

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the progressive loss of motor neurons, leading to muscle weakness, paralysis, and death, most often from respiratory failure. Over 200 pyrimidine-2,4,6-trione (PYT) small molecules, which prevent aggregation and reduce the associated toxicity of mutant superoxide dismutase 1 (SOD1) found in patients with familial ALS, have been synthesized and tested. One of the compounds (1,3-bis(2-phenylethyl)pyrimidine-2,4,6(1H,3H,5H)-trione, (1) was previously found to have an excellent combination of potency efficacy, and some desirable pharmacokinetic properties. To improve the solubility and metabolic stability properties of this compound, deuterium and fluorine were introduced into 1. New analogs with better solubility, plasma stability, and human microsome stability were identified.
Keywords: Pyrimidine-2,4,6-triones (PYT); amyotrophic lateral sclerosis (ALS); fluorination; deuteration; ADME; microsome stability; pharmacokinetics
Amyotrophic lateral sclerosis (ALS), an orphan disease, is estimated to afflict about 87,000 people worldwide, but its prevalence would be much higher were it not for the fact that ALS patients survive only 3 to 5 years, on average, after diagnosis.1 Riluzole, which decreases glutamate excitotoxicity,2 is the only FDA-approved therapeutic drug for ALS, but extends the median survival by only 2–3 months.3, 4
A cultured cell model, the PC12-G93A-YFP cell line, developed by Morimoto and coworkers,5 was utilized in a high throughput assay to identify compounds that protect against mutant SOD1-induced cytotoxicity. Two assays were established, a cytotoxicity protection assay, in which compounds were screened for their ability to protect cells from the cytotoxic effects of aggregated mutant SOD1, and a protein aggregation assay, in which compounds that are active in the cytotoxicity protection screen were tested for their ability to reduce the aggregation of mutant SOD1.6
Pyrimidine-2,4,6-triones (PYT) were identified among the active compounds and selected as one of the scaffolds for chemistry optimization.7 More than 200 PYT compounds were synthesized, and some general observations about the SAR were concluded. As shown in Figure 1, compound 1 from our previous work was identified as one of the best analogues, having good potency, low toxicity (maximum tolerated dose is 100 mg/kg), and good oral and brain absorption; however, the solubility, microsome stability, and plasma stability of this compound was not very satisfactory (see also Supporting Information SP-1 for details), and further improvement was needed.
Figure 1.

Pharmacokinetic properties of Compound 1
Considering the poor pharmacokinetic properties of 1, attempts to determine the major metabolites were performed (see Supporting Information SP-2 for details). As shown in Figure 2, two major mass spectral peaks were detected, corresponding to the incorporation of an oxygen atom (m/z = 353) and the loss of two hydrogen atoms (m/z = 335), suggesting hydroxylation of either the phenyl ring or the side chain and the oxidation of the side chain to the corresponding alkene; no other major metabolites were observed. The alkene metabolite could have come from the corresponding alcohol by elimination, which supports a metabolite with the hydroxyl group attached to the side chain. The same metabolic products were also observed with human microsomes. With this hypothesis, we directed our efforts at modification of 1 to decrease this potentially harmful metabolism and to increase its solubility.
Figure 2.

Metabolites of 1 in mouse microsomes
Deuteration of 1
One approach that is exploited to slow cytochrome P450-dependent drug metabolism is deuteration of the suspected site of C-H bond cleavage.8 The C-D bond has a lower zero point energy and, therefore, is stronger; if the rate-determining step involves C-H bond cleavage, then deuteration at that site should slow the rate of metabolism. The emergence of companies such as Concert Pharmaceuticals and Auspex Pharmaceuticals, which incorporate deuterium into existing drugs with poor metabolic stability, have established this strategy as a viable low-risk approach to drug development.
To protect both carbons on the chain of 1, several deuterated analogues were synthesized, as shown in Scheme 1. 2-Phenylethanamines, deuterated on either or both carbons, were synthesized from commercially available benzyl cyanide as HCl salts in excellent yields. Following well-established procedures,9 deuterated compounds 2, 3, and 4 were obtained in very high overall yields. As expected, the potencies of all of the compounds were very similar (within the error of the measurement).
Scheme 1.

