

Abstract
Drug combinations that include the psychostimulant methylphenidate plus a selective serotonin reuptake inhibitor (SSRI) such as fluoxetine are increasingly used in children and adolescents. For example, this combination is indicated in the treatment of attention-deficit/hyperactivity disorder and depression comorbidity and other mental disorders. Such co-exposure also occurs in patients on SSRIs that use methylphenidate as a cognitive enhancer. The neurobiological consequences of these drug combinations are poorly understood. Methylphenidate alone can produce gene regulation effects that mimic addiction-related gene regulation by cocaine, consistent with its moderate addiction liability. We have previously shown that combining SSRIs with methylphenidate potentiates methylphenidate-induced gene regulation in the striatum. The present study investigated which striatal output pathways are affected by the methylphenidate+fluoxetine combination, by assessing effects on pathway-specific neuropeptide markers, and which serotonin receptor subtypes may mediate these effects. Our results demonstrate that a 5-day repeated treatment with fluoxetine (5 mg/kg) potentiates methylphenidate (5 mg/kg)-induced expression of both dynorphin (direct pathway marker) and enkephalin (indirect pathway). These changes were accompanied by correlated increases in the expression of the 5-HT1B, but not 5-HT2C, serotonin receptor in the same striatal regions. A further study showed that the 5-HT1B receptor agonist CP94253 (3-10 mg/kg) mimics the fluoxetine potentiation of methylphenidate-induced gene regulation. These findings suggest a role for the 5-HT1B receptor in the fluoxetine effects on striatal gene regulation. Given that 5-HT1B receptors are known to facilitate addiction-related gene regulation and behavior, our results suggest that SSRIs may enhance the addiction liability of methylphenidate by increasing 5-HT1B receptor signaling.
Keywords: methylphenidate, psychostimulant, SSRI, striatum, dynorphin, enkephalin
1. Introduction
Use of psychotropic medications in children and adolescents is increasing. This is of concern because preclinical studies indicate that such drugs can induce maladaptive neuronal changes suggestive of an increased risk for drug addiction and other neuropsychiatric disorders later in life (for reviews, see Carlezon and Konradi, 2004; Carrey and Wilkinson, 2011; Marco et al., 2011).
The most often used psychotropic drugs in pediatric populations include psychostimulants such as methylphenidate and selective serotonin reuptake inhibitor (SSRI) antidepressants such as fluoxetine. Methylphenidate is widely employed in the treatment of attention-deficit/hyperactivity disorder (ADHD), which is diagnosed in up to 7% of school-age children in the US (DSMMD, 2000; Kollins, 2008). In addition, methylphenidate is increasingly used as a recreational drug or as a so-called cognitive enhancer (Greely et al., 2008) to improve concentration and performance in certain tasks or to study harder (Kollins, 2008; Swanson and Volkow, 2008; Wilens et al., 2008). For example, the 2011 National Survey on Drug Use and Health (NSDUH) reported that approximately 1 million persons age 12 or older in the US admitted current nonmedical use of prescription psychostimulants (SAMHSA, 2012). SSRIs are first-line treatments for major depressive disorder (MDD) and are also helpful to treat anxiety disorders, obsessive compulsive disorder and others. The SSRI fluoxetine is specifically approved for the treatment of pediatric MDD (Iversen, 2006).
While the potential for adverse developmental effects of individual psychotropic drugs is well recognized (see above), possible interactions between different drugs have received little attention, despite the fact that co-exposure to more than one drug is quite common. For example, methylphenidate plus SSRI combinations are indicated in the treatment of ADHD/MDD co-morbidity (Rushton and Whitmire, 2001; Safer et al., 2003), which occurs in up to 40% of pediatric ADHD cases (Waxmonsky, 2003; Spencer, 2006). Methylphenidate is also added to SSRI treatments, for example, as augmentation therapy in MDD (e.g., Nelson, 2007; Ishii et al., 2008; Ravindran et al., 2008), as acceleration treatment (e.g., Lavretsky et al., 2003), or to treat sexual dysfunction related to SSRIs (e.g., Csoka et al., 2008). It is unknown how much accidental co-exposure occurs in patients on antidepressants who use methylphenidate recreationally or as a cognitive enhancer.
Despite prevalent use, the neurobiological consequences of methylphenidate plus SSRI combination treatments are little understood. The psychostimulant methylphenidate acts by blocking dopamine transporters (Volkow et al., 2002), thus causing dopamine overflow, among other effects, similar to cocaine (Yano and Steiner, 2007). As one consequence of the ensuing dopamine receptor overstimulation (Yano et al., 2006; Alburges et al., 2011), methylphenidate produces altered gene regulation, predominantly in dopamine terminal areas such as the striatum and cortex (Steiner and Van Waes, 2013). However, while methylphenidate has the potential to change the expression of numerous genes (Adriani et al., 2006), other genes that are robustly affected by drugs such as cocaine were minimally or not impacted by methylphenidate treatments (Steiner and Van Waes, 2013). Cocaine, in contrast to methylphenidate (e.g., Kuczenski and Segal, 1997; Segal and Kuczenski, 1999; see Yano and Steiner, 2007), also blocks the serotonin transporter, and serotonin plays a facilitatory role in cocaine-induced gene regulation (e.g., Bhat and Baraban, 1993). Combining methylphenidate (dopamine action) with an SSRI (serotonin action) may thus produce more “cocaine-like” molecular changes than methylphenidate alone.
Our recent series of studies in adolescent rats supports this hypothesis. We showed that adding the SSRIs fluoxetine or citalopram, in doses that by themselves did not affect gene expression, potentiated gene regulation effects of methylphenidate. This potentiation was first shown for acute induction of immediate-early genes (IEGs) such as c-Fos and Zif268 (Steiner et al., 2010; Van Waes et al., 2010), as well as the neuropeptides substance P and dynorphin (Van Waes et al., 2012). However, effects of repeated drug treatments are more relevant for long-term neurobehavioral changes. Repeated treatments with psychostimulants, including methylphenidate, produce several alterations in gene expression, for example, blunting (repression) of IEG induction (Steiner and Van Waes, 2013). We recently showed that IEG blunting is also potentiated by SSRIs. Thus, fluoxetine given in conjunction with methylphenidate for 5 days in adolescent rats potentiated blunting of Zif268 and Homer1a induction by a subsequent cocaine challenge (Van Waes et al., 2013).
In the present study, we assessed the impact of the same repeated treatment on another well-established effect of repeated exposure to psychostimulants, increases in the expression of the opioid peptides dynorphin and enkephalin in the striatum (Steiner and Gerfen, 1998). These neuropeptides are useful cell-type markers due to their differential expression in striatal projection neurons. Neurons of the direct (striatonigral) pathway express dynorphin, whereas neurons of the indirect (striatopallidal) pathway contain enkephalin (Steiner and Gerfen, 1998). Our previous study showed that fluoxetine potentiated acute methylphenidate-induced expression of dynorphin, but not enkephalin (Van Waes et al., 2012), thus suggesting that gene regulation by this drug combination may be restricted to direct pathway neurons. Our present results of repeated combination treatment demonstrate that fluoxetine potentiates gene regulation for both neuropeptides, indicating that indeed both pathways are affected. Moreover, we also addressed the potential underlying mechanisms by investigating associated changes in the expression of serotonin (5-HT) receptor subtypes in the striatum that may mediate these effects. Research shows that 5-HT1B receptor signaling regulates various behavioral responses to cocaine including self-administration (e.g., Parsons et al., 1998; Neumaier et al., 2002; Przegaliński et al., 2004; Przegaliński et al., 2008; Pentkowski et al., 2012; Neisewander et al., 2014, for review), as well as cocaine-induced gene regulation (e.g., Lucas et al., 1997; Castanon et al., 2000). For comparison, we assessed treatment effects on the 5-HT2C receptor, which also modifies cocaine effects in several ways (Bubar and Cunningham, 2008; Devroye et al., 2013). Our results show increased expression of 5-HT1B by repeated methylphenidate treatment, an effect that is also potentiated by co-treatment with fluoxetine. Furthermore, a role for 5-HT1B in such gene regulation is suggested by our finding that stimulation of 5-HT1B receptors mimics the fluoxetine potentiation of acute Zif268 induction by methylphenidate.
2. Materials and methods
2.1. Subjects
Male Sprague–Dawley rats (35 days old at the beginning of the drug treatment; Harlan, Madison, WI, USA) were housed 2–3 per cage under standard laboratory conditions (12:12h light/dark cycle; lights on at 07:00h) with food and water available ad libitum. Experiments were performed between 13:00 and 17:00h. Prior to the drug treatment, the rats were allowed one week of acclimation during which they were repeatedly handled. All procedures met the NIH guidelines for the care and use of laboratory animals and were approved by the Rosalind Franklin University Animal Care and Use Committee.
