

Abstract
Synthesis and SAR investigation of 2-guanidinoquinazolines, initially identified in a high content screen for selective STAT3 pathway inhibitors, led to a more potent analog (11c) that demonstrated improved anti-proliferative activity against a panel of HNSCC cell lines.
Keywords: STAT3 pathway, Guanidinoquinazolines, Skraup synthesis, Structure–activity relationships, Cancer cell line screening
Despite recent advances in early detection methods and treatment regimens, cancer continues to be a major health threat, responsible for over 25% of deaths annually in the U.S. alone.1 Head and neck squamous cell carcinomas (HNSCC) are particularly challenging therapeutic targets2 as evidenced by the fact that the monoclonal antibody Cetuximab (Erbitux), an epidermal growth factor receptor (EGFR) inhibitor,3 was the only new drug approved for this indication in the last several decades. To address this issue, a wide range of signaling pathways that control cell proliferation have been interrogated as potential therapeutic strategies for HNSCC, including the family of signal transducers and activators of transcription (STATs).4-6 STAT3 is a tumor promoting transcription factor that has been shown to be constitutively activated in numerous cancers, and suppression of STAT3 leads to inhibition of tumor growth in both in vitro and in vivo experiments. In contrast, the related transcription factor, STAT1, activates genes that promote tumor suppression. Therefore, molecules that selectively inhibit STAT3-mediated pathways with no effect on STAT1 pathways, have the potential to be highly effective anti-tumor agents.
Several small organic molecules that inhibit the STAT3 pathway have been reported in the literature.7 One strategy has been to design molecules that directly target the Src homology 2 (SH2) domain in STAT3 (1–4, Fig. 1).8 Other approaches include focusing on inhibiting kinases operative in the STAT3 pathway, such as Janus activated kinases (JAKs), and identified quinolones, pyridones, and the pyridine carboxamide, sorafenib (5, 6 and 7, respectively, Fig. 1).9 Additionally, natural products, including STA-21 (8), curcumin (9), and cucurbitacin Q (10), inhibit the STAT3 pathway; however, specific inhibitory mechanisms are still being elucidated (Fig. 2).5b Finally, anti-sense oligonucleotides (AZD9150) and decoy nucleotides directed at STAT3 also exhibit promising anti-proliferative activities in cellular assays.5,10
Figure 1.

SH2 targeted phosphopeptide mimetics and JAK inhibitors of the STAT3 pathway.
Figure 2.
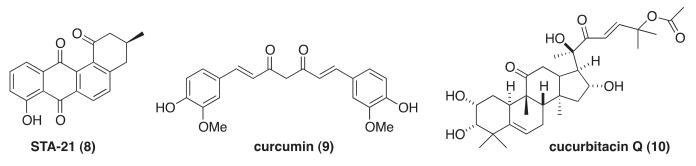
Natural product STAT3 inhibitors.
By using a high content phenotypic screen (HCS) to identify selective inhibitors of IL-6 induced activation of the STAT3 pathway,11 we identified the quinazoline 11a (Fig. 3). In Cal33 head and neck tumor cells, 11a inhibited IL-6-induced STAT3 tyrosine phosphorylation and nuclear translocation (IC50 = 15.7 μM), but had no effect on IFNγ-induced activation of the STAT1 pathway at 50 μM (Fig. 3). Western blot analysis indicated a 69% decrease in phospho-STAT3 (pSTAT3) levels upon treatment of 11a at 39.6 μM concentration (Fig. 4, A and B). Unlike the JAK inhibitor 6 that displayed nanomolar potencies against both STAT3 and STAT1 (data not shown),11 compound 11a selectively inhibited STAT3 compared to STAT1 and displayed no effects on JAK1/JAK2 as determined by Western blot analysis (Figs. 3 and 4, panels C and D). In addition, 11a exhibited anti-proliferative activities (IC50’s = 17-37 lM in four HNSCC cell lines (CAL33, FADU, 686 LN, OSC19, Fig. 3). Examination of the literature and PubChem revealed limited examples of biological effects for this chemotype, and Lipinski and Veber parameters fell into the generally desirable ranges (Fig. 3).12-15 While the specific mode of action of 11a was not determined, its apparent lack of activity in the STAT1 assay likely rules out direct binding to SH2 domains. Furthermore, this hit compound did not exhibit any significant activity against a panel of >80 kinases (data not shown). The promising selectivity for STAT3, the notable anti-proliferative activity and desirable physical properties made this compound an attractive lead structure for further medicinal chemistry optimization, and herein we report the results of these efforts.
