (***WT cells N=8 P<0.001; Cav3 KO cells N=3, P=N.S.). Within paired observations, only untreated WT hearts and myocytes show statistically significant changes with stretch.
In micropatterned cultures of ventricular cardiomyocytes isolated from neonatal Cav3 KO mice, CV in unstretched conditions was not significantly different (503±96 mm.s−1) than in WT cells (P=N.S., N=3), but there was no significant change in CV during stretch of Cav3 KO mouse myocyte cultures (553±83 mm.s−1, P=N.S.) (Fig. 3B) indicating that Cav3 is required for stretch-dependent conduction slowing in vitro.
Hence stretch-dependent conduction slowing was absent both in hearts and cardiomyocyte monolayers from Cav3 KO mice, and in WT hearts treated with MβCD.
3.4 Stretch In Vitro Significantly Increases Whole Cell Capacitance, Independent of SACs
Patch clamp studies in micropatterned WT NMVMs showed that membrane capacitance (Cm) increased dramatically and significantly with stretch from 22±3 pF to 43±8 pF (N=30, P<0.001), as seen in Figure 4A. SAC blockade using 3 μM GsMTx-4 for 30 minutes did not alter this effect (16±2 pF in unstretched cells vs. 40±4 pF in stretched cultures, N=19, P<0.01.).
Figure 4.
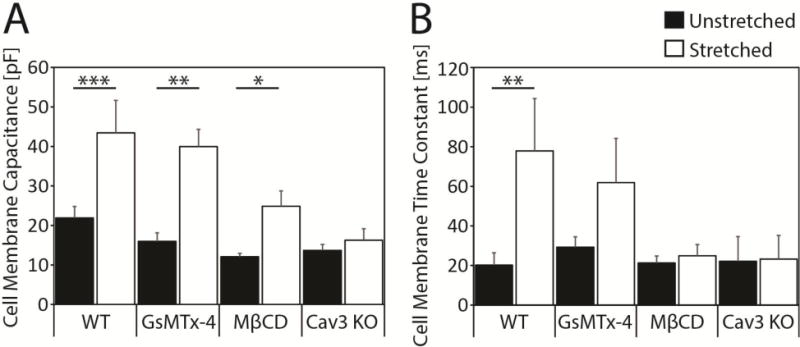
Cell membrane capacitance increases with stretch in a caveolae-dependent manner, resulting in a stretch-dependent and caveolae-dependent increase in membrane time constant. (A) Patch clamp recordings of sparsely-plated, micropatterned NMVMs significantly increased membrane capacitance with stretch in WT (N=15,15, ***P<0.001) and GsMTx-4-treated (N=11,8, **P<0.01) myocytes. There was a 41% smaller increase in MβCD-treated (N=18,22, *P<0.05) myocytes, and no significant change with stretch in Cav3 KO myocytes (N=13,12, P=N.S.). (B) There was a significant stretch-dependent increase in membrane time constant in untreated WT myocytes (**P<0.01), a doubling of time constant in GsMTx-4 treated WT myocytes (P=N.S.), and no significant change in MβCD-treated and Cav3 KO myocytes (P=N.S.).
3.5 Caveolae are Required for Stretch-Dependent Increase in Membrane Capacitance and Time Constant
Cell membrane capacitance was not significantly altered in unstretched cells treated with 1 mM MβCD for 30 minutes (12±1 pF, N=18, P=N.S. vs. WT), but the effect of stretch on Cm was significantly lower in treated than untreated cells (25±4 pF, N=22, P<0.01 vs. WT). MβCD treatment inhibited but did not fully block the increase in membrane capacitance during stretch (13±4 pF, P<0.05). However, genetic ablation of caveolae completely inhibited the effect of stretch on Cm. The mean increase in Cm with stretch in myocytes from Cav3 KO mice was only 3±4 pF (P=N.S., N=25) from 14±2 pF (N=13, P=N.S. vs. WT) (Fig. 4A).
There were no significant changes with stretch, treatment or between genotypes in membrane resistance (Table S2). However, membrane electrical time constants shown in Figure 4B increased almost four-fold in WT cells during stretch (from 20±6 ms to 78±26 ms, P<0.01). GsMTx-4 treatment did not significantly alter time constants in unstretched (29±5 ms, P=N.S.) or stretched (62±22 ms, P=N.S.) myocytes from WT mice. However, in MβCD-treated and Cav3 KO myocytes, the effect of stretch on cell membrane electrical time constant was eliminated but time constants in the unstretched MβCD-treated and Cav3 KO myocytes were not significantly different from WT (21±4 ms and 22±12 ms, respectively, P=N.S.), Therefore, caveolae depletion inhibited the stretch-induced increase in cell membrane electrical time constant, but SAC-blockade had no effect.