Synthesis of Deuterated PYT Analogs
Unfortunately, 1 and the three deuterated PYT analogues had very similar solubilities, microsome stabilities, and plasma stabilities (Table 1; see Supporting Information SP-1 for details). There was no deuterium isotope effect on the metabolism of 1, suggesting that side chain C-H(D) cleavage is not a rate-determining step of metabolism.
Table 1.
| Potency (EC50) |
Solubility | Human Metabolic Potential (T1/2) |
Mouse Metabolic Potential (T1/2) |
Plasma Stability (Calc T1/2) |
|
|---|---|---|---|---|---|
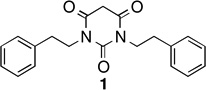 |
1.68 uM | 31.3 uM | 64 min | 16 min | 63.5 min |
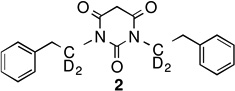 |
1.70 uM | 31.3 uM | 75 min | 16 min | 49.8 min |
 |
1.57 uM | 31.3 uM | 64 min | 15 min | 54.0 min |
 |
1.05 uM | 62.5 uM | 64 min | 14 min | 49.4 min |
Fluorination of 1
As deuteration was not effective in slowing metabolism, a more stable bond was sought. Fluorine substitution has been extensively investigated in drug research as a means of enhancing biological activity and increasing chemical or metabolic stability.10 Important characteristics of fluorine-containing compounds are: 1) the size of the fluorine atom compared to hydrogen; 2) the highly electron-withdrawing character of fluorine; 3) the greater stability of the C-F bond compared to the C-H bond; and 4) the solubility of fluorine-containing compounds.
Despite fluorine’s slightly larger van der Waals radius than hydrogen, several studies have demonstrated that it is a reasonable hydrogen mimic and is expected to cause minimal steric perturbations with respect to binding to a receptor or enzyme.11 Metabolic stability is important to bioavailability of compounds, and the C-F bond is stable to oxidative cleavage. Fluorine substitution also protects adjacent or distal sites from metabolism because of its strong electron-withdrawing properties. Fluorine also can reduce the basicity of compounds, which can result in better membrane permeation of the compound.
To improve the microsome stability of PYT compounds, fluorinated analogues of 1 were synthesized (Scheme 2). The fluorine atoms were introduced into the carbon chains of 1 with DAST followed by standard urea synthesis and PYT ring closing7. The fluorine atoms decreased the potency of the PYT analogues in comparison to 1 and 5. Further introduction of electron-withdrawing groups, such as F or Cl on the phenyl rings, also slightly decreased the potency of 5 compared with that of 6, 7, and 8. The added halogen substitution on the phenyl rings did not affect the potency.
Scheme 2.