Figure 3.

Guanidinoquinazoline hit 11a.
Figure 4.

Inhibition of STAT3 phosphorylation using Western blot analysis of compound 11a versus vehicle in interleukin 6 (IL6, 50 ng/mL)-stimulated CAL33 cells (A & B). Compound 11a did not show any effects on pJAK1/JAK1 (C) or pJAK2/JAK2 (D).
Our initial strategy was to incorporate modest structural modifications onto the 2-guanidinoquinazoline core in order to establish preliminary structure-activity relationships. Using established synthetic procedures,16 the dihydroquinolines 13 were generated through the treatment of the substituted anilines 12 with acetone under modified Skraup conditions (Scheme 1).17 Conversion to the guanidines 14 occurred by reaction with cyano-guanidine under aqueous acidic conditions.18 The final products, dihydropyrimidinyl-aminoquinazolines 11a-d, were formed via thermal cyclodehydrations using mesityl oxide in DMSO. The structure of 11b was confirmed by X-ray analysis (Scheme 1).16 In this subset of analogs (Table 1), it was apparent that structural modification was tolerated and modulated the biological profile; the C6-methyl (11b) and C6-,C8-dimethyl (11c) analogs exhibited improved potency (4- and 30-fold, respectively) while maintaining selectivity versus STAT1 compared to the original hit 11a. Unlike 11a, 11b and 11d, which failed to achieve ≥50% inhibition of IFNγ-induced STAT1 activation at 50 μM, 11c exhibited an IC50 of 5.9 μM for STAT1 but still maintained a good selectivity index (Table 1).
Scheme 1.

Preparation of 2-guanidinoquinazolines 11a–d and X-ray structure of 11b (CCDC 1020633).
Table 1.
STAT3 and STAT1 Activity of 2-guanidinoquinazolines 11a–d
| Compd# | R1 | R2 | R3 | STAT3 IC5011 (μM) |
STAT1 IC5011 (μM) |
SIa (STAT1/STAT3) |
|---|---|---|---|---|---|---|
| 11a | OEt | H | H | 15.7 ± 5.4 | >50 | >3 |
| 11b | Me | H | H | 7.9 ± 2.8 | >50 | >6 |
| 11c | Me | H | Me | 0.8 ± 0.6 | 5.9 ± 0.9 | >7 |
| 11d | H | OEt | H | 20.7 ± 3.0 | >50 | >2 |
Selectivity index ratio.
Efforts to examine a simplified pharmacophore focused on the preparation of 2-aminoquinazolines of general structure 17 (Scheme 2). The pivotal 2-chloro intermediate 16 was prepared in three steps19 and subjected to amination under microwave conditions to provide the corresponding quinazoline derivatives 17a–i. All of these compounds were devoid of activity and therefore established the importance of quinazoline substitutions as well as the C2-linked nitrogenous heterocycle (Table 2).11
Scheme 2.
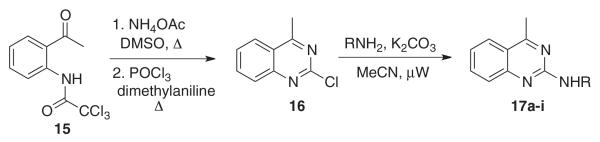
Preparation of 2-aminoquinazolines (17a–j).
Table 2.
Inhibitory potencies of 2-aminoquinazolines (17a–j)
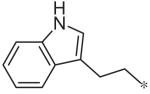
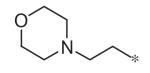
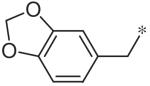
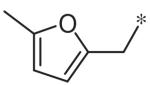
To investigate substituent effects on the pyrimidine, we also synthesized pyrimidinones of general structure 19. The reaction of guanidines 14 with substituted acetoacetic esters (Scheme 3)20,21 afforded the dihydropyrimidones 19a–t. Representative compounds in this series and their activities are shown in Table 3. Several substitutions (e.g., entries 19a, b, g, h) slightly improved potency and selectivity compared to the original hit (11a), whereas other substitution patterns (e.g., 19e, o, p) completely abolished activity. With the exception of 19p, all 5,6-dimethylpyrimidinones (19b, 19f, 19l, 19n) demonstrated inhibition of pSTAT3 at less than 10 lM concentration and maintained at least a 3-fold selectivity against pSTAT1. All new compounds were fully characterized, and all tested compounds had LC/MS/ELSD purities exceeding 91%.