3.6 LV Pressure Loading Reduces Subsarcolemmal Caveolae
Electron microscopy revealed significant changes in the density and localization of caveolae with increased ventricular pressure load, and by interventions (MβCD-perfusion, Cav3 KO) intended to deplete caveolae (Fig. 5A). In WT, total caveolae density (Fig. 5B) was significantly lower in the pressure loaded heart (0.78±0.11 caveolae/μm, N=10) compared to the unloaded heart (1.23±0.14, N=10, P<0.05). Initial caveolae density was restored in the heart fixed after load had been applied then removed, for 1 minute per load condition change (1.27±0.10, N=10, P<0.05 vs. loaded, P=N.S. vs. unloaded). The density of caveolar structures in the unloaded MβCD-perfused and Cav3 KO hearts were 0.42±0.4 and 0.12±0.02 caveolae/μm respectively, as shown in Fig. 5C, significantly fewer than in the unloaded WT heart (1.23±0.14 caveolae/μm, N=10, P<0.001 for both). The ratio of membrane length to absolute length, a quantification of slack membrane folds, also decreased in the loaded state (Supplement Figure S2). Density of caveolae is quantified with regard to membrane length, measured manually in ImageJ as illustrated in Fig. S2.
Figure 5.
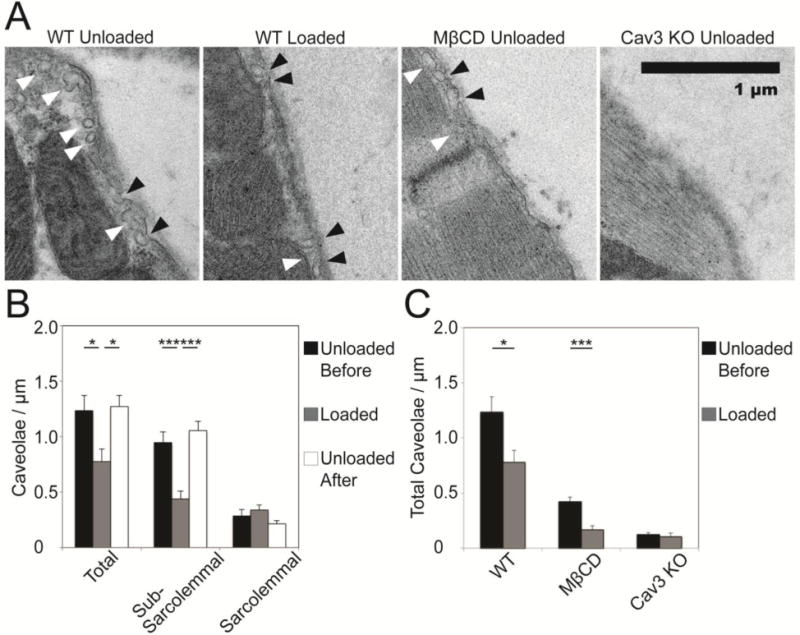
Stretch reversibly recruits sub-sarcolemmal caveolae in loaded hearts. Caveolae are reduced after treatment with MβCD or genetic deletion of caveolin-3. (A) Pressure load alters caveolae density and localization in the wild-type heart. Sample images of sub-sarcolemmal caveolae (white arrows) and caveolae integrated into the sarcolemma (black arrows) in the loaded and unloaded WT hearts, and the unloaded Cav3 KO and MβCD treated hearts (scale: 1 µm). (B) Caveolae are reversibly recruited from the sub-sarcolemmal region during loading in WT hearts (P<0.001), and appear to integrate with the sarcolemma, though material contributed by caveolae is difficult to identify once integrated. Density was quantified with respect to membrane length. (C) Total caveolae density is reversibly lower in the loaded state than before or after loading in WT hearts. Caveolae in unloaded MβCD-treated hearts are significantly less frequent than in WT (P<0.001), and are reduced (P<0.001) during loading. In Cav3 KO hearts, caveolae are almost completely absent (P<0.001), and are not significantly affected by stretch (P=N.S.). (N=10 images per condition, comparisons by unpaired t-test).
Pressure-loading recruited caveolae away from the sub-sarcolemmal space in WT. In the unloaded heart, 0.95±0.09 and 0.29±0.06 caveolae/μm were observed in the sub-sarcolemmal and sarcolemmal categories, respectively (N=10). In the loaded heart, there were significantly fewer caveolae per membrane length in the sub-sarcolemmal space (0.44±0.07 caveolae/μm, N=10, P<0.001 vs. unloaded) and a slight but not significant increase in the density of caveolae fused with the membrane (0.34±0.05 caveolae/μm, N=10, P=N.S. vs. unloaded). The WT heart that was pressure-loaded and then unloaded prior to fixing revealed caveolae density and localization similar to the original unloaded heart (1.05±0.09 sub-sarcolemmal and 0.22±0.03 sarcolemmal caveolae/μm, N=10, both P=N.S. compared with the initial measurement). All electron microscopy statistical comparisons use unpaired t-tests.