2.2. Drug treatment
In experiment 1, rats received 5 daily injections of vehicle (V, i.p.), methylphenidate HCl (MP, 5 mg/kg; in 0.02% ascorbic acid, 1 ml/kg; Sigma, St. Louis, MO, USA), fluoxetine HCl (FLX, 5 mg/kg; Sigma), or methylphenidate plus fluoxetine (MP+FLX) in their home cage (n=6-9). These rats were killed with CO2 2 h after the last injection. In experiment 2, rats received an injection of vehicle, or the 5-HT1B receptor agonist CP94253 (CP, 3 or 10 mg/kg; Tocris/R&D Systems, Minneapolis, MN, USA) (Borycz et al., 2008; Przegaliński et al., 2008), followed 15 min later by an injection of vehicle, methylphenidate (5 mg/kg), or methylphenidate plus fluoxetine (5 mg/kg) (n=5-9 each) and were killed 40 min later.
2.3. Tissue preparation and in situ hybridization histochemistry
The brain was rapidly removed, frozen in isopentane cooled on dry ice and then stored at −30 °C until cryostat sectioning. Coronal sectio ns (12 μm) were thaw-mounted onto glass slides (Superfrost/Plus, Daigger, Wheeling, IL, USA), dried on a slide warmer and stored at −30 °C. In preparation for the in situ hy bridization histochemistry, the sections were fixed in 4% paraformaldehyde/0.9% saline for 10 min at room temperature, incubated in a fresh solution of 0.25% acetic anhydride in 0.1 M triethanolamine/0.9% saline (pH 8.0) for 10 min, dehydrated, defatted for 2 × 5 min in chloroform, rehydrated, and air-dried. The slides were then stored at −30 °C until hybridization.
Oligonucleotide probes (48-mers; Invitrogen, Rockville, MD, USA) were labeled with [33P]-dATP as described earlier (Steiner and Kitai, 2000). The probes had the following sequence: dynorphin, complementary to bases 862–909, GenBank accession number M10088; enkephalin, bases 436–483, M28263; 5-HT1B (Htr1b), bases 62–109, NM022225; 5-HT2C (Htr2c), bases 363–410, NM012765; Zif268 (Egr1), bases 352–399, M18416.
One hundred μl of hybridization buffer containing labeled probe (~3 × 106 cpm) was added to each slide. The sections were coverslipped and incubated at 37 °C overnight. After incubation, the slides were first rinsed in four washes of 1X saline citrate (150 mM sodium chloride, 15 mM sodium citrate), and then washed 3 times 20 min each in 2X saline citrate/50% formamide at 40 °C, followed by 2 washes of 30 min each in 1X saline citrate at room temperature. After a brief water rinse, the sections were air-dried and then apposed to X-ray film (BioMax MR-2, Kodak) for 3–14 days.
2.4. Analysis of autoradiograms
Striatal gene expression was assessed in sections from three rostrocaudal levels, rostral (approximately +1.6 mm relative to bregma, Paxinos and Watson, 1998), middle (+0.4) and caudal (−0.8), in a total of 23 sectors (Fig. 1) that are mostly defined by their predominant cortical inputs (see Willuhn et al., 2003; Yano and Steiner, 2005b). Eighteen of these sectors represent the caudate-putamen and 5 the nucleus accumbens (Fig. 1).
Figure 1.

Topography of potentiated neuropeptide expression in the striatum after repeated methylphenidate plus fluoxetine treatment. Maps depict the distribution of the increases (vs. V) in dynorphin (A) and enkephalin expression (B) in the rostral, middle and caudal striatum after 5 daily injections of methylphenidate (5 mg/kg, i.p.; MP), fluoxetine (5 mg/kg; FLX) or methylphenidate+fluoxetine (5 mg/kg each; MP+FLX). Potentiation (POT) denotes the difference between methylphenidate+fluoxetine and methylphenidate groups. The increases are expressed relative to the maximal increase for each neuropeptide (% of max.). Sectors with significant differences [vs. vehicle (V) controls, or methylphenidate+fluoxetine vs. methylphenidate (POT)] (P<0.05) are coded as indicated. Sectors without significant effects are in white. Illustrations of film autoradiograms depicting the expression of dynorphin (left) and enkephalin (right) in coronal sections from the middle striatum after repeated treatment with vehicle (V) or methylphenidate+fluoxetine (MP+FLX) are shown below the maps. Abbreviations: caudate-putamen: c, central; d, dorsal*; dc, dorsal central; dl, dorsolateral*; dm, dorsomedial; m, medial; v, ventral; vc, ventral central; vl, ventrolateral*; nucleus accumbens: mC, medial core; lC, lateral core; mS, medial shell; vS, ventral shell; lS, lateral shell; *sensorimotor sectors (see Yano and Steiner, 2005a).
Hybridization signals on film autoradiograms were measured by densitometry (NIH Image; Wayne Rasband, NIMH, Bethesda, MD, USA). The films were captured using a light table (Northern Light, Imaging Research, St. Catharines, Ontario, Canada) and a Sony CCD camera (Imaging Research). The “mean density” value of a region of interest was measured by placing a template over the captured image. Mean densities were corrected for background by subtracting mean density values measured over white matter (corpus callosum). Values from corresponding regions in the two hemispheres were then averaged. The illustrations of film autoradiograms are computer-generated images and are contrast-enhanced. Maximal hybridization signal is black.
2.5. Statistics
Treatment effects were determined by two-factor (experiment 1) or one-factor ANOVAs (experiment 2). Newman-Keuls post hoc tests were used to describe differences between individual groups (Statistica, StatSoft, Tulsa, OK, USA). For the maps illustrating the distribution of changes in gene expression, the difference in signals between a drug treatment group and the vehicle controls was expressed relative to the maximal difference observed for that probe (% max.), for each sector. For the maps showing basal expression of receptors, the signal in each sector was expressed relative to the maximal signal. Changes in neuropeptide expression (dynorphin, enkephalin) across these 23 striatal sectors were compared with those in 5-HT receptor expression (present study) and changes in IEG expression (Zif268; as reported before Van Waes et al., 2010; Van Waes et al., 2013), by Pearson correlations.
3. Results
3.1. Repeated treatment with methylphenidate+fluoxetine produces increased expression of dynorphin and enkephalin in the striatum
The 5-day repeated treatment (experiment 1) with the moderate dose of methylphenidate (5 mg/kg) alone did not induce changes in dynorphin expression in any of the 23 striatal sectors (P>0.05 for all sectors; Figs. 1A and 2A). Similarly, repeated treatment with fluoxetine (5 mg/kg) alone had no statistically significant effects on the expression of this neuropeptide. In contrast, the drug combination of methylphenidate+fluoxetine produced a widespread increase in the expression of dynorphin (vs. vehicle controls, P<0.05 in 9 of the 18 sectors of the caudate-putamen; Figs. 1A and 2A). This effect occurred on all three rostrocaudal levels, but was most robust in the middle to caudal striatum. On all levels, dynorphin expression was increased in dorsal and lateral (sensorimotor) sectors. On the middle level, a significant increase was also found in the medial and central sectors (Fig. 2A). No effect was seen in the 5 sectors of the nucleus accumbens (Fig. 1A).
Figure 2.

Potentiation of striatal neuropeptide expression after repeated methylphenidate plus fluoxetine treatment. The changes in dynorphin (A) and enkephalin expression (B) in the 6 sectors of the middle striatum are shown. Mean density values (mean±SEM, arbitrary units) are given for rats that received 5 daily injections of vehicle (V), methylphenidate (5 mg/kg; MP), fluoxetine (5 mg/kg; FLX), or methylphenidate+fluoxetine (MP+FLX) (n=6-9 per group). Sectors: m, medial; d, dorsal; dl, dorsolateral; v, ventral; c, central; vl, ventrolateral. * P<0.05, ** P<0.01, *** P<0.001 vs. V controls or as indicated; # P<0.05, ## P<0.01, ### P<0.001, MP+FLX vs. MP (“potentiation”).
Similar but less widespread effects were found for enkephalin expression (Fig. 1B and 2B). Again, neither methylphenidate alone nor fluoxetine alone produced significant changes in expression in the striatum. In contrast, the combination treatment increased enkephalin expression in 3 sectors (medial, dorsal, central) on the middle level (Fig. 2B). Compared to the distribution of the changes in dynorphin expression, the changes in enkephalin expression were thus more centered in the middle and medial striatum (Fig. 1), and there was no significant correlation between dynorphin and enkephalin for “potentiation” (i.e., difference MP+FLX minus MP) in the 23 sectors (r=0.148, P>0.05).