Scheme 3.
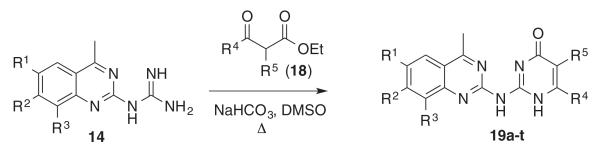
Preparation of 2-aminoquinozoline dihydropyrimidones (19a–t).
Table 3.
Inhibitory potencies of 2-amino-quinazolinyl-dihydropyridimidones 19a–t
| Compd# | R1 | R2 | R3 | R4 | R5 | STAT3 IC5011 (μM) | STAT1 IC5110 (μM) | SIb (STAT1/STAT3) |
|---|---|---|---|---|---|---|---|---|
| 19a | Me | H | Me | Me | H | 5.0 ± 1.8 | 12.1 ± 0.9 | >2 |
| 19b | Me | H | Me | Me | Me | 6.4 ± 1.6 | >50 | >7 |
| 19c | Me | H | Me | Me | Allyl | 14.3 ± 0.9 | >50 | >3 |
| 19d | Me | H | Me | Ph | H | 11.2 ± 2.1 | >50 | >4 |
| 19e | OEt | H | H | Me | H | >50 | >50 | – |
| 19f | OEt | H | H | Me | Me | 11.7 ± 5.7 | >50 | >4 |
| 19g | OEt | H | H | Ph | H | 3.4 ± 0.6 | >50 | >14 |
| 19h | OEt | H | H | Me | Bn | 2.7 ± 2.5 | >50 | >18 |
| 19i | OMe | H | H | Me | H | 34.9 ± 2.7 | >50 | >1 |
| 19j | OMe | H | H | Me | Bn | 34.7a | 50 | >1 |
| 19k | H | Me | H | Me | H | 19.1 ± 1.3 | 39.7 ± 1.6 | >1 |
| 19l | H | Me | H | Me | Me | 10.5 ± 0.2 | 35.2 ± 1.1 | >3 |
| 19m | Me | Me | H | Me | H | 24.7 ± 0.5 | >50 | >2 |
| 19n | Me | Me | H | Me | Me | 10.4 ± 1.3 | >50 | >4 |
| 19o | −OCH2O− | H | Me | H | >50 | >50 | – | |
| 19p | −OCH2O− | H | Me | Me | >50 | >50 | – | |
| 19q | −OCH2O− | H | Ph | H | 13.0 ± 1.4 | >50 | >3 | |
| 19r | −OCH2O− | H | Me | Bn | >50 | >50 | – | |
| 19t | H | OMe | H | Me | H | 33.9 ± 1.8 | >50 | >1 |
Results from a single experiment.
Selectivity index ratio.
The ability of the most potent analogs to inhibit the proliferation of a panel of HNSCC cell lines is shown in Table 4. While a strict correlation between STAT3 inhibition and cell growth inhibition potency was not evident, compound 11c, which exhibited the most potent activity in our STAT3 assays, also displayed the most potent anti-proliferative effects.
Table 4.
Growth inhibition of selected analogs on HNSCC cell lines 686LN, CAL33, FADU and OSC19
| Compd# | 686LN IC50 (μM) |
CAL33 IC50 (μM) |
FADU IC50 (μM) |
OSC19 IC50 (μM) |
|---|---|---|---|---|
| 11a | 37.5 ± 11.1 | 29.4 ± 5.9 | 18.8 ± 5.3 | 17.0 ± 4.6 |
| 11b | 49.3a | 19.7 ± 0.5 | 11.0 ± 0.1 | 12.8 ± 0.9 |
| 11c | 2.4 ± 0.6 | 1.2 ± 0.3 | 1.9 ± 0.1 | 3.2 ± 0.4 |
| 19a | 18.0 ± 10.4 | 12.9 ± 0.9 | 30.9 ± 14.9 | 11.2a |
| 19g | 37.8a | 11.1a | 17.9a | 19.0a |
| 19h | 43.7 ± 0.7 | >50 | 27.4 ± 13.3 | >50 |
| 6 | >5 | 1.3 ± 0.4 | 2.4 ± 0.6 | 7.7 ± 4.7 |
Results from a single experiment.