3.7 Sarcolemmal Lipid Density Increases with Stretch in a Caveolae-Dependent Manner
Cells labeled with lipophilic fluorescent tracer showed a significant caveolae-dependent increase in fluorescent density with stretch, suggesting the addition of material from sub-sarcolemmal caveolar stores to the sarcolemma (Fig. 6). In WT cells, fluorescent intensity per area increased nearly two-fold with stretch, from 303±42 to 556±90 in arbitrary units (A.U.) (N=9, P<0.01). There was no significant change in Cav3 KO myocytes (1078±343 A.U. in unstretched vs. 1061±353 A.U. in stretched cells; N=6, P=N.S.). Comparing the ratio of stretched to unstretched fluorescence, the difference between the response of WT and Cav3 KO cells to stretch was also statistically significant (P<0.05).
Figure 6.
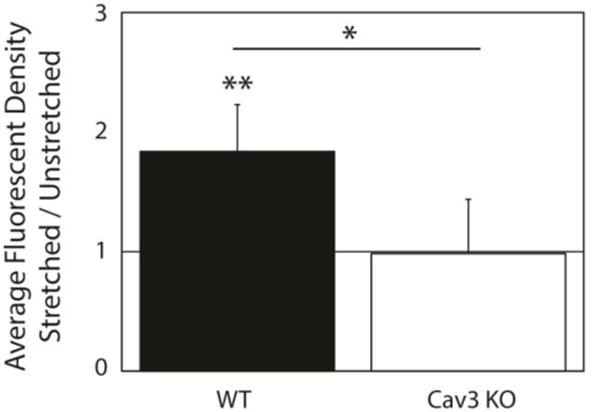
Stretch is associated with significant Cav3-dependent increase in lipophilic fluorescence density. Fluorescent intensity per cell unit area increased by 84±39% with stretch in WT experiments (N=9, P<0.05) and did not change significantly with stretch in Cav3 KO cells (−2±45%, N=6, P=N.S.).
4. DISCUSSION
Mechanoelectric feedback has long been thought to contribute to arrhythmia susceptibility,1 but most studies to date have focused on stretch-dependent alterations in ionic currents and intracellular calcium handling.23,24 Here we showed for the first time that brief stretch slows conduction in isolated myocytes and the intact heart by increasing membrane capacitance via a mechanism that requires caveolae.
Previous investigators have observed that caveolae are recruited and integrated into the sarcolemma of cardiomyocytes during brief stretch,25 and proposed that caveolae may modulate cell electrophysiology5 via enrichment of subpopulations of ion channels or signaling receptors, increased t-tubular convection, or by addition of cell area which could impact excitability.5 Increased cell membrane capacitance slows conduction by increasing the amount of inward current required to accumulate charge at a rate sufficient to depolarize the cell membrane beyond the threshold for excitation.26 Cell membrane capacitance is typically assumed as a constant in modern models of cardiac electrophysiology, even as the various gating parameters of individual ion channel subtypes are varied precisely.27,28 Conduction slowing via a stretch-dependent increase in cell membrane capacitance is consistent with a previous analysis from our lab of increased electrical time constants calculated during ventricular filling in isolated, perfused rabbit hearts.3 Capacitance has been observed to increase when membrane tension is increased in a range of cell types.29 In this study we report new findings suggesting that this mechanism is dependent on caveolae in ventricular myocytes, and that depletion of caveolae inhibits the slowing effect of stretch on myocardial impulse conduction. This finding was obtained with two preparations, a micropatterned neonatal mouse ventricular myocyte monolayer and isolated adult mouse hearts, and with two different mechanisms of caveolae depletion, genetic ablation of Cav3 and pharmacologic treatment with MβCD. We confirmed by electron microscopy that caveolae were reversibly affected by stretch within minutes in control preparations, and depleted in KO mice and with MβCD treatment. Caveolae were present in micropatterned cells (Fig. S3) at a density similar to native tissue (Fig. S4).
Stretch is applied for several minutes, a period sufficient for viscoelastic changes, in this model of hemodynamic load alterations which precludes chronic responses. This is consistent with the duration of tension previously shown to recruit and “flatten” caveolae into the cell membrane, and to re-form caveolae following tension removal.6,30 This relatively fast recruitment of caveolae to the cell membrane has been suggested to be the work of passive mechanical processes in a study by Sinha and colleagues, with mechanisms of caveolae recruitment shown to not include dynamin GTPase, clathrin-coated pits, actin, or ATP, though cortical actin and ATP were indicated to play a role in caveolae reassembly.6
Transmission electron microscopy (TEM) studies were consistent with previous electron microscopy studies in the rabbit heart suggesting that stretch recruits subsarcolemmal caveolae to the membrane5. Although it is not possible to make paired observations by electron microscopy before and after stretch, it was intriguing that in one heart that had been loaded for one minute then unloaded and fixed one minute later, there was evidence suggesting reappearance of caveolae. This time-course needs further confirmation, but is consistent with studies using total internal reflection fluorescence microscopy in live HeLa, endothelial, fibroblast, and muscle cells.6 We also observed that loading appeared to straighten folds in the membrane (Fig. S2). However, given that paired measurements were not possible with TEM, and that there is the possibility of residual crossbridge interactions in the arrested preparations, we can not rule out that differences in contractile state between the preparations may have contributed to this observation.