We also compared the distribution of the present changes in dynorphin expression with other neuronal changes induced by acute and repeated methylphenidate+fluoxetine treatment. For example, there was a positive correlation between the potentiation of dynorphin expression after repeated treatment (present results) and the previously reported fluoxetine potentiation of acute IEG (Zif268) induction (Van Waes et al., 2010) across the 23 striatal sectors (r=0.493, P<0.02). Moreover, our correlation analysis (Fig. 3) demonstrates that the potentiation of Zif268 blunting found after repeated methylphenidate+fluoxetine treatment (Van Waes et al., 2013) was also positively correlated with the present increases in dynorphin expression across the 23 striatal sectors (r=0.591, P<0.005). These molecular changes thus had a similar regional distribution across the striatum. Robust changes preferentially occurred in, but were not limited to, sectors of the sensorimotor striatum (Fig. 3).
Figure 3.

Striatal distribution of the potentiation of increases in dynorphin expression (present study) vs. that of blunting of Zif268 induction (Van Waes et al., 2013) after a 5-day repeated methylphenidate plus fluoxetine treatment. The scatterplot shows the correlation between the present dynorphin potentiation (MP+FLX minus MP) and the previously reported potentiation of Zif268 blunting in the 23 striatal sectors (r=0.591; open diamonds, sensorimotor sectors; full circles, non-sensorimotor sectors). The data are expressed as the percentage of the maximal potentiation for each gene. Values for dynorphin expression were obtained 2h after the last injection (present study); those for Zif268 blunting represent values of Zif268 induction by a cocaine (25 mg/kg) challenge 24h after a 5-day repeated methylphenidate plus fluoxetine treatment (Van Waes et al., 2013). Potentiation was most robust in sectors of the sensorimotor striatum (open diamonds). ** P<0.005.
3.2. Distribution of 5-HT1B and 5-HT2C receptor expression in the striatum
The distribution of 5-HT1B and 5-HT2C receptor mRNAs in striatum and nucleus accumbens was determined in vehicle-treated controls and is depicted in Figure 4. The distribution of 5-HT1B expression (Fig. 4A) is fairly uniform throughout the rostral, middle and caudal striatum, with somewhat higher levels in the lateral striatum that peak on the middle level (lateral half of dorsolateral and ventrolateral sectors; Fig. 4A). In the nucleus accumbens (rostral level), the highest 5-HT1B expression is seen in the lateral part of the shell (Fig. 4A).
Figure 4.

Expression of 5-HT1B (A) and 5-HT2C (B) receptor mRNAs in the striatum. Illustrations of film autoradiograms (left) depict “basal” 5-HT1B and 5-HT2C expression (i.e., in vehicle-treated rats, n=6) in coronal sections from the rostral, middle and caudal striatum. The maps (right) show the distribution of gene expression across the 23 striatal sectors. Values are given as percentages of maximal expression and are coded as indicated. The maximal hybridization signal is black.
In marked contrast, the distribution of 5-HT2C mRNA in the striatum is distinctly uneven (Fig. 4B). Overall, there is a rostrocaudal gradient, with high expression rostrally that fades towards background levels in most of the caudal striatum. The expression is considerably higher in ventral and medial (limbic, associative) than in dorsal/lateral (sensorimotor) sectors. On all three rostrocaudal levels, there are distinct small areas of high-density labeling present, probably reflecting the patches of the patch/matrix compartments (Eberle-Wang et al., 1997). In the nucleus accumbens, 5-HT2C expression is by far highest in the rostral pole (data not shown), fairly high in the medial shell and medial core and lowest in the lateral shell (Fig. 4B).
3.3. Fluoxetine potentiates repeated methylphenidate-induced increases in the expression of 5-HT1B, but not 5-HT2C
The present drug treatments (experiment 1) had differential effects on 5-HT1B vs. 5-HT2C expression in the striatum (Figs. 5 and 6). The 5-day repeated treatment with methylphenidate alone produced significantly increased 5-HT1B expression in 6 sectors of the caudate-putamen (Fig. 5A), with similar tendencies in several additional sectors (Fig. 6A). This effect occurred in the middle and caudal striatum and was maximal in the dorsal sector on the caudal level (Fig. 5A). No changes in 5-HT1B expression were seen in the nucleus accumbens after repeated methylphenidate treatment (Fig. 5A).
Figure 5.
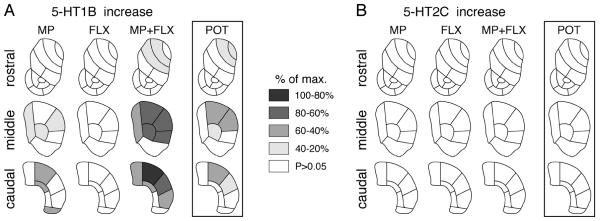
Topography of changes in 5-HT receptor expression in the striatum after repeated methylphenidate plus fluoxetine treatment. Maps depict the distribution of the increases in 5-HT1B expression (A) and the lack of changes in 5-HT2C expression (B) in the rostral, middle and caudal striatum after the 5-day treatment with methylphenidate (5 mg/kg, i.p.; MP), fluoxetine (5 mg/kg; FLX) or methylphenidate+fluoxetine (5 mg/kg each; MP+FLX). The potentiation (POT) denotes the difference between methylphenidate+fluoxetine and methylphenidate alone. The differences are expressed relative to the maximal difference for each gene (% of max.). Sectors with significant differences (P<0.05) are coded as indicated. Sectors without significant effects are in white.
Figure 6.

Changes in striatal 5-HT receptor expression after repeated methylphenidate plus fluoxetine treatment. The changes in 5-HT1B expression (A) and the lack of changes in 5-HT2C expression (B) in the 6 middle striatal sectors are shown. Mean density values (mean±SEM) are given for rats that received 5 daily injections of vehicle (V), methylphenidate (5 mg/kg; MP), fluoxetine (5 mg/kg; FLX), or methylphenidate+fluoxetine (MP+FLX) (n=6-9 per group). Sectors: m, medial; d, dorsal; dl, dorsolateral; v, ventral; c, central; vl, ventrolateral. * P<0.05, ** P<0.01, *** P<0.001 vs. V controls or as indicated; # P<0.05, ## P<0.01, ### P<0.001, MP+FLX vs. MP (potentiation).
In contrast to methylphenidate, repeated treatment with fluoxetine alone did not alter 5-HT1B expression in any of these striatal sectors (Figs. 5A and 6A). However, fluoxetine given in conjunction with methylphenidate potentiated methylphenidate-induced increases in 5-HT1B expression (Figs. 5A and 6A). Thus, the combination treatment (MP+FLX) produced significantly increased expression (vs. vehicle controls) in 12 of the 18 sectors of the caudate-putamen (compared to 6 sectors for MP alone) and in none of the nucleus accumbens (Fig. 5A). Moreover, MP+FLX animals displayed significantly higher levels of expression than MP animals (potentiation) in 7 sectors. Increased 5-HT1B expression again occurred on all three rostrocaudal levels, but predominantly on middle and caudal levels (Fig. 5A). Increases were present in medial to lateral sectors, but they were most robust dorsally and laterally. Thus, potentiation of 5-HT1B expression was most pronounced in the dorsal/dorsolateral striatum on middle and caudal levels (Fig. 5A).
We also compared the regional distribution of these drug-induced changes in 5-HT1B expression with those in dynorphin and enkephalin expression, by correlation analysis. There was a significant positive correlation between the potentiation of increases in 5-HT1B expression and that of dynorphin expression (r=0.634, P<0.002; Fig. 7), but not that of enkephalin expression (r=0.378, P>0.05). Overall, this analysis confirmed preferential potentiation of changes in 5-HT1B and dynorphin expression in the sensorimotor striatum (Fig. 7).
Figure 7.

Striatal distribution of the potentiation of increases in 5-HT1B expression vs. that in dynorphin expression after the 5-day repeated methylphenidate plus fluoxetine treatment. The scatterplot depicts the correlation between the 5-HT1B potentiation and the dynorphin potentiation (MP+FLX minus MP) in the 23 striatal sectors (r=0.634). The data are expressed as the percentage of the maximal potentiation for each gene. Potentiation was most robust in sectors of the sensorimotor striatum (open diamonds). ** P<0.002.
In contrast to 5-HT1B expression, 5-HT2C expression was not affected by these drug treatments. Neither methylphenidate alone, nor fluoxetine alone or the methylphenidate+fluoxetine combination altered 5-HT2C expression in any of these striatal sectors (P>0.05; Figs. 5B and 6B).