In summary, 6-ethoxy-4-methylquinazoline-2-amino-dihydro-trimethylpyrimidine 11a, a structurally novel, selective inhibitor of STAT3-mediated signaling, was further optimized through three rounds of SAR studies. Modest structural changes established the preferred quinazoline substitutions. Most notably, a 20-fold improvement in STAT3 inhibition, while maintaining selectivity over STAT1, was obtained with the 6,8-dimethyl substituted analog 11c. More substantial changes to the dihydropyrimidine moiety led to complete loss of activity; however, substitution of the quinazoline with dihydropyrimidinones also retained potency and selectivity. The most promising lead structures, 11b, 11c, and 19a exhibited low-micromolar potency against STAT3, at least a 2-fold selectivity over STAT1, and anti-proliferative activity in the 1–50 μM range. Additional SAR iterations will be needed to increase the activity of these compounds to the sub-micromolar level; however, 11b, 11c, and 19a represent attractive tool compounds to investigate a kinase- and STAT1-independent downregulation of the STAT3 pathway. Studies on the mechanism of action of this class of compounds as well as the biological data and medicinal chemistry of other STAT3 HCS hits will be reported in due course.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Steven J. Geib for the X-ray analysis of 11b, Mr. Pete Chambers for LCMS/ELS analyses, Dr. Tong Ying Shun for data processing, and Ms. Mary Liang and Ms. Shelby Anderson for sample handling, procurement, registration, and distribution. We also thank Drs. Mark Schurdak and D. Lansing Taylor (University of Pittsburgh Specialized Applications Center), Drs. Bill Moore (Leidos), Beverly Teicher (NCI) and Shizuko Sei (Leidos) for their helpful discussions. This project was funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Chemical Biology Consortium Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2014.09.001.
References and notes
- 1.Siegel R, Naishadham D, Jemal A. Cancer Stat. 2012;62:10. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Song JI, Grandis JR. Oncogene. 2000;19:2489. doi: 10.1038/sj.onc.1203483. [DOI] [PubMed] [Google Scholar]
- 3.Specenier P, Vermorken JB. Biol. Targets Ther. 2013;7:77. doi: 10.2147/BTT.S43628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. Cell. 2011;144:646. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.For recent reviews of STAT3 and inhibitors, see: Peyser ND, Grandis JR. Onco Targets Ther. 2013;6:999. doi: 10.2147/OTT.S47903. Lin L, Hutzen B, Zuo M, Ball S, Deangelis S, Foust E, Pandit B, Ihnat MA, Shenoy SS, Kulp S, Li P-K, Li C, Fuchs J, Lin J. Cancer Res. 2010;70:2445. doi: 10.1158/0008-5472.CAN-09-2468.
- 6.(a) Masciocchi D, Gelain A, Villa S, Meneghetti F, Barlocco D. Future Med. Chem. 2011;3:567. doi: 10.4155/fmc.11.22. [DOI] [PubMed] [Google Scholar]; (b) Walker SR, Frank DA. JAK-STAT. 2012;1:292. doi: 10.4161/jkst.22662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For recent reviews, see: Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP, Tan BKH, Sethi G, Bishayee A. Biochim. Biophys. Acta. 1845;2014:136. doi: 10.1016/j.bbcan.2013.12.005. Wang X, Crowe PJ, Goldstein D, Yang J-L. Int. J. Oncol. 2012;41:1181. doi: 10.3892/ijo.2012.1568.