Similar levels of conduction slowing occurred with comparable magnitudes of stretch in the cultured monolayers and in isolated, Langendorff-perfused murine hearts—in which no electromechanical uncoupling reagents or mechanical restraint was used. Mechanical restraint or pharmacological uncoupling was not necessary, as the cardiac action potential propagated over the region of the heart observed before ventricular wall motion affected its measurement. The use of cultured myocytes allowed us to measure the effects of stretch on membrane capacitance directly. This strongly suggests that our earlier measurements in the rabbit heart showing increased time constant3 of subthreshold impulse decay are very likely to be a direct effect of stretch on the myocyte cell membrane capacitance. The micropatterned culture system also enabled us to apply different patterns of biaxial stretch. Even though the culture itself and the measured electrical activation patterns were anisotropic, the response to non-equibiaxial stretch was not. The amount of conduction slowing was not different whether myofibrils were aligned with the direction of maximum or minimum stretch. This finding is also consistent with a mechanism that depends on membrane capacitance and surface area rather than specific localized structures or ion channels.
The stretches applied to isolated hearts and cell cultures in this study were brief (but not rapid) and relatively large. The duration of stretch required to slow conduction and the time taken for recovery of conduction velocity were longer than a single beat but less than a minute. We did not test whether there is an adaptation to stretch over time, though we did find that cell cultures stretched for as long as 90 mins still showed increased membrane capacitance. The magnitude of loading was comparable to that which might occur during acute heart failure. As such, these stretches were supraphysiological. However, in the isolated rabbit heart we previously found that conduction velocity changes were a continuous progressive function of stretch magnitude3, hence this response is not necessarily relevant only to pathophysiological conditions. More studies are needed to elucidate the effects of varying strain magnitudes on membrane properties and electrical conduction and to understand better the time-courses of the structural and functional responses to mechanical loading.
Treatment with the stretch-activated channel blockers Gd3+ and GsMTx-4 had no effect on the change in conduction velocity caused by stretch. This is consistent with earlier observations in the rabbit heart using Gd3+ and streptomycin.2,3 We also observed that changes in action potential duration accompany stretch or ventricular filling were inhibited by these blockers suggesting that they did act to inhibit stretch activate currents. However, as evidence of stretch-dependent channel gating accumulates, we remain limited by the relative paucity of specific SAC inhibitors. Hence we cannot rule out the possibility that potassium, chloride,31,32 or unknown mechanosensitive33 channels at the cell surface may be recruited by caveolae during stretch. However, these observations have only been reported in atrial not ventricular cells,31 and in response to fluid shear stress,31 or by patch suction or probe indentation (through approximately 50% or more of cell diameter) in isolated myotubes33. While stretched-activated channel blockade with Gd3+ in isolated hearts and GsMTx-4 in isolated myocytes did not alter the effects of stretch on conduction velocity, both inhibitors did affect baseline conduction velocity in our preparations. These observations have not been reported before to our knowledge and the explanation for them is uncertain. However, studies of stretch-activated channel blockers and electromechanical decouplers in myocytes co-cultured with myofibroblasts34,35 did show that SAC blockade increased CV. Although we attempted to reduce the number of fibroblasts in our cultures, they could not be completely eliminated and it is possible that GsMTx-4 had a similar effect on interactions between cardiomyocytes and remaining fibroblasts in our cell culture preparation. In the intact hearts, Gd3+ has non-specific effects on calcium and potassium channels that might have affected conduction velocity.36,37 In the rabbit heart,3 we saw that Gd3+ did not inhibit the slowing of CV due to stretch as we reported here in the mouse. However, unlike in the present study, 50 μM Gd3+ in the isolated rabbit heart did not significantly alter CV when the LV was not loaded. Hence the finding in the present study may be a non-SAC specific effect of Gd3+ in the mouse heart.
Our two models for probing whether caveolae mediate stretch-dependent conduction slowing each have advantages and disadvantages. Caveolin-3 is a cholesterol binding protein expressed in muscles that is instrumental in shaping caveolae and in t-tubule genesis, and is implicated in myopathies including cardiomyopathies.38,39,40 Isolated adult hearts and NMVMs isolated from Cav3 KO mice were used in this study. MβCD could be used in isolated hearts only, because it prevented a detectable signaling from the voltage-sensitive fluorescent dye from being obtained from the monolayers.
Caveolin-3 knock-out mouse lines provide a useful tool to assess the impact of cardiac caveolae on physiology.9,10,41 These mice develop cardiac hypertrophy at six-nine months of age.38,40 All of our studies were performed in mice at 8–12 weeks of age when cardiac function and gross morphology were not different from WT animals, but we cannot rule out the effects of possible adaptive responses or of caveolar depletion on myocyte signaling mechanisms that may be involved in the regulating excitability and conduction. However we obtained similar and consistent results with depletion of caveolae using MβCD.