3.4. 5-HT1B receptor stimulation potentiates acute methylphenidate-induced expression of Zif268
Experiment 2 assessed whether the 5-HT1B receptor could modify methylphenidate-induced gene regulation. Our results show that the 5-HT1B receptor agonist CP94253 (3-10 mg/kg) indeed potentiated acute methylphenidate-induced expression of Zif268 in the striatum in a dose-dependent manner (Fig. 8). This effect was present in many striatal regions, but was maximal in the lateral striatum. Stimulating 5-HT1B receptors thus mimicked fluoxetine effects on gene regulation, consistent with a role for 5-HT1B in the fluoxetine potentiation of methylphenidate-induced gene regulation.
Figure 8.

Stimulation of 5-HT1B receptors potentiates acute induction of Zif268 by methylphenidate. Illustrations of film autoradiograms depict Zif268 expression in the middle striatum in rats that were treated with vehicle (V) (upper left), methylphenidate (5 mg/kg, MP) (upper middle), and methylphenidate (5 mg/kg) plus 5-HT1B agonist CP94253 (10 mg/kg, MP+CP10) (upper right) and were killed 40 min later. For comparison, Zif268 expression after methylphenidate plus fluoxetine (5 mg/kg each, MP+FLX) treatment (lower right) is also shown. The graph lower left presents mean density values (mean±SEM) in the dorsolateral and ventrolateral sectors pooled for rats that were treated with vehicle, methylphenidate and/or CP94253 (3 or 10 mg/kg), or methylphenidate plus fluoxetine (n=5-9). *** P<0.001 vs. V controls; # P<0.05, ## P<0.01, ### P<0.001, as indicated.
4. Discussion
The goal of the present study was to determine whether repeated combination treatment with methylphenidate plus fluoxetine would produce potentiated changes in gene regulation in both striatal output pathways, by assessing effects on the cell-type markers dynorphin and enkephalin. We here demonstrate that the 5-day repeated treatment with methylphenidate alone or fluoxetine alone with the present subthreshold doses had no effect on either neuropeptide, but that the combined treatment produced increases in the expression for both markers. These results indicate that gene regulation in both pathways is affected by this combination treatment. In addition, we show that these increases in neuropeptide expression were associated with increases in the expression of the 5-HT1B, but not 5-HT2C, receptor subtype in the same striatal regions, and that 5-HT1B receptor stimulation mimicked fluoxetine effects on gene regulation.
4.1. Fluoxetine potentiation of gene regulation by methylphenidate in the striatum: pathways affected and potential significance
Illicit psychostimulants such as cocaine and amphetamine alter gene regulation in the striatum preferentially in the subtype of medium spiny projection neurons that express D1 dopamine receptors and project to the substantia nigra/internal pallidum (direct pathway) (Steiner, 2010; Lobo and Nestler, 2011; Steiner and Van Waes, 2013). Gene regulation in the projection neurons that target the globus pallidus (external pallidum) and contain mostly D2 receptors (first link of the indirect pathway) is also affected, but generally less, and these effects tend to be more context-dependent (Steiner, 2010). This selectivity, confirmed with various double-labeling approaches (Steiner, 2010), was first demonstrated by studies that assessed drug actions on neuropeptide markers that are differentially localized in the two striatal output pathways (Steiner and Gerfen, 1998). Direct pathway neurons predominantly express the neuropeptides substance P and dynorphin, whereas indirect pathway neurons express enkephalin. While not absolute, the principal segregation of these neuropeptides between the two pathways has been demonstrated by a variety of molecular techniques (e.g., Gerfen and Young, 1988; Gerfen et al., 1990; Gerfen et al., 1991; Surmeier et al., 1996; Heiman et al., 2008).
Numerous studies have shown that drugs such as cocaine and amphetamine produce pronounced increases in the expression of substance P and dynorphin (direct pathway), while expression of enkephalin (indirect pathway) is increased to a considerably lesser extent (see Yano and Steiner, 2007; Steiner, 2010, for reviews). (Note that, in contrast, enkephalin expression is very responsive to antipsychotic/D2 receptor antagonist treatment or dopamine loss (Steiner and Gerfen, 1998)). Importantly, increased dynorphin mRNA and peptide levels have also been demonstrated in human cocaine abusers (Hurd and Herkenham, 1993; Frankel et al., 2008).
In our previous studies, we found that acute and repeated treatment with methylphenidate induced robust changes in substance P expression (Brandon and Steiner, 2003; Yano and Steiner, 2005a; Van Waes et al., 2012). Dynorphin expression was marginally affected by acute methylphenidate (Yano and Steiner, 2005a; Van Waes et al., 2012), but increased levels of striatal dynorphin mRNA (Brandon and Steiner, 2003) and dynorphin peptide in striatum and substantia nigra (Alburges et al., 2011) were found after repeated treatment with high doses of methylphenidate (10 mg/kg). In contrast, in these studies, enkephalin expression was minimally or not altered (Brandon and Steiner, 2003; Yano and Steiner, 2005a; Van Waes et al., 2012). These differential effects between the two pathways are consistent with findings by others. For example, Kim et al. demonstrated that repeated methylphenidate treatment increased deltaFosB expression in direct, but not in indirect, pathway neurons, as shown by double-labeling techniques (Kim et al., 2009). These findings thus indicate that methylphenidate exposure alone more selectively affects the direct pathway than cocaine or amphetamine do.
Our more recent studies show that adding an SSRI (serotonin action) to methylphenidate (dopamine action) potentiates striatal gene regulation by methylphenidate (see Introduction; Steiner and Van Waes, 2013). This effect is robust in direct pathway neurons, but, as indicated by our neuropeptide marker in the present study, also occurs in indirect pathway neurons with repeated treatment. Thus, repeated methylphenidate plus fluoxetine treatment increases the expression of dynorphin and, to some lesser extent, also enkephalin. This pattern is reminiscent of the effects of cocaine and amphetamine, which have significant effects (if more moderate compared with dynorphin) also on enkephalin expression (e.g., Hurd and Herkenham, 1993; Steiner and Gerfen, 1993; Wang and McGinty, 1996; Spangler et al., 1997). In this sense, the methylphenidate plus fluoxetine combination treatment produces more “cocaine-like” gene regulation than methylphenidate alone.
The present findings also confirm and extend our earlier observations (Van Waes et al., 2010; Van Waes et al., 2012; Van Waes et al., 2013) that this drug-induced gene regulation predominantly occurs in, but is not limited to, sensorimotor sectors mostly in the middle to caudal striatum. Among the affected non-sensorimotor sectors are medial and central striatal sectors that receive inputs from the prefrontal and cingulate cortex (see Willuhn et al., 2003; Steiner and Van Waes, 2013) and are important for goal-directed behavior. The lateral (sensorimotor) striatum, on the other hand, is critical for `automatic' and habitual behavior (Yin and Knowlton, 2006). Psychostimulant-induced changes in gene regulation (and resulting structural plasticity; Jedynak et al., 2007) in lateral striatal circuits are implicated in habitual and obsessive aspects of drug addiction (Berke and Hyman, 2000; Everitt et al., 2001; Everitt and Robbins, 2013) as well as in relapse to drug taking after abstinence (Vanderschuren et al., 2005; Fuchs et al., 2006; See et al., 2007).
Although overlapping, the regional distribution of methylphenidate plus fluoxetine-induced changes in enkephalin expression was not identical to that of dynorphin expression (not correlated). The basis for this apparent dissociation in distribution (and magnitude) is presently unknown. However, there are various differences in the regulation of gene expression in neurons of these two pathways. These including the dopamine receptors involved (D1 vs. D2) and their respective second messenger signaling pathways, a differential sensitivity to and possibly origin of cortical inputs that drive these changes, differential thalamic inputs and others (see Steiner, 2010; Van Waes et al., 2012, for discussion). For example, stimulation of D1 receptors (direct pathway neurons) facilitates gene regulation driven by cortical (or thalamic) inputs in these neurons, while stimulation of D2 receptors (indirect pathway neurons) dampens such gene regulation in those neurons (Steiner, 2010). This latter effect may account for the more limited changes in enkephalin expression, especially in the lateral striatum, where D2 receptor levels are considerably higher than in the medial striatum (see Steiner and Gerfen, 1999). Future studies will have to elucidate the exact mechanisms involved.
