

Abstract
Progesterone receptors (PRs) are phosphorylated on multiple sites, and a variety of roles for phosphorylation have been suggested by cell-based studies. Previous studies using PR-null mice have shown that PR plays an important role in female fertility, regulation of uterine growth, the uterine decidualization response, and proliferation as well as ductal side-branching and alveologenesis in the mammary gland. To study the role of PR phosphorylation in vivo, a mouse was engineered with homozygous replacement of PR with a PR serine-to-alanine mutation at amino acid 191. No overt phenotypes were observed in the mammary glands or uteri of PR S191A treated with progesterone (P4). In contrast, although PR S191A mice were fertile, litters were 19% smaller than wild type and the estrous cycle was lengthened slightly. Moreover, P4-dependent gene regulation in primary mammary epithelial cells (MECs) was altered in a gene-selective manner. MECs derived from wild type and PR S191A mice were grown in a three-dimensional culture. Both formed acinar structures that were morphologically similar, and proliferation was stimulated equally by P4. However, P4 induction of receptor activator of nuclear factor-κB ligand and calcitonin was selectively reduced in S191A cultures. These differences were confirmed in freshly isolated MECs. Chromatin immunoprecipitation analysis showed that the binding of S191A PR to some of the receptor activator of nuclear factor-κB ligand enhancers and a calcitonin enhancer was substantially reduced. Thus, the elimination of a single phosphorylation site is sufficient to modulate PR activity in vivo. PR contains many phosphorylation sites, and the coordinate regulation of multiple sites is a potential mechanism for selective modulation of PR function.
Phosphorylation regulates diverse functions of proteins, including steroid receptors, either as a result of changes in conformation or a charge of the protein, both of which can alter activity and/or protein-protein interactions; phosphorylation also serves as a signal for other protein posttranslational modifications. Steroid receptors are hormone-activated transcription factors; thus, the role of phosphorylation is thought to be more modulatory than for some other transcription factors whose activities are regulated primarily by posttranslational modification. We have identified more than 10 phosphorylation sites in the human progesterone receptor (PR) (1, 2), and numerous sites have been identified in other steroid receptors (3). Most of the phosphorylation sites in PRs are serine (Ser) residues in the amino-terminal domain (NTD). Studies seeking to assess the role of specific phosphorylation sites have relied on functional analyses of receptors that contain an alanine (Ala) substitution to prevent phosphorylation. The wild type (WT) or mutant receptors are ectopically expressed in cell lines that typically lack expression of the endogenous receptor. Because most cells used for this purpose are transformed immortalized cells or cancer cells, they may well lack cell-specific factors that play a role in tissue-specific activities. Despite these experimental limitations, these kinds of studies have shown that specific phosphorylation events can alter the nuclear translocation, protein stability, DNA binding, and gene-specific transcriptional activity (3, 4).
Only a few studies have sought to identify the role of phosphorylation of any transcription factor or transcriptional coactivator in vivo under more physiological conditions by selectively mutating one or more phosphorylation sites in a mouse model. For example, homozygous substitution of Ala for two threonine (Thr) phosphorylation sites, T51 and T53, in mouse activating transcription factor-2 resulted in pups that died shortly after birth (5). No such studies have been reported for steroid receptors. However, a coactivator knock-in mouse was developed that contains Ala substitutions for four Ser/Thr phosphorylation sites in steroid receptor coactivator-3 (6). The steroid receptor coactivator-3 mutant mouse exhibited an increase in body weight, altered peripheral insulin sensitivity, increased IGFBP-3 expression, and increased IGF-1 signaling.
The human PR is expressed as two protein isoforms, PR-A and PR-B, which are derived from alternate promoters of a single gene (7). PR-A is identical to PR-B except that it lacks the first 164 amino acids in the N-terminal domain. Mouse PR is homologous to human PR, although the lengths of the receptors differ slightly (933 for human and 923 for mouse, with the start of PR-A at amino acid 166). The phenotype of the PR-null knockout female mice (PRKO) has shown that PR is required for fertility as well as for development and differentiation of the uterus and mammary gland. Mice with PR isoform-specific deletions have also been constructed and their phenotypes demonstrate that PR-A plays a more important role in the uterus and ovary, whereas PR-B is the predominant functional isoform in the mammary gland (8, 9). To evaluate the role of site-specific PR phosphorylation in vivo, a PR knock-in mouse was created in which Ala was substituted for Ser 191, the site that corresponds to human PR serine 190. We report a modest but detectable phenotype of PR S191A mice for a subset of progesterone (P4)-dependent responses including subfertility, an altered length of estrous cycles, and impaired P4-regulation of selected PR target genes in the mammary epithelium.