An increased propensity for arrhythmia has also been observed in other Cav3 mutant mouse lines. The potassium ion channel Kv11.1 (ERG1), but not a long-QT associated mutant of the channel, localize to cholesterol- and sphingolipid-enriched microdomains including caveolae.42 Additionally, depletion of membrane cholesterol using MβCD was observed to accelerate the kinetics of the channel, potentially a capacitance-driven effect.42 Similarly, a Cav3 mutation has been linked to long-QT syndrome through increasing late sodium current through a channel localized to caveolae43 Hence there are clearly other electrophysiological phenotypes associated with Cav3 loss of function, but it is unclear how any of these observations would suggest an alternative interpretation of the data in the present study. On the other hand, alterations of stretch-dependent changes in membrane capacitance in combination with altered ion channel regulation in caveolin mutants could well affect the resultant electrophysiological phenotype including arrhythmia propensity.
Cholesterol sequestration using MβCD has also been used to obtain insights into the mechanisms by which caveolae are involved in regulating cell physiology. The insertion of cholesterol and other sterols in a plasma membrane can increase lipid packing density, reducing proton and cation leak currents by a factor of three at physiologic concentrations of cholesterol when compared with lipid bilayers lacking sterols44 Those findings suggest a role for cholesterol in maintaining membrane pH and electrochemical gradients against passive currents, and our current study demonstrates a role for cholesterol-rich caveolae in augmenting membrane electrochemical capacitance during stretch, with physiologically meaningful slowing effects on cardiac conduction.
The two-fold increase in capacitance we observed in isolated myocytes during stretch is comparable in magnitude to that estimated in the intact heart,3 and is much greater than the expected cell area change due to cell shape change alone. The nearly two-fold increase in lipophilic dye fluorescence we saw was comparable to the magnitude of increase in cell membrane capacitance that we measured. These findings suggest that myocyte stretch recruits substantial new membrane to the sarcolemma and increasing cell capacitance, and that this response requires caveolae.
Disproportionate effects of cholesterol-rich membrane material (rafts) associated with caveolae and T-tubules on cell membrane capacitance have been reported. Reports of skeletal muscle cell membrane capacitance using a glycerol infusion technique for disconnecting t-tubules have calculated two-fold higher (4 μF.cm−2) membrane capacitance in the t-tubule system than in the surface plasma membrane, which itself measured two-fold higher (2 μF.cm−2) than expected for the cell membranes of less caveolae-rich cells (0.7-1 μF.cm−2).45 This finding concurs with capacitive transients previously recorded in Purkinje fibers, which suggested a 2.4 μF.cm−2 cell surface capacitance, compared with a 7 μF.cm−2 element suggested to be the t-tubular network.46 T-tubules, which are also enriched in Cav3, may contain as much as four-fold more cholesterol than the plasma membrane.38 Our findings that cholesterol sequestration and Cav3 deletion lower baseline cell membrane capacitance by approximately 40% support the hypothesis that mechanically-dynamic, caveolae-rich cell types have an elevated cell membrane-capacitance. The absence of the usual contributions of Cav3 to trafficking and concentrating lipids and cholesterol at the cell membrane47 may, in this context, explain the faster baseline conduction velocities in the Cav3 KO NMVMs. While no significant difference was observed in the adult Cav3 KO hearts, this could be explained by the availability of circulating cholesterol to replenish membrane cholesterol48 (but not caveolae) in cardiomyocytes. MβCD treatment also raised conduction velocity in hearts, but this difference was not significant, perhaps due to cholesterol replenishment through non-caveolar pathways.
4.1 Conclusions
In conclusion, these data support a caveolae-dependent mechanism for stretch-slowing of conduction coinciding with increased cell membrane capacitance. This increased capacitance appears due in part to addition of membrane material to the sarcolemma by recruitment and integration of caveolae, but might also be augmented by the high cholesterol content of the caveolar material. Stretch-activated ion currents do not appear to contribute significantly to conduction slowing. The effective increase in cardiomyocyte membrane capacitance via caveolae delays depolarization, slowing cardiac conduction. Such findings may have implications for therapeutically targeting cardiac arrhythmias under pathological conditions such as heart failure that increase ventricular loading and myocardial stretch.
Supplementary Material
{kind=link}
Highlights.
Brief stretch reversibly slows ventricular impulse propagation in mouse
Conduction slowing with stretch requires caveolae but not stretch-activated current
Brief stretch opens caveolae and recruits lipid to the sarcolemma
Stretch substantially increases ventricular myocyte membrane capacitance in vitro
Acknowledgments
We gratefully acknowledge the contribution of peptide GsMTx-4 from Dr. Fred Sachs in Buffalo, NY, and technical assistance of Barbara Murienne, Michael Yang, Kyle Buchholz, Tammy Soo-hoo, Tik-Chee “Jenny” Cheng, and Mathivadhani Panneerselvam, and the animal management of Selma Garcia and others in the laboratory of Professors Roth and Patel and VA San Diego Hospital vivariums, as well as in the UCSD vivariums. Microfabrication of molds for PDMS patterning was carried out in the Nano3-Calit2 cleanroom facility at UCSD.