The functional consequences of increased dynorphin and enkephalin expression after psychostimulant treatments remain to be established. However, dynorphin and enkephalin are neurotransmitters released from these neurons. There is evidence that both neuropeptides act, at least in part, as negative feedback systems (`brake') (Steiner and Gerfen, 1998) to limit dopamine and glutamate (e.g., Atwood et al., 2014) input to striatal neurons and help maintain systems homeostasis (Hyman and Nestler, 1996; Steiner and Gerfen, 1998; Steiner, 2010). Increased dynorphin function in these neurons has been implicated in addiction processes. Thus, there is good evidence that dynorphin inhibits dopamine release via kappa opioid receptors on dopamine terminals and dendrites/cell bodies (e.g., Di Chiara and Imperato, 1988; Spanagel et al., 1992; Marinelli et al., 1998; see Shippenberg et al., 2007, for review). Increased dynorphin signaling after psychostimulant treatments may thus excessively inhibit such inputs to the striatum (Hyman and Nestler, 1996; Steiner and Gerfen, 1998; Shippenberg et al., 2007). Based on these and other findings, it has been proposed that increased dynorphin function (in the ventral striatum) may contribute to somatic signs of withdrawal, such as dysphoria, anxiety, anhedonia and depression (Nestler and Carlezon, 2006; Shippenberg et al., 2007). The behavioral consequences of increased dynorphin (and enkephalin) signaling in the dorsal striatum remain to be investigated.
4.2. Role for the 5-HT1B receptor in the fluoxetine potentiation?
In the present study, we started to investigate the mechanisms underlying the fluoxetine potentiation of methylphenidate-induced gene regulation, by assessing a potential role for specific serotonin receptor subtypes. It is clear that dopamine is critical for gene regulation by psychostimulants such as cocaine (Steiner and Van Waes, 2013) as well as methylphenidate (Yano et al., 2006; Alburges et al., 2011). However, serotonin facilitates these effects of cocaine. For example, it has been shown that attenuation of the serotonin neurotransmission by transmitter depletion (Bhat and Baraban, 1993) or receptor antagonism (e.g., Lucas et al., 1997; Castanon et al., 2000) reduces IEG induction by cocaine in the striatum. A similar effect has been demonstrated for cocaine action on striatal neuropeptide expression (Morris et al., 1988; Walker et al., 1996; Horner et al., 2005).
Serotonin is known to enhance activity in the mesostriatal and mesolimbic dopamine pathways by complex interactions in both the dopamine terminal regions and the somatodendritic areas in the midbrain (for reviews, see Muller and Huston, 2006; Weikop et al., 2007; Bubar and Cunningham, 2008). Therefore, the SSRI potentiation of methylphenidate-induced gene regulation in the striatum could reflect potentiated dopamine action mediated by serotonin receptors in the striatum and/or other brain areas. It is unclear that dopamine neurons express serotonin receptors (Hoyer et al., 1994; Barnes and Sharp, 1999), so these interactions are likely indirect. For example, several serotonin receptor subtypes are expressed by striatal projection neurons themselves (Hoyer et al., 1994; Barnes and Sharp, 1999). Among the most highly expressed are 5-HT1B and 5-HT2C. We first investigated whether the effects of the repeated methylphenidate+fluoxetine treatment on the neuropeptide expression were associated with the distribution of and/or changes in 5-HT1B or 5-HT2C expression.
Expression of 5-HT2C (formerly named 5-HT1C) shows a distinctly uneven distribution throughout the striatum, with a rostrocaudal gradient and preferential expression in medial and ventral (“limbic”) regions (e.g., Mengod et al., 1990; Eberle-Wang et al., 1997). This distribution thus does not match the observed distribution of the fluoxetine potentiation of methylphenidate-induced gene regulation in our studies (most robust in dorsal/lateral, sensorimotor regions). Furthermore, our present results show that 5-HT2C expression was not affected by either drug treatment.
In contrast, in agreement with previous studies (e.g., Voigt et al., 1991; Bruinvels et al., 1994), we found that the distribution of 5-HT1B expression is relatively homogeneous throughout the striatum. There is a somewhat higher expression in the lateral striatum on the middle level (Voigt et al., 1991), roughly matching the localization of maximally potentiated IEG expression after acute methylphenidate+fluoxetine treatment (Van Waes et al., 2010).
Moreover, we here demonstrate for the first time that methylphenidate also alters 5-HT1B expression in the striatum. The 5-day repeated treatment with methylphenidate (5 mg/kg) alone was sufficient to increase the expression of 5-HT1B, mostly in the middle to caudal striatum. In contrast, fluoxetine (5 mg/kg, 5 days) alone had no effect [note that a more aggressive treatment (8 mg/kg, 21 days) did increase striatal 5-HT1B expression (Le Poul et al., 2000)]. Adding fluoxetine (5 mg/kg) to methylphenidate (5 mg/kg), however, potentiated the methylphenidate-induced increases. Overall, this potentiation of 5-HT1B expression occurred in the same striatal regions as (i.e., was correlated with) the potentiation of dynorphin expression (maximal in sensorimotor striatum).
These findings of methylphenidate+fluoxetine-induced increases in 5-HT1B expression are consistent with previous studies showing increased 5-HT1B expression in the striatum after repeated cocaine exposure (Hoplight et al., 2007; Neumaier et al., 2009). Increased 5-HT1B expression thus represents a further example of mimicked cocaine effects of the methylphenidate+fluoxetine exposure. In summary, both basal distribution and our drug-induced changes for 5-HT1B expression are consistent with a role for striatal 5-HT1B in the fluoxetine effects on methylphenidate-induced gene regulation.
We therefore further investigated whether 5-HT1B receptor stimulation could modify methylphenidate-induced gene regulation. Indeed, our results show that the 5-HT1B receptor agonist CP94253 potentiated acute Zif268 induction by methylphenidate, and that this effect was maximal in the lateral striatum. 5-HT1B receptor activation thus mimicked fluoxetine effects on gene regulation. These findings are the first to demonstrate that 5-HT1B receptors can facilitate methylphenidate-induced gene regulation. They extend previous findings showing that 5-HT1B receptor stimulation enhances methylphenidate-induced locomotor activity (Borycz et al., 2008). Overall, these findings are consistent with previous results demonstrating that 5-HT1B receptors contribute to cocaine-induced gene expression (Lucas et al., 1997; Castanon et al., 2000) and regulate behavioral responses to cocaine (e.g., Neisewander et al., 2014).
At the cellular level, the 5-HT1B receptor subtype is predominantly located on axon terminals to regulate (inhibit) neurotransmitter release (Boschert et al., 1994). There is evidence that 5-HT1B receptors expressed by direct pathway (striatonigral) neurons mediate serotonin-induced inhibition of GABA release from their terminals, and that this effect results in disinhibition of mesostriatal dopamine neurons and increased striatal dopamine release (c.f. Castanon et al., 2000; Hoplight et al., 2007). Alternatively, 5-HT1B-mediated inhibition of GABA release from local striatal axon terminals of striatal projection neurons (Gerfen and Bolam, 2010) may directly disinhibit striatal neurons. Either mechanism could thus be expected to produce potentiated (disinhibited) gene induction in striatal neurons. However, given their fairly widespread distribution in the brain (e.g., cortex; Bruinvels et al., 1994), it is conceivable that 5-HT1B signaling in other brain areas might contribute to the SSRI potentiation of methylphenidate-induced gene regulation in the striatum. Future studies with local experimental manipulations will have to clarify which 5-HT1B receptors are involved.
5. Conclusion
Repeated methylphenidate-induced changes in gene regulation in striatal circuits (Brandon and Steiner, 2003; see Steiner and Van Waes, 2013, for review) are associated with a facilitation of subsequent cocaine seeking and taking in the cocaine self-administration model (Brandon et al., 2001; Schenk and Izenwasser, 2002; Crawford et al., 2011). Potentiated gene regulation by fluoxetine may thus enhance this effect. It has been shown that methylphenidate+fluoxetine co-exposure in juvenile rats enhances their sensitivity to cocaine and natural reward (among other behavioral effects) in adulthood (Warren et al., 2011). Future studies will have to determine whether such co-exposure to fluoxetine, either in the treatment of mental disorders or, more likely, during medication abuse which typically involves higher-level drug exposure (Steiner and Van Waes, 2013), will increase the abuse/addiction liability of methylphenidate, and whether the 5-HT1B receptor may offer a pharmacological target to attenuate these effects.
Highlights.
Fluoxetine potentiates methylphenidate-induced gene regulation in the striatum.
Gene regulation is altered in both direct and indirect striatal output pathways.
Methylphenidate treatment increases the expression of 5-HT1B serotonin receptors.
Stimulation of 5-HT1B receptors potentiates methylphenidate-induced gene regulation.
Acknowledgments
This work was supported in part by National Institutes of Health Grants DA011261 and DA031916 (H. S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that they have no financial interests or other conflicts of interest.