Materials and Methods
Animals and hormone treatments
The targeting vector was constructed by first performing PCR oligonucleotide directed mutagenesis to change the mouse PR Ser191 phosphorylation site from a serine to alanine (Supplemental Figure 1). The construct contained the 129SV mouse PR genomic locus corresponding to exons 1 and 2, with a neomycin resistance cassette (PGK-NEO) flanked by FRT sites within intron 1 as well as a herpes simplex virus thymidine kinase region upstream of the PR locus. We also engineered a new NheI restriction site within exon 1, which does not alter the amino acid coding sequence, to facilitate Southern blot screening of recombinant embryonic stem (ES) cells. The targeting plasmid was electroporated into R1 ES cells and selected for resistance to geneticin (G418) and to 1-(2′-deoxy-2′-fluoro-D-arabinofuranosyl)-5-iodouracil to ensure homologous recombination, eliminating the thymidine kinase sequence. The recombinant clones were confirmed by Southern blot analysis and six independent clones of the ES cells were injected into the inner cell mass of blastocysts from C57BL/6 mice and implanted into the uteri of recipient pseudopregnant ICR mice. The 129/C57BL chimeric mice were readily identified by the agouti/black coat color and male offspring containing greater than 60% agouti (contributed by the ES cells) were mated to C57BL/6 females.
We maintained two distinct lines of mutant mice for our studies. Offspring containing agouti coat color, as an indication of germline transmission, were then bred to the Gt(ROSA)26Sortm1(FLP1)Dym mice (The Jackson Lab) to remove the neo cassette. Mice containing 50% 129SV and 50% C57BL/6 were designated as the 129/C57BL mixed background. These mice were also backcrossed to pure C57BL/6 mice for a total of 5 (N5) generations to obtain a 97% C57BL/6 pure line. The genotype of the PR S191A mice was determined by PCR amplification with the forward primer 5′-AGCCTGCGAGGCCATCACTT-3′ and the reverse primer 5′-AGTGAGCACTCCCCGAGGTAGG-3′ to produce a 218-bp band. Restriction digestion with Nhe1 generates 169- and 49-bp bands specific for the PR S191A allele, whereas the WT allele is uncut. The PRKO allele was detected by PCR genotyping. Mice were maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and experiments were performed as described in a protocol approved by the Baylor College of Medicine Institutional Animal Care and Use Committee.
The artificial decidual response has been described previously (30). Briefly, 6-week mice were ovariectomized (OVX) and rested for 2 weeks prior to treatment with three daily injections of 100 ng estradiol (E2) per mouse. After 2 days of rest, mice were then treated with daily sc injections of 6.7 ng E2 and 1 mg P4 per mouse for 3 days. One uterine horn was traumatized with a burred needle 6 hours after the last injection. The contralateral horn was not traumatized and served as a control. Mice were given daily sc injections of 1 mg P4 and 6.7 ng E2 for 5 additional days and then killed as described above. Uterine tissues were placed in 4% paraformaldehyde or flash frozen.
For uterine PR target gene induction studies, mice were OVX at 6 weeks, rested for 2 weeks, and then treated with sesame oil or 1 mg P4 by sc injection for 6 hours. Mice were killed and tissues harvested as described above. Fertility studies were carried out over a period of 6 months. Briefly, WT males were placed in a breeding cage with three 6-week-old females, either two WT and one PR S191A or one WT and two PR S191A. Initial matings were verified by the presence of a vaginal plug. Females were transferred to a new cage prior to birth and immediately returned to the breeding cage after birth. Pups were killed by deep anesthesia under carbon dioxide and then decapitated. The cyclicity study was carried out over a period of 60 days, beginning on the day of the vaginal opening. Vaginal cells were collected each morning by pipetting saline onto the surface of the vagina. Cells were placed onto a microscope slide, allowed to dry, stained with 0.5% crystal violet in 25% methanol for 30 seconds, and washed in distilled water. Smears were examined under a light microscope and classified into one of four phases of the estrous cycle (proestrus, estrus, metestrus, or diestrus) as described elsewhere (31).
Hormone treatments for mammary gland studies
To observe the mammary gland response to chronic hormone treatment studies, adult (9–12 wk), intact females were implanted sc with 40 mg beeswax pellets containing 20 μg of E2 and 20 mg P4. The pellets were replaced after 2 weeks for a total treatment period of 4 weeks. For mammary gland response to acute treatment, young female mice (6 wk) were OVX, rested for 2 weeks, and then treated for 76 hours with daily sc injections of estrogen and progesterone (EP) [10 μg E2 (Sigma-Aldrich) + 1 mg P4 (Sigma-Aldrich)] dissolved in sesame oil or oil alone. Mice were anesthetized with Avertin (2,2-tribromoethyl alcohol; Sigma-Aldrich) and then sacrificed by cervical dislocation. Mammary tissues were prepared for whole mount as described below.
For isolation of P4-treated mammary epithelial cells (MECs), 23-week-old, ovary-intact, nulliparous WT or S191A mice were treated for 76 hours with three interscapular sc injections of progesterone (1 mg) in 100 μL of sesame oil (Sigma-Aldrich). Two hours before the animals were killed, they were injected ip with bromodeoxyuridine (BrdU; 0.03 mg/g of body weight; Sigma-Aldrich). All mammary glands (four mice per genotype) were removed and pooled to prepare primary MECs, as described below, to isolate total RNA. A small piece of each number 3 thoracic mammary gland was also fixed in 10% neutral buffered formalin for 2 hours at 4°C. After embedding in paraffin, the tissues were sectioned (3–4 μm) onto Plus slides (Fisher Scientific).
Immunoblot assays
Uterine tissues were homogenized using a Polytron 2000 homogenizer in 10 mM Tris HCl (pH 7.5), 300 mM NaCl, 0.125% Nonidet P-40, 1 mM Na3VO4, 20 mM NaF, 10 mM Na molybdate, and protease inhibitor tablets (Roche Diagnostics). Samples were kept on ice for 30 minutes, and cellular debris was removed by centrifugation at 14 000 rpm for 10 minutes at 4°C. Protein concentration was determined by Bradford's method using BSA as the standard. Samples containing 20 ug of protein for PR or 60 μg protein for phospho-PR Ser190 were subjected to sodium dodecyl sulfate 7.5% PAGE. The separated proteins were then transferred onto a nitrocellulose membrane (PerkinElmer). Membranes were blocked with 5% (wt/vol) nonfat dry milk (Bio-Rad Laboratories) in Tris-buffered saline with 10 mM Tris-HCL, 150 mM NaCl, 0.1% Tween 20 (pH 7.5). Total PR was detected with mouse anti-hPRa7 (0.5 μg/ml; Lab Vision/Neomarkers), and PR phospho-191 was detected with mouse antiphospho-PR Ser190 [10 μg/mL (11)], which cross-reacts with the phosphorylated mouse PR Ser191 (Figure 1B). The rabbit antimouse IgG secondary antibody (1:5000; Zymed) and the antirabbit horseradish peroxidase (1:5000; GE Healthcare) were used, followed by chemiluminescence with the ECL Plus Western blot detection system (GE Healthcare) according to the manufacturer's protocol to detect the signal.
Figure 1.

Conserved phosphorylation sites in human and mouse PR. A, A comparison of human and mouse sequences surrounding known phosphorylation sites in human PR. Identity between amino acids shown by bars. B, HEK 293T cells were transduced with human or mouse hemagglutinin (HA)-PR-B lentivirus (WT or phosphomutant) and treated for 2 hours with 0.01% ethanol or 10 nM R5020 before collecting whole-cell extracts. Total PR was immunoprecipitated using an HA polyclonal antibody, run on an SDS-PAGE gel and total PR (HA antibody) and P-Ser191 (Ser190 PR antibody) detected by Western analysis. IP, immunoprecipitation; WB, Western blot.
Whole-mount staining
The number 4 inguinal mouse mammary glands were stretched onto a microscope slide and allowed to dry for 5–10 minutes. Glands were fixed overnight at room temperature in 3:1 ethanol/glacial acetic acid. The following day, they were washed in 70% ethanol and then water, followed by Carmine Red staining overnight at room temperature (0.2% Carmine Red, 0.5% aluminum potassium sulfate; Sigma-Aldrich). Glands were then dehydrated in a graded series of ethanol, cleared in toluene until the fat pads were clear, and then stored in methyl salicylate until mounted in Permount (Fisher Scientific) for microscopy.
Isolation of MECs for three-dimensional primary cultures
Mammary glands from WT or S191A C57Bl/6 mice were isolated to prepare primary MEC cultures, as previously described (18). Glands from four to five animals were pooled for each experiment, and the experiment was performed three times. Briefly, after mincing the tissue with scalpels and digesting with collagenase B at room temperature for 1.5 hours (Roche; 2 mg/mL), mammary organoids were separated from fat, fibroblasts, and red blood cells by centrifugation. Single cell primary MEC cultures were obtained by additional treatment with 0.05% Trypsin-EDTA (Gibco) for 10 minutes followed by filtration through a 70-μm mesh screen (BD Biosciences). After cell counting, 200 000 MECs were resuspended in 150 μL of Matrigel (BD Biosciences) and plated in eight-well chamber slides (BD Biosciences). After incubating 30 minutes at 37°C, the Matrigel hardened and 400 μL of growth medium was added to each well (DMEM/F12, 5% fetal bovine serum, 200 U/mL PenStrep, 100 μg/mL gentamicin, 10 μg/mL insulin, 2 μg/mL hydrocortisone, and 10 ng/mL mouse epithelial growth factor). Growth medium contained either 0.01% ethanol as vehicle or 100 nM R5020 for the 13 days of culture and was replaced every 2–3 days. After 13 days, the medium was removed, and duplicate wells of acini were fixed for 15 minutes in methacarn (60% methanol, 30% chloroform, 10% glacial acetic acid) (32). The fixative was removed, and the Matrigel plug was transferred to a plastic cryochamber containing 250 μL of melted Histogel (Thermo Scientific). After solidifying, the fixed matrigel plugs were transferred to tissue cassettes, placed in 70% ethanol, and processed for paraffin embedding.
RNA isolation and quantitative real-time PCR
Total RNA from individual uterine tissues was extracted using the QIAGEN RNeasy total RNA isolation kit (QIAGEN) after homogenization. Triplicate samples of Matrigel-embedded MEC acini cultured for 13 days or 1° MECs enriched directly from hormone-treated mammary glands were resuspended in QIAGEN RLT lysis buffer, with total RNA isolated using RNeasy minicolumns (QIAGEN). cDNA was then prepared from 100 ng of uterine RNA or 25 ng of mammary gland RNA using Superscript III reverse transcriptase according to the manufacturer's protocol (Invitrogen). Expression of target genes was analyzed by quantitative real-time PCR using either the TaqMan universal PCR master mix (Applied Biosystems) or the SYBR green PCR master mix (Applied Biosystems). Primers and probe set information can be found in Supplemental Table 1. An analysis was carried out using the ABI Prism 7500 sequence detector system according to the manufacturer's instructions (Applied Biosystems). Transcript levels were normalized to 18S RNA or glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and data were analyzed using Q-Gene software (BioTechniques Software Library) (33).
Immunohistochemistry (IHC)
Paraffin-embedded tissue sections were deparaffinized in xylene and then rehydrated through a graded ethanol series. Antigen retrieval was performed for 10 minutes in a pressure cooker using 0.1 M citrate (pH 6) (BrdU) or 0.1 M Tris (pH 9) (Ki67, PR). Sections were blocked in 3% H2O2 for 10 minutes, followed by a 1-hour incubation at room temperature with the following primary antibodies: biotinylated BrdU mouse monoclonal at 1:10 (BD Pharmingen), Ki67 rabbit polyclonal at 1:2000 (Novocastra), and PR rabbit polyclonal at 1:10 (A0098; Dako). Antibody detection used either ready-to-use streptavidin (15 min; Vector Laboratories) with DABdefine+ (30 min; Dako; 3,3-diaminobenzidine) or Envision+ horseradish peroxidase-labeled antirabbit polymer (30 min; Dako).
Image capture, cell counting, and statistical analysis
An Olympus BX40 light microscope connected to a MagnaFire digital camera was used to capture images from immunohistochemical staining. At least 15–20 individual ×40 fields per genotype and treatment group were captured for counting from each three-dimensional culture sample. The number of positively stained MEC nuclei was expressed as a percentage of the total number of luminal epithelial cells. Data are expressed as mean ± SEM. A two-way ANOVA test was used to determine the significant differences between groups using the computer software Prism 5 (GraphPad Software). Statistical significance is indicated by one to three asterisks corresponding to P ≤ .05 or P ≤ .01 or P ≤ .001, respectively, or P = NS (not significant).
Chromatin immunoprecipitation (ChIP) assay
A ChIP assay of enriched primary MECs was carried out as previously described (18), in which the mammary glands from four mice were harvested and pooled for each hormone treatment group. MECs were isolated from WT C57BL/6 or mutant PR S191A mice, as above. Either vehicle [ethanol (EtOH)] or progesterone (100 nM) was added throughout the in vitro processing of the MECs (∼4.5 h). Enriched MECs were then fixed with 1% paraformaldehyde for 10 minutes at room temperature, and cross-linking was terminated with 1.25 mM glycine for 5 minutes at room temperature. Enriched mouse primary MEC pellets were sonicated as previously described (18). Ten percent of the sonicated chromatin was taken for input controls and processed with immunoprecipitated samples, beginning with the cross-linking reversal step. The remaining sonicated chromatin was incubated with 2.5 ug of anti-PR (Santa Cruz Biotechnology, Inc; sc-7208X) or 1 ug control IgG (ICN Biomedicals; 55456) conjugated to Protein A magnetic beads (Invitrogen) overnight at 4°C. Beads were washed repeatedly to remove nonspecifically bound DNA. The DNA was eluted from the beads (55°C for 10 min) and cross-links were removed overnight at 65°C. Immunoprecipitated DNA was purified according to the manufacturer's protocol (QIAGEN, Inc) and amplified by quantitative PCR with an ABI Prism 7500 sequence detector system (PE Applied Biosystems) using SYBR green PCR master mix (Applied Biosystems). Immunoprecipitated DNA was amplified with PCR primers for the enhancer regions of the receptor activator of nuclear factor-κB ligand (RANKL) or calcitonin genes and a nonspecific intragenic region defined in the Supplemental Data and analyzed and normalized to input DNA amplification product.
Results
Generation of the PR S191A phosphomutant mouse
Multiple phosphorylation sites have been identified in human PR, but the sites have not been mapped in the mouse PR. Human and mouse PR exhibit 94.5% amino acid sequence identity in the DNA binding and ligand binding domains, but there is only 70.1% identity in the NTD in which most of the Ser phosphorylation sites reside. An alignment of nine phosphorylation sites in the NTD of human and mouse PR reveals that the position of most of these Ser residues is conserved between species (eight of nine). The sequences immediately flanking these serine residues are also similar or identical, indicating that most sites identified in human PR likely are also phosphorylation sites in the mouse (Figure 1A). In this study, we analyzed the effects of mutating the Ser 191 site of mouse PR (equivalent to Ser 190 of human PR). The rationale for this choice was as follows. First, a means to confirm the authenticity of the putative mouse site was needed. Second, to increase the likelihood of a phenotype in multiple tissues, a site common to both PR-A and PR-B would be optimal. Finally, substituting Ala for Ser 190 was previously reported to substantially reduce transcriptional activity measured using reporter genes (10). Phosphorylation of the site in mouse PR was confirmed by expressing hemagglutinin-tagged human or mouse PR in human embryonic kidney (HEK) 293 cells and immunoblotting with an antibody specific for human phospho-Ser190 (11). This antibody recognized both human and mouse WT PR-B but did not interact with either of the receptors containing Ala substitutions for the phosphorylation site (Figure 1B). Note that the sequences surrounding the site are not identical in mouse and human, so the weaker signal in mouse may be due to reduced interaction with the phosphorylated mouse PR and does not necessarily imply reduced phosphorylation.
The targeting vector and genetic engineering strategy to produce a PR S191A knock-in mouse is shown in Supplemental Figure 1. The mouse PR allele inserted in the 129Sv bacterial artificial chromosome was used for ES targeting. PCR oligo-directed mutagenesis was used to create a PR Ser191 to Ala substitution (S191A). In addition, we created a silent mutation to engineer a NheI restriction site near the point substitution for Southern blot screening and PCR genotyping. We derived and maintained two independent targeted lines on both the 129Sv/C57BL/6 (129/C57BL) mixed genetic background and the N5 C57BL/6 pure genetic background (five backcrosses to WT C57BL/6J mice to generate a 97% pure strain). Initial studies were performed on the 129/C57BL mixed background, while the N5 C57BL/6 pure genetic background mice were being generated.
Confirmation of PR S191A protein expression in the mouse
To confirm the expression of the PR S191A protein, immunoblots of protein extracts from a whole uterus were performed with antibodies to total mouse PR and to phospho-PR (S191). Equal amounts of total intact PR were detected in WT and PR S191A mice and the relative amounts of the PR-A and PR-B isoforms were also similar, with a higher ratio of PR-A to PR-B in uteri from both WT and S191A mice (Figure 2). We also observed a partial slowing in mobility of PR on immunoblots from uteri of mice pretreated with P4 for 6 hours, indicating that hormone stimulates an overall increase in PR phosphorylation that is similar for WT