SOURCES OF FUNDING
Research was supported by NIH grants HL105242, GM094503, 5P01HL098053 and HL96544 (ADM), 5P01HL46345, P41 GM103426, 5T32EB009380, HL091071 (HHP), HL107200 (HHP), and HL066941 (DMR), National Science Foundation Graduate Research Fellowship DGE1144086 (ERP), and VA Merit BX001963 (HHP) and BX000783 (DMR).
ABBREVIATIONS
- AL
absolute length
- Cav3 KO
caveolin-3 knockout mice
- Cm
membrane capacitance
- CV
conduction velocity
- CVmax
maximum conduction velocity
- CVmin
minimum conduction velocity
- Gd3+
gadolinium
- LV
left ventricular
- LVP
left ventricular filling pressure
- MβCD
methyl-β-cyclodextrin
- ML
membrane length
- NMVM
neonatal murine ventricular myocyte
- PDMS
polydimethylsiloxane
- Rm
membrane resistance
- SAC
stretch-activated current
- Tm
membrane time constant
- WT
wild-type
GLOSSARY
- anisotropic biaxial
Stretch magnitude is directionally depended along the two axes of a plane. In other words, stretch in the direction of a major axis is greater than stretch in the direction of a minor axis
- dichroic
An optical filter which selectively transmits light above a specific wavelength and reflects light below that wavelength
- equibiaxial
Stretch magnitude is the same in all directions within a plane
- long-pass
An optical filter which excludes light below a specific wavelength
- mechanoelectric feedback
The processes by which mechanical alterations influence cardiac electrical activity
- optical mapping
Optical measurement of electrophysiological changes in cardiac tissue or cells, as marked using a voltage-sensitive fluorophore
- photolithography
A cleanroom process used to manufacture (micropattern) micrometer-scale topography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
None.
References
- 1.Quinn TA, Bayliss RA, Kohl P. Mechano-electric feedback in the heart: effects on heart rate and rhythm. In: Tripathi ON, Ravens U, Sanguinetti MC, editors. Heart Rate and Rhythm. Vol. 2011. Berlin, Heidelberg: Springer Berlin Heidelberg; pp. 133–151. [Google Scholar]
- 2.Sung D, Mills RW, Schettler J, Narayan SM, Omens JH, McCulloch AD. Ventricular filling slows epicardial conduction and increases action potential duration in an optical mapping study of the isolated rabbit heart. J Cardiovasc Electrophysiol. 2003;14(7):739–749. doi: 10.1046/j.1540-8167.2003.03072.x. [DOI] [PubMed] [Google Scholar]
- 3.Mills RW, Narayan SM, McCulloch AD. Mechanisms of conduction slowing during myocardial stretch by ventricular volume loading in the rabbit. Am J Physiol – Hear Circ Physiol. 2008;295(3):H1270–H1278. doi: 10.1152/ajpheart.00350.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McNary TG, Sohn K, Taccardi B, Sachse FB. Experimental and computational studies of strain-conduction velocity relationships in cardiac tissue. Prog Biophys Mol Biol. 2008;97(2–3):383–400. doi: 10.1016/j.pbiomolbio.2008.02.023. [DOI] [PubMed] [Google Scholar]
- 5.Kohl P, Cooper PJ, Holloway H. Effects of acute ventricular volume manipulation on in situ cardiomyocyte cell membrane configuration. Prog Biophys Mol Biol. 2003;82(1–3):221–227. doi: 10.1016/s0079-6107(03)00024-5. [DOI] [PubMed] [Google Scholar]
- 6.Sinha B, Köster D, Ruez R, et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell. 2011;144(3):402–413. doi: 10.1016/j.cell.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kosawada T, Inoue K, Schmid-Schonbein GW. Mechanics of curved plasma membrane vesicles: resting shapes, membrane curvature, and in-plane shear elasticity. J Biomech Eng. 2005;127(2):229. doi: 10.1115/1.1865197. [DOI] [PubMed] [Google Scholar]
- 8.Sens P, Turner MS. Budded membrane microdomains as regulators for cellular tension. Phys Rev E. 2006;73(3):031918. doi: 10.1103/PhysRevE.73.031918. [DOI] [PubMed] [Google Scholar]
- 9.Oshikawa J, Otsu K, Toya Y, et al. Insulin resistance in skeletal muscles of caveolin-3-null mice. Proc Natl Acad Sci. 2004;101(34):12670–5. doi: 10.1073/pnas.0402053101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horikawa YT, Patel HH, Tsutsumi YM, et al. Caveolin-3 expression and caveolae are required for isoflurane-induced cardiac protection from hypoxia and ischemia/reperfusion injury. J Mol Cell Cardiol. 2008;44(1):123–30. doi: 10.1016/j.yjmcc.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bayly PV, KenKnight BH, Rogers JM, Hillsley RE, Ideker RE, Smith WM. Estimation of conduction velocity vector fields from epicardial mapping data. IEEE Trans Biomed Eng. 1998;45(5):563–71. doi: 10.1109/10.668746. [DOI] [PubMed] [Google Scholar]