Synthesis of new fluorinated PYT analogues
By blocking the potential metabolism site, these new analogues should have much longer metabolic half-lives. The fluorinated analogues of 1 exhibited similar potency as the parent compound (Table 2; see Supporting Information SP-3 for details). The pharmacokinetic properties of the fluorinated PYT compounds were much improved (compare 1 and 5). m-Fluorine substitution on the Ph ring of the parent compound lowered the potency and stability slightly (compare 1 and 97 or 5 and 8). Interestingly, fluorine substitution on the phenyl ring lowered solubility (compare 1 and 9), but fluorine substitution on the side chain enhanced solubility (compare 1 and 5); when fluorine was added to the phenyl rings of 5 to give 8, the solubility decreased relative to that of 5. Compounds 5 and 8 had much greater human microsome stability and plasma stability relative to 1, but the mouse microsome stability of 8 was less than that of 1.
Table 2.
| Potency (EC50) |
Solubility | Caco-2 (A->B) 10−6 cm s−1 |
Caco-2 (B->A) 10−6 cm s−1 |
Efflux ratio |
Human Metabolic Potential (T1/2) |
Mouse Metabolic Potential (T1/2) |
Plasma Stability (Calc T1/2) |
|
|---|---|---|---|---|---|---|---|---|
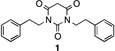 |
1.68 uM | 31.3 uM | 73.4 | 26.0 | 0.35 | 64 min | 16 min | 63.5 min |
 |
2.19 uM | >500 uM | 60.7 | 14.0 | 0.23 | >180 min | 43 min | >1000 min |
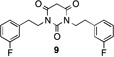 |
3.23 uM | 31.3 uM | 66.3 | 17.1 | 0.26 | 31 min | 22 min | 131.2 min |
 |
2.54 uM | 250 uM | 51.7 | 10.5 | 0.20 | > 180 min | 20 min | >1000 min |
Modification of 1 was carried out to improve the pharmacokinetic properties of the PYT analogues. Although deuterium incorporation did not affect the rate of metabolism, fluorination on the carbon chain greatly improved the solubility, human microsome stability, and plasma stability of the PYT analogues. Possibly, C-H bond cleavage of the side chain is not rate determining, leading to no effect by deuterium, but is an important site of metabolism. Fluorination on the Ph ring, however, decreased both the metabolic stability and the potency.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health [1R43NS057849], the ALS Association (TREAT program), the Department of Defense [AL093052], and the ALS Therapy Alliance for their generous support of the research project.
ABBREVIATIONS
- ADME
absorption, distribution, metabolism, excretion
- ALS
amyotrophic lateral sclerosis
- PYT
pyrimidine 2,4,6-trione
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Brown RWJ. In: Harrison's Principles of Internal Medicine. Fauchi AS, Braunwald E, Isselbacher KJ, WIlson JD, Martin JB, Kasper DL, Hauser SL, Longo DL, editors. New York: McGraw-Hill; 1998. p. 2368. [Google Scholar]
- 2.Jimonet P, Audiau F, Barreau M, Blanchard J-C, Boireau A, Bour Y, Coléno M-A, Doble A, Doerflinger G, Do Huu C, Donat M-H, Duchesne JM, Ganil P, Guérémy C, Honoré E, Just B, Kerphirique R, Gontier S, Hubert P, Laduron PM, Le Blevec J, Meunier M, Miquet J-M, Nemecek C, Pasquet M, Piot O, Pratt J, Rataud J, Reibaud M, Stutzmann J-M, Mignani S. J. Med. Chem. 1999;42:2828. doi: 10.1021/jm980202u. [DOI] [PubMed] [Google Scholar]
- 3.Bensimon G, Lacomblez L, Meininger V. N. Engl. J. Med. 1994;330:585. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- 4.Francis K, Bach JR, DeLisa JA. Arch. Phys. Med. Rehabil. 1999;80:951. doi: 10.1016/s0003-9993(99)90089-8. [DOI] [PubMed] [Google Scholar]
- 5.Morimoto RI. Genes. Dev. . 2008;22(11):1427. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benmohamed R, Arvanites AC, Kim J, Ferrante RJ, Silverman RB, Morimoto RI, Kirsch DR. Amyotroph. Lat. Scler. 2010;12:87. doi: 10.3109/17482968.2010.522586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia G, Benmohamed R, Kim J, Arvanites AC, Morimoto RI, Ferrante RJ, Kirsch DR, Silverman RB. J. Med. Chem. 2011;54:2409. doi: 10.1021/jm101549k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shao L, Hewitt MC. Drug News Perspect. 2010;23:398. doi: 10.1358/dnp.2010.23.6.1426638. [DOI] [PubMed] [Google Scholar]
- 9.Messe OC, Ebner T. J. Labelled. Comp. Rad. 1988;25:335. [Google Scholar]
- 10.Shah P, Westwell AD. J. Enz. Inhib. Med. Chem. 2007;22:527. doi: 10.1080/14756360701425014. [DOI] [PubMed] [Google Scholar]
- 11.Park BK, Kitteringham NR, O’Neil PM. Annu. Rev. Pharmacol. Toxicol. 2001;41:443. doi: 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.