- 8.(a) Mandal PK, Gao F, Lu Z, Ren Z, Ramesh R, Birtwistle JS, Kaluarachchi KK, Chen X, Bast RC, Jr., Liao W, McMurray JS. J. Med. Chem. 2011;54:3549. doi: 10.1021/jm2000882. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen J, Bai L, Bernard D, Nikolovska-Coleska Z, Gomez C, Zhang J, Yi H, Wang S. ACS Med. Chem. Lett. 2010;1:85. doi: 10.1021/ml100010j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Haftchenary S, Luchman HA, Jouk AO, Veloso AJ, Page BDG, Cheng XR, Dawson SS, Grinshtein N, Shahani VM, Kerman K, Kaplan DR, Griffin C, Aman AM, Al-awar R, Weiss S, Gunning PT. ACS Med. Chem. Lett. 2013;4:1102. doi: 10.1021/ml4003138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Pedranzini L, Dechow T, Berishaj M, Comenzo R, Zhou P, Azare J, Bornmann W, Bromberg J. Cancer Res. 2006;66:9714. doi: 10.1158/0008-5472.CAN-05-4280. [DOI] [PubMed] [Google Scholar]; (b) Swiatek-Machado K, Mieczkowski J, Ellert-Miklaszewska A, Swierk P, Fokt I, Szymanski S, Skora S, Szeja W, Grynkiewicz G, Lesyng B, Priebe W, Kaminska B. Canc. Biol. Ther. 2012;13:657. doi: 10.4161/cbt.20083. [DOI] [PubMed] [Google Scholar]; (c) Yang F, Jove V, Buettner R, Xin H, Wu J, Wang Y, Nam S, Xu Y, Ara T, DeClerck YA, Seeger R, Yu H, Jove R. Cancer Biol. Ther. 2012;13:534. doi: 10.4161/cbt.19603. [DOI] [PubMed] [Google Scholar]
- 10.(a) Hayakawa F, Sugimoto K, Harada Y, Hashimoto N, Ohi N, Naoe T. Blood Cancer J. 2013;3:e166. doi: 10.1038/bcj.2013.63. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim M-J, Nam H-J, Kim H-P, Han S-W, Im S-A, Kim T-Y, Oh D-Y, Bang Y-J. Cancer Lett. 2013;335:145. doi: 10.1016/j.canlet.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 11.Johnston PA, Sen M, Hua Y, Camarco D, Shun TY, Lazo JS, Grandis JR. Assay Drug Dev. Technol. 2014;12:55. doi: 10.1089/adt.2013.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Holloway GA, Charman WN, Fairlamb AJ, Brun R, Kaiser M, Kostewicz E, Novellow PM, Parisot JP, Richardson J, Street IP, Watson KG, Baell JB. Antimicrob. Agents Chemother. 2009;53:2824. doi: 10.1128/AAC.01568-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ciustea M, Silverman JEY, Shudofsky AMD, Ricciardi RP. J. Med. Chem. 2008;51:6563. doi: 10.1021/jm800366g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Webb TR, Lvovskiy D, Kim S-A, Ji X-D, Melman N, Linden J, Jacobson KA. Bioorg. Med. Chem. 2003;11:77. doi: 10.1016/s0968-0896(02)00323-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pubchem (http://pubchem.ncbi.nlm.nih.gov) activity for 11a indicates 21 active assays (6-confirmatory) in 666 bioassays tested: Down Regulation Insulin Promoter (AID 1628); Cytotoxicity MIN6 cells (AID 2122); Cruzain Inhibition (AIDs 1476, 1478); FEN1 Endonuclease inhibition (AIDs 588795, 720498).
- 14.(a) Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv. Drug Delivery Rev. 1997;23:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]; (b) Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. J. Med. Chem. 2002;45:2615. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 15.Physicochemical Properties were Calculated Using: Maestro 8.5.207-QikProp 2.1. Schrödinger, LLC; New York, NY, USA: [Google Scholar]
- 16.Shikhaliev KS, Falaleev AV, Ermolova GI, Solov’ev AS. Chem. Heterocycl. Compd. 2002;38:210. [Google Scholar]
- 17.Fotie J, Kaiser M, Delfin DA, Manley J, Reid CS, Paris J-M, Wenzler T, Maes L, Mahasenan KV, Li C, Werbovetz KA. J. Med. Chem. 2010;53:966. doi: 10.1021/jm900723w. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 18.Brown JP. J. Chem. Soc. 1964:3012. [Google Scholar]
- 19.(a) Arienzo R, Cramp S, Dyke HJ, Lockey PM, Norman D, Roach AG, Smith P, Wong M, Wren SP. Bioorg. Med. Chem. Lett. 2007;17:1403. doi: 10.1016/j.bmcl.2006.11.092. [DOI] [PubMed] [Google Scholar]; (b) Armarego WLF, Smith JIC. J. Chem. Soc. 1966:234. [Google Scholar]
- 20.Shikhaliev KS, Falaleev AV, Krylsky DV, Solov’ev AS, Shatalov GV. Chem. Heterocycl. Compd. 2002;38:1368. [Google Scholar]
- 21.Guiles J, Sun X, Critchley IA, Ochsner U, Tregay M, Stone K, Bertino J, Green L, Sabin R, Dean F, Dallmann HG, McHenry CS, Janjic N. Bioorg. Med. Chem. Lett. 2009;19:800. doi: 10.1016/j.bmcl.2008.12.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.