- 12.Camelliti P, Gallagher JO, Kohl P, McCulloch AD. Micropatterned cell cultures on elastic membranes as an in vitro model of myocardium. Nat Protoc. 2006;1(3):1379–91. doi: 10.1038/nprot.2006.203. [DOI] [PubMed] [Google Scholar]
- 13.Camelliti P, McCulloch AD, Kohl P. Microstructured cocultures of cardiac myocytes and fibroblasts: a two-dimensional in vitro model of cardiac tissue. Microsc Microanal. 2005;11(3):249–59. doi: 10.1017/S1431927605050506. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Sekar RB, McCulloch AD, Tung L. Cell cultures as models of cardiac mechanoelectric feedback. Prog Biophys Mol Biol. 2008;97(2–3):367–382. doi: 10.1016/j.pbiomolbio.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sreejit P, Kumar S, Verma RS. An improved protocol for primary culture of cardiomyocyte from neonatal mice. Vitr Cell Dev Biol – Anim. 2008;44(3–4):45–50. doi: 10.1007/s11626-007-9079-4. [DOI] [PubMed] [Google Scholar]
- 16.de Bakker JMT, van Capelle FJL, Janse MJ, et al. Slow conduction in the infarcted human heart. “Zigzag” course of activation. Circulation. 1993;88(3):915–26. doi: 10.1161/01.cir.88.3.915. [DOI] [PubMed] [Google Scholar]
- 17.Tung L, Zhang Y. Optical imaging of arrhythmias in tissue culture. J Electrocardiol. 2006;39(4 Suppl):S2–6. doi: 10.1016/j.jelectrocard.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 18.Warren MD, Spitzer KW, Steadman BW, et al. High-precision recording of the action potential in isolated cardiomyocytes using the near-infrared fluorescent dye di-4-ANBDQBS. Am J Physiol – Hear Circ Physiol. 2010;299(4):H1271–81. doi: 10.1152/ajpheart.00248.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terracciano CMN, Harding SE, Adamson D, et al. Changes in sarcolemmal Ca entry and sarcoplasmic reticulum Ca content in ventricular myocytes from patients with end-stage heart failure following myocardial recovery after combined pharmacological and ventricular assist device therapy. Eur Heart J. 2003;24(14):1329–1339. doi: 10.1016/s0195-668x(03)00242-2. [DOI] [PubMed] [Google Scholar]
- 20.Zaniboni M, Cacciani F, Groppi M. Effect of input resistance voltage-dependency on DC estimate of membrane capacitance in cardiac myocytes. Biophys J. 2005;89(3):2170–81. doi: 10.1529/biophysj.105.062828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nuss HB, Marbán E. Electrophysiological properties of neonatal mouse cardiac myocytes in primary culture. J Physiol. 1994;479(2):265–79. doi: 10.1113/jphysiol.1994.sp020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christian AE, Haynes MP, Phillips MC, George H. Use of cyclodextrins for manipulating cellular cholesterol content. J Lipid Res. 1997;38:2264–2272. [PubMed] [Google Scholar]
- 23.Calaghan SC, White E. Mechanical modulation of intracellular ion concentrations: mechanisms and electrical consequences. In: Kamkin A, Kiseleva I, editors. Mechanosensitivity in Cells and Tissues. Moscow: Academia; 2005. pp. 230–254. [PubMed] [Google Scholar]
- 24.ter Keurs HEDJ. Electromechanical coupling in the cardiac myocyte; stretch-arrhythmia feedback. Pflügers Arch Eur J Physiol. 2011;462(1):165–75. doi: 10.1007/s00424-011-0944-3. [DOI] [PubMed] [Google Scholar]
- 25.Sens P, Turner MS. Budded membrane microdomains as regulators for cellular tension. Phys Rev E. 2006;73(3):031918. doi: 10.1103/PhysRevE.73.031918. [DOI] [PubMed] [Google Scholar]
- 26.King JH, Huang CL-H, Fraser JA. Determinants of myocardial conduction velocity: implications for arrhythmogenesis. Front Physiol. 2013;4:154. doi: 10.3389/fphys.2013.00154. Published online Jun 28, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clayton RH, Bernus O, Cherry EM, et al. Models of cardiac tissue electrophysiology: progress, challenges and open questions. Prog Biophys Mol Biol. 2011;104(1–3):22–48. doi: 10.1016/j.pbiomolbio.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 28.Bondarenko VE, Szigeti GP, Bett GCL, Kim S-J, Rasmusson RL. Computer model of action potential of mouse ventricular myocytes. Am J Physiol – Hear Circ Physiol. 2012;287:H1378–H1403. doi: 10.1152/ajpheart.00185.2003. [DOI] [PubMed] [Google Scholar]
- 29.Morris C, Homann U. Cell surface area regulation and membrane tension. J Membr Biol. 2001;179:79–102. doi: 10.1007/s002320010040. [DOI] [PubMed] [Google Scholar]