References
- Adriani W, Leo D, Greco D, Rea M, di Porzio U, Laviola G, Perrone-Capano C. Methylphenidate administration to adolescent rats determines plastic changes in reward-related behavior and striatal gene expression. Neuropsychopharmacology. 2006;31:1946–1956. doi: 10.1038/sj.npp.1300962. [DOI] [PubMed] [Google Scholar]
- Alburges ME, Hoonakker AJ, Horner KA, Fleckenstein AE, Hanson GR. Methylphenidate alters basal ganglia neurotensin systems through dopaminergic mechanisms: A comparison with cocaine treatment. J. Neurochem. 2011;117:470–478. doi: 10.1111/j.1471-4159.2011.07215.x. [DOI] [PubMed] [Google Scholar]
- Atwood BK, Kupferschmidt DA, Lovinger DM. Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nat. Neurosci. 2014;17:540–548. doi: 10.1038/nn.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083–1152. doi: 10.1016/s0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- Berke JD, Hyman SE. Addiction, dopamine, and the molecular mechanisms of memory. Neuron. 2000;25:515–532. doi: 10.1016/s0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Baraban JM. Activation of transcription factor genes in striatum by cocaine: role of both serotonin and dopamine systems. J. Pharmacol. Exp. Ther. 1993;267:496–505. [PubMed] [Google Scholar]
- Borycz J, Zapata A, Quiroz C, Volkow ND, Ferré S. 5-HT(1B) receptor-mediated serotoninergic modulation of methylphenidate-induced locomotor activation in rats. Neuropsychopharmacology. 2008;33:619–626. doi: 10.1038/sj.npp.1301445. [DOI] [PubMed] [Google Scholar]
- Boschert U, Amara DA, Segu L, Hen R. The mouse 5-hydroxytryptamine1B receptor is localized predominantly on axon terminals. Neuroscience. 1994;58:167–182. doi: 10.1016/0306-4522(94)90164-3. [DOI] [PubMed] [Google Scholar]
- Brandon CL, Steiner H. Repeated methylphenidate treatment in adolescent rats alters gene regulation in the striatum. Eur. J. Neurosci. 2003;18:1584–1592. doi: 10.1046/j.1460-9568.2003.02892.x. [DOI] [PubMed] [Google Scholar]
- Brandon CL, Marinelli M, Baker LK, White FJ. Enhanced reactivity and vulnerability to cocaine following methylphenidate treatment in adolescent rats. Neuropsychopharmacology. 2001;25:651–661. doi: 10.1016/S0893-133X(01)00281-0. [DOI] [PubMed] [Google Scholar]
- Bruinvels AT, Landwehrmeyer B, Gustafson EL, Durkin MM, Mengod G, Branchek TA, Hoyer D, Palacios JM. Localization of 5-HT1B, 5-HT1D alpha, 5-HT1E and 5-HT1F receptor messenger RNA in rodent and primate brain. Neuropharmacology. 1994;33:367–386. doi: 10.1016/0028-3908(94)90067-1. [DOI] [PubMed] [Google Scholar]
- Bubar MJ, Cunningham KA. Prospects for serotonin 5-HT2R pharmacotherapy in psychostimulant abuse. Prog. Brain Res. 2008;172:319–346. doi: 10.1016/S0079-6123(08)00916-3. [DOI] [PubMed] [Google Scholar]
- Carlezon WAJ, Konradi C. Understanding the neurobiological consequences of early exposure to psychotropic drugs: linking behavior with molecules. Neuropharmacology. 2004;47:47–60. doi: 10.1016/j.neuropharm.2004.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrey N, Wilkinson M. A review of psychostimulant-induced neuroadaptation in developing animals. Neurosci. Bull. 2011;27:197–214. doi: 10.1007/s12264-011-1004-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanon N, Scearce-Levie K, Lucas JJ, Rocha B, Hen R. Modulation of the effects of cocaine by 5-HT1B receptors: a comparison of knockouts and antagonists. Pharmacol. Biochem. Behav. 2000;67:559–566. doi: 10.1016/s0091-3057(00)00389-0. [DOI] [PubMed] [Google Scholar]
- Crawford CA, Baella SA, Farley CM, Herbert MS, Horn LR, Campbell RH, Zavala AR. Early methylphenidate exposure enhances cocaine self-administration but not cocaine-induced conditioned place preference in young adult rats. Psychopharmacology. 2011;213:43–52. doi: 10.1007/s00213-010-2011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csoka A, Bahrick A, Mehtonen OP. Persistent sexual dysfunction after discontinuation of selective serotonin reuptake inhibitors. J. Sex. Med. 2008;5:227–233. doi: 10.1111/j.1743-6109.2007.00630.x. [DOI] [PubMed] [Google Scholar]
- Devroye C, Filip M, Przegaliński E, McCreary AC, Spampinato U. Serotonin2C receptors and drug addiction: focus on cocaine. Exp. Brain Res. 2013;230:537–545. doi: 10.1007/s00221-013-3593-2. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. USA. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DSMMD . Diagnostic and Statistical Manual of Mental Disorders. Fourth Edition American Psychiatric Association; Washington, DC: 2000. [Google Scholar]
- Eberle-Wang K, Mikeladze Z, Uryu K, Chesselet MF. Pattern of expression of the serotonin2C receptor messenger RNA in the basal ganglia of adult rats. J. Comp. Neurol. 1997;384:233–247. [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. From the ventral to the dorsal striatum: devolving views of their roles in drug addiction. Neurosci. Biobehav. Rev. 2013;37:1946–1954. doi: 10.1016/j.neubiorev.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Dickinson A, Robbins TW. The neuropsychological basis of addictive behaviour. Brain Res. Rev. 2001;36:129–138. doi: 10.1016/s0165-0173(01)00088-1. [DOI] [PubMed] [Google Scholar]
- Frankel PS, Alburges ME, Bush L, Hanson GR, Kish SJ. Striatal and ventral pallidum dynorphin concentrations are markedly increased in human chronic cocaine users. Neuropharmacology. 2008;55:41–46. doi: 10.1016/j.neuropharm.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs RA, Branham RK, See RE. Different neural substrates mediate cocaine seeking after abstinence versus extinction training: A critical role for the dorsolateral caudate–putamen. J. Neuroscience. 2006;26:3584–3588. doi: 10.1523/JNEUROSCI.5146-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Young WS., III Distribution of striatonigral and striatopallidal peptidergic neurons in both patch and matrix compartments: an in situ hybridization histochemistry and fluorescent retrograde tracing study. Brain Res. 1988;460:161–167. doi: 10.1016/0006-8993(88)91217-6. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Bolam JP. The neuroanatomical organization of the basal ganglia. In: Steiner H, Tseng KY, editors. Handbook of Basal Ganglia Structure and Function. Academic Press/Elsevier; London: 2010. pp. 3–28. [Google Scholar]
- Gerfen CR, McGinty JF, Young WS., III Dopamine differentially regulates dynorphin, substance P, and enkephalin expression in striatal neurons: in situ hybridization histochemical analysis. J. Neurosci. 1991;11:1016–1031. doi: 10.1523/JNEUROSCI.11-04-01016.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr., Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- Greely H, Sahakian B, Harris J, Kessler RC, Gazzaniga M, Campbell P, Farah MJ. Towards responsible use of cognitive-enhancing drugs by the healthy. Nature. 2008;456:702–705. doi: 10.1038/456702a. [DOI] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, Greengard P, Heintz N. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoplight BJ, Vincow ES, Neumaier JF. Cocaine increases 5-HT1B mRNA in rat nucleus accumbens shell neurons. Neuropharmacology. 2007;52:444–449. doi: 10.1016/j.neuropharm.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Horner KA, Adams DH, Hanson GR, Keefe KA. Blockade of stimulant-induced preprodynorphin mRNA expression in the striatal matrix by serotonin depletion. Neuroscience. 2005;131:67–77. doi: 10.1016/j.neuroscience.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Hoyer D, Clarke DE, Fozard JR, Hartig PR, Martin GR, Mylecharane EJ, Saxena PR, Humphrey PP. International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (Serotonin) Pharmacol. Rev. 1994;46:157–203. [PubMed] [Google Scholar]
- Hurd YL, Herkenham M. Molecular alterations in the neostriatum of human cocaine addicts. Synapse. 1993;13:357–369. doi: 10.1002/syn.890130408. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Nestler EJ. Initiation and adaptation: a paradigm for understanding psychotropic drug action. Am. J. Psychiatry. 1996;153:151–162. doi: 10.1176/ajp.153.2.151. [DOI] [PubMed] [Google Scholar]
- Ishii M, Tatsuzawa Y, Yoshino A, Nomura S. Serotonin syndrome induced by augmentation of SSRI with methylphenidate. Psychiatry Clin. Neurosci. 2008;62:246. doi: 10.1111/j.1440-1819.2008.01767.x. [DOI] [PubMed] [Google Scholar]
- Iversen L. Neurotransmitter transporters and their impact on the development of psychopharmacology. Br. J. Pharmacol. 2006;147(Suppl1):S82–88. doi: 10.1038/sj.bjp.0706428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedynak JP, Uslaner JM, Esteban JA, Robinson TE. Methamphetamine-induced structural plasticity in the dorsal striatum. Eur. J. Neurosci. 2007;25:847–853. doi: 10.1111/j.1460-9568.2007.05316.x. [DOI] [PubMed] [Google Scholar]
- Kim Y, Teylan MA, Baron M, Sands A, Nairn AC, Greengard P. Methylphenidate-induced dendritic spine formation and DeltaFosB expression in nucleus accumbens. Proc. Natl. Acad. Sci. U S A. 2009;106:2915–2920. doi: 10.1073/pnas.0813179106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollins SH. ADHD, substance use disorders, and psychostimulant treatment: current literature and treatment guidelines. J. Atten. Disord. 2008;12:115–125. doi: 10.1177/1087054707311654. [DOI] [PubMed] [Google Scholar]
- Kuczenski R, Segal DS. Effects of methylphenidate on extracellular dopamine, serotonin, and norepinephrine: comparison with amphetamine. J. Neurochem. 1997;68:2032–2037. doi: 10.1046/j.1471-4159.1997.68052032.x. [DOI] [PubMed] [Google Scholar]
- Lavretsky H, Kim MD, Kumar A, Reynolds CF. Combined treatment with methylphenidate and citalopram for accelerated response in the elderly: an open trial. J. Clin. Psychiatry. 2003;64:1410–1414. doi: 10.4088/jcp.v64n1202. [DOI] [PubMed] [Google Scholar]
- Le Poul E, Boni C, Hanoun N, Laporte AM, Laaris N, Chauveau J, Hamon M, Lanfumey L. Differential adaptation of brain 5-HT1A and 5-HT1B receptors and 5-HT transporter in rats treated chronically with fluoxetine. Neuropharmacology. 2000;39:110–122. doi: 10.1016/s0028-3908(99)00088-x. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Nestler EJ. The striatal balancing act in drug addiction: distinct roles of direct and indirect pathway medium spiny neurons. Front. Neuroanat. 2011;5:41. doi: 10.3389/fnana.2011.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas JJ, Segu L, Hen R. 5-Hydroxytryptamine1B receptors modulate the effect of cocaine on c-fos expression: converging evidence using 5-hydroxytryptamine1B knockout mice and the 5-hydroxytryptamine1B/1D antagonist GR127935. Mol. Pharmacol. 1997;51:755–763. doi: 10.1124/mol.51.5.755. [DOI] [PubMed] [Google Scholar]
- Marco EM, Adriani W, Ruocco LA, Canese R, Sadile AG, Laviola G. Neurobehavioral adaptations to methylphenidate: the issue of early adolescent exposure. Neurosci. Biobehav. Rev. 2011;35:1722–1739. doi: 10.1016/j.neubiorev.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Marinelli M, Barrot M, Simon H, Oberlander C, Dekeyne A, Le Moal M, Piazza PV. Pharmacological stimuli decreasing nucleus accumbens dopamine can act as positive reinforcers but have a low addictive potential. Eur. J. Neurosci. 1998;10:3269–3275. doi: 10.1046/j.1460-9568.1998.00340.x. [DOI] [PubMed] [Google Scholar]
- Mengod G, Nguyen H, Le H, Waeber C, Lübbert H, Palacios JM. The distribution and cellular localization of the serotonin 1C receptor mRNA in the rodent brain examined by in situ hybridization histochemistry. Comparison with receptor binding distribution. Neuroscience. 1990;35:577–591. doi: 10.1016/0306-4522(90)90330-7. [DOI] [PubMed] [Google Scholar]
- Morris BJ, Reimer S, Hollt V, Herz A. Regulation of striatal prodynorphin mRNA levels by the raphe-striatal pathway. Brain Res. 1988;464:15–22. doi: 10.1016/0169-328x(88)90013-7. [DOI] [PubMed] [Google Scholar]
- Muller CP, Huston JP. Determining the region-specific contributions of 5-HT receptors to the psychostimulant effects of cocaine. Trends Pharmacol. Sci. 2006;27:105–112. doi: 10.1016/j.tips.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Neisewander JL, Cheung TH, Pentkowski NS. Dopamine D3 and 5-HT1B receptor dysregulation as a result of psychostimulant intake and forced abstinence: Implications for medications development. Neuropharmacology. 2014;76:301–319. doi: 10.1016/j.neuropharm.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JC. Augmentation strategies in the treatment of major depressive disorder. Recent findings and current status of augmentation strategies. CNS Spectr. 2007;12(Suppl 22):6–9. doi: 10.1017/s1092852900016011. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WAJ. The mesolimbic dopamine reward circuit in depression. Biol. Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Neumaier JF, McDevitt RA, Polis IY, Parsons LH. Acquisition of and withdrawal from cocaine self-administration regulates 5-HT mRNA expression in rat striatum. J. Neurochem. 2009;111:217–227. doi: 10.1111/j.1471-4159.2009.06313.x. [DOI] [PubMed] [Google Scholar]
- Neumaier JF, Vincow ES, Arvanitogiannis A, Wise RA, Carlezon WAJ. Elevated expression of 5-HT1B receptors in nucleus accumbens efferents sensitizes animals to cocaine. J. Neurosci. 2002;22:10856–10863. doi: 10.1523/JNEUROSCI.22-24-10856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons LH, Weiss F, Koob GF. Serotonin1B receptor stimulation enhances cocaine reinforcement. J. Neurosci. 1998;18:10078–10089. doi: 10.1523/JNEUROSCI.18-23-10078.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; New York: 1998. [Google Scholar]
- Pentkowski NS, Cheung TH, Toy WA, Adams MD, Neumaier JF, Neisewander JL. Protracted withdrawal from cocaine self-administration flips the switch on 5-HT(1B) receptor modulation of cocaine abuse-related behaviors. Biol. Psychiatry. 2012;72:396–404. doi: 10.1016/j.biopsych.2012.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przegaliński E, Gołda A, Filip M. Effects of serotonin (5-HT)(1B) receptor ligands on cocaine-seeking behavior in rats. Pharmacol. Rep. 2008;60:798–810. [PubMed] [Google Scholar]
- Przegaliński E, Papla I, Siwanowicz J, Filip M. Effects of 5-HT1B receptor ligands microinjected into the ventral tegmental area on the locomotor and sensitizating effects of cocaine in rats. Eur. Neuropsychopharmacol. 2004;14:217–225. doi: 10.1016/S0924-977X(03)00106-8. [DOI] [PubMed] [Google Scholar]
- Ravindran AV, Kennedy SH, O'Donovan MC, Fallu A, Camacho F, Binder CE. Osmotic-release oral system methylphenidate augmentation of antidepressant monotherapy in major depressive disorder: results of a double-blind, randomized, placebo-controlled trial. J. Clin. Psychiatry. 2008;69:87–94. doi: 10.4088/jcp.v69n0112. [DOI] [PubMed] [Google Scholar]
- Rushton JL, Whitmire JT. Pediatric stimulant and selective serotonin reuptake inhibitor prescription trends: 1992 to 1998. Arch. Pediatr. Adolesc. Med. 2001;155:560–565. doi: 10.1001/archpedi.155.5.560. [DOI] [PubMed] [Google Scholar]
- Safer DJ, Zito JM, DosReis S. Concomitant psychotropic medication for youths. Am. J. Psychiatry. 2003;160:438–449. doi: 10.1176/appi.ajp.160.3.438. [DOI] [PubMed] [Google Scholar]
- SAMHSA (NSDUH Series H-44).Results from the 2011 National Survey on Drug Use and Health: Summary of National Findings. 2012 HHS Publication No. (SMA) 12-4713, http://www.samhsa.gov/data/NSDUH/2011SummNatFindDetTables/Index.aspx.
- Schenk S, Izenwasser S. Pretreatment with methylphenidate sensitizes rats to the reinforcing effects of cocaine. Pharmacol. Biochem. Behav. 2002;72:651–657. doi: 10.1016/s0091-3057(02)00735-9. [DOI] [PubMed] [Google Scholar]
- See RE, Elliott JC, Feltenstein MW. The role of dorsal vs ventral striatal pathways in cocaine-seeking behavior after prolonged abstinence in rats. Psychopharmacology. 2007;194:321–331. doi: 10.1007/s00213-007-0850-8. [DOI] [PubMed] [Google Scholar]
- Segal DS, Kuczenski R. Escalating dose-binge treatment with methylphenidate: role of serotonin in the emergent behavioral profile. J. Pharmacol. Exp. Ther. 1999;291:19–30. [PubMed] [Google Scholar]
- Shippenberg TS, Zapata A, Chefer VI. Dynorphin and the pathophysiology of drug addiction. Pharmacol. Ther. 2007;116:306–321. doi: 10.1016/j.pharmthera.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R, Herz A, Shippenberg TS. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc. Natl. Acad. Sci. USA. 1992;89:2046–2050. doi: 10.1073/pnas.89.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangler R, Zhou Y, Maggos CE, Schlussman SD, Ho A, Kreek MJ. Prodynorphin, proenkephalin and kappa opioid receptor mRNA responses to acute “binge” cocaine. Mol. Brain Res. 1997;44:139–142. doi: 10.1016/s0169-328x(96)00249-5. [DOI] [PubMed] [Google Scholar]
- Spencer TJ. ADHD and comorbidity in childhood. J. Clin. Psychiatry. 2006;67(Suppl 8):27–31. [PubMed] [Google Scholar]
- Steiner H. Psychostimulant-induced gene regulation in corticostriatal circuits. In: Steiner H, Tseng KY, editors. Handbook of Basal Ganglia Structure and Function. Academic Press/Elsevier; London: 2010. pp. 501–525. [Google Scholar]
- Steiner H, Gerfen CR. Cocaine-induced c-fos messenger RNA is inversely related to dynorphin expression in striatum. J. Neurosci. 1993;13:5066–5081. doi: 10.1523/JNEUROSCI.13-12-05066.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Gerfen CR. Role of dynorphin and enkephalin in the regulation of striatal output pathways and behavior. Exp. Brain Res. 1998;123:60–76. doi: 10.1007/s002210050545. [DOI] [PubMed] [Google Scholar]
- Steiner H, Gerfen CR. Enkephalin regulates acute D2 dopamine receptor antagonist-induced immediate-early gene expression in striatal neurons. Neuroscience. 1999;88:795–810. doi: 10.1016/s0306-4522(98)00241-3. [DOI] [PubMed] [Google Scholar]
- Steiner H, Kitai ST. Regulation of rat cortex function by D1 dopamine receptors in the striatum. J. Neurosci. 2000;20:5449–5460. doi: 10.1523/JNEUROSCI.20-14-05449.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Van Waes V. Addiction-related gene regulation: Risks of exposure to cognitive enhancers vs. other psychostimulants. Prog. Neurobiol. 2013;100:60–80. doi: 10.1016/j.pneurobio.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Van Waes V, Marinelli M. Fluoxetine potentiates methylphenidate-induced gene regulation in addiction-related brain regions: Concerns for use of cognitive enhancers? Biol. Psychiatry. 2010;67:592–594. doi: 10.1016/j.biopsych.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Song W-J, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J. Neurosci. 1996;16:6579–6591. doi: 10.1523/JNEUROSCI.16-20-06579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson JM, Volkow ND. Increasing use of stimulants warns of potential abuse. Nature. 2008;453:586. doi: 10.1038/453586a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Waes V, Beverley J, Marinelli M, Steiner H. Selective serotonin reuptake inhibitor antidepressants potentiate methylphenidate (Ritalin)-induced gene regulation in the adolescent striatum. Eur. J. Neurosci. 2010;32:435–447. doi: 10.1111/j.1460-9568.2010.07294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Waes V, Carr B, Beverley JA, Steiner H. Fluoxetine potentiation of methylphenidate-induced neuropeptide expression in the striatum occurs selectively in direct pathway (striatonigral) neurons. J. Neurochem. 2012;122:1054–1064. doi: 10.1111/j.1471-4159.2012.07852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Waes V, Vandrevala M, Beverley J, Steiner H. Selective serotonin re-uptake inhibitors potentiate gene blunting induced by repeated methylphenidate treatment: Zif268 versus Homer1a. Addict. Biol. Epub. 2013 Jun; doi: 10.1111/adb.12067. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderschuren LJ, Di Ciano P, Everitt BJ. Involvement of the dorsal striatum in cue-controlled cocaine seeking. J. Neurosci. 2005;25:8665–8670. doi: 10.1523/JNEUROSCI.0925-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt MM, Laurie DJ, Seeburg PH, Bach A. Molecular cloning and characterization of a rat brain cDNA encoding a 5-hydroxytryptamine1B receptor. EMBO J. 1991;10:4017–4023. doi: 10.1002/j.1460-2075.1991.tb04977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Franceschi D, Maynard L, Ding YS, Gatley SJ, Gifford A, Zhu W, Swanson JM. Relationship between blockade of dopamine transporters by oral methylphenidate and the increases in extracellular dopamine: therapeutic implications. Synapse. 2002;43:181–187. doi: 10.1002/syn.10038. [DOI] [PubMed] [Google Scholar]
- Walker PD, Capodilupo JG, Wolf WA, Carlock LR. Preprotachykinin and preproenkephalin mRNA expression within striatal subregions in response to altered serotonin transmission. Brain Res. 1996;732:25–35. doi: 10.1016/0006-8993(96)00483-0. [DOI] [PubMed] [Google Scholar]
- Wang JQ, McGinty JF. D1 and D2 receptor regulation of preproenkephalin and preprodynorphin mRNA in rat striatum following acute injection of amphetamine or methamphetamine. Synapse. 1996;22:114–122. doi: 10.1002/(SICI)1098-2396(199602)22:2<114::AID-SYN4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Warren BL, Iñiguez SD, Alcantara LF, Wright KN, Parise EM, Weakley SK, Bolaños-Guzmán CA. Juvenile administration of concomitant methylphenidate and fluoxetine alters behavioral reactivity to reward- and mood-related stimuli and disrupts ventral tegmental area gene expression in adulthood. J. Neurosci. 2011;31:10347–10358. doi: 10.1523/JNEUROSCI.1470-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxmonsky J. Assessment and treatment of attention deficit hyperactivity disorder in children with comorbid psychiatric illness. Curr. Opin. Pediatr. 2003;15:476–482. doi: 10.1097/00008480-200310000-00006. [DOI] [PubMed] [Google Scholar]
- Weikop P, Yoshitake T, Kehr J. Differential effects of adjunctive methylphenidate and citalopram on extracellular levels of serotonin, noradrenaline and dopamine in the rat brain. Eur. Neuropsychopharmacol. 2007;17:658–671. doi: 10.1016/j.euroneuro.2007.02.014. [DOI] [PubMed] [Google Scholar]
- Wilens TE, Adler LA, Adams J, Sgambati S, Rotrosen J, Sawtelle R, Utzinger L, Fusillo S. Misuse and diversion of stimulants prescribed for ADHD: a systematic review of the literature. J. Am. Acad. Child Adolesc. Psychiatry. 2008;47:21–31. doi: 10.1097/chi.0b013e31815a56f1. [DOI] [PubMed] [Google Scholar]
- Willuhn I, Sun W, Steiner H. Topography of cocaine-induced gene regulation in the rat striatum: Relationship to cortical inputs and role of behavioural context. Eur. J. Neurosci. 2003;17:1053–1066. doi: 10.1046/j.1460-9568.2003.02525.x. [DOI] [PubMed] [Google Scholar]
- Yano M, Steiner H. Topography of methylphenidate (Ritalin)-induced gene regulation in the striatum: differential effects on c-fos, substance P and opioid peptides. Neuropsychopharmacology. 2005a;30:901–915. doi: 10.1038/sj.npp.1300613. [DOI] [PubMed] [Google Scholar]
- Yano M, Steiner H. Methylphenidate (Ritalin) induces Homer 1a and zif 268 expression in specific corticostriatal circuits. Neuroscience. 2005b;132:855–865. doi: 10.1016/j.neuroscience.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Yano M, Steiner H. Methylphenidate and cocaine: the same effects on gene regulation? Trends Pharmacol. Sci. 2007;28:588–596. doi: 10.1016/j.tips.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Yano M, Beverley JA, Steiner H. Inhibition of methylphenidate-induced gene expression in the striatum by local blockade of D1 dopamine receptors: Interhemispheric effects. Neuroscience. 2006;140:699–709. doi: 10.1016/j.neuroscience.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nat. Rev. Neurosci. 2006;7:464–476. doi: 10.1038/nrn1919. [DOI] [PubMed] [Google Scholar]