- 30.Gervásio OL, Phillips WD, Cole L, Allen DG. Caveolae respond to cell stretch and contribute to stretch-induced signaling. J Cell Sci. 2011 doi: 10.1242/jcs.084376. [DOI] [PubMed] [Google Scholar]
- 31.Kozera L, White E, Calaghan SC. Caveolae act as membrane reserves which limit mechanosensitive I(Cl,swell) channel activation during swelling in the rat ventricular myocyte. PLoS One. 2009;4(12):e8312. doi: 10.1371/journal.pone.0008312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boycott HE, Barbier CSM, Eichel Ca, et al. Shear stress triggers insertion of voltage-gated potassium channels from intracellular compartments in atrial myocytes. Proc Natl Acad Sci. 2013:1–10. doi: 10.1073/pnas.1309896110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang H, Bae C, Sachs F, Suchyna TM. Caveolae regulation of mechanosensitive channel function in myotubes. PLoS One. 2013;8(8):e72894. doi: 10.1371/journal.pone.0072894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson SA, Copeland CR, Reich DH, Tung L. Mechanical coupling between myofibroblasts and cardiomyocytes slows electric conduction in fibrotic cell monolayers. Circulation. 2011;123(19):2083–93. doi: 10.1161/CIRCULATIONAHA.110.015057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson Sa, Blazeski A, Copeland CR, et al. Acute slowing of cardiac conduction in response to myofibroblast coupling to cardiomyocytes through N-cadherin. J Mol Cell Cardiol. 2014;68:29–37. doi: 10.1016/j.yjmcc.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lacampagne A, Gannier F, Argibay J, Garnier D, Le Guennec J-Y. The stretch-activated ion channel blocker gadolinium also blocks L-type calcium channels in isolated ventricular myocytes of the guinea-pig. Biochim Biophys Acta. 1994;1191:205–208. doi: 10.1016/0005-2736(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 37.Hongo K, Pascarel C, Cazorla O, Gannier F, Le Guennec JY, White E. Gadolinium blocks the delayed rectifier potassium current in isolated guinea-pig ventricular myocytes. Exp Physiol. 1997;82(4):647–56. doi: 10.1113/expphysiol.1997.sp004053. [DOI] [PubMed] [Google Scholar]
- 38.Le Lay S, Kurzchalia TV. Getting rid of caveolins: phenotypes of caveolin-deficient animals. Biochim Biophys Acta. 2005;1746(3):322–33. doi: 10.1016/j.bbamcr.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 39.Al-Qusairi L, Laporte J. T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skelet Muscle. 2011;1:26. doi: 10.1186/2044-5040-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fridolfsson HN, Patel HH. Caveolin and caveolae in age associated cardiovascular disease. J Geriatr Cardiol. 2013;10:66–74. doi: 10.3969/j.issn.1671-5411.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsutsumi YM, Kawaraguchi Y, Horikawa YT, et al. Role of caveolin-3 and glucose transporter-4 in isoflurane-induced delayed cardiac protection. Anesthesiology. 2010;112(5):1136–45. doi: 10.1097/ALN.0b013e3181d3d624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balijepalli RC, Delisle BP, Foell JD, Slind JK, Kamp TJ, January CT. Kv11.1 (ERG1) K+ channels localize in cholesterol and sphingolipid enriched membranes and are modulated by membrane cholesterol. Channels. 2007;1(4):263–272. doi: 10.4161/chan.4946. [DOI] [PubMed] [Google Scholar]
- 43.Vatta M, Ackerman MJ, Ye B, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114(20):2104–12. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- 44.Haines TH. Do sterols reduce proton and sodium leaks through lipid bilayers? Prog Lipid Res. 2001;40(4):299–324. doi: 10.1016/s0163-7827(01)00009-1. [DOI] [PubMed] [Google Scholar]
- 45.Gage P, Eisenberg R. Capacitance of the surface and transverse tubular membrane of frog sartorius muscle fibers. J Gen Physiol. 1969;53(3):265–278. doi: 10.1085/jgp.53.3.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fozzard H. Membrane capacity of the cardiac Purkinje fibre. J Physiol. 1966;182:255–267. doi: 10.1113/jphysiol.1966.sp007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salanueva IJ, Cerezo A, Guadamillas MC, del Pozo Ma. Integrin regulation of caveolin function. J Cell Mol Med. 2007;11(5):969–80. doi: 10.1111/j.1582-4934.2007.00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Defesche JC. Low-density lipoprotein receptor—its structure, function, and mutations. Semin Vasc Med. 2004;4(1):5–11. doi: 10.1055/s-2004-822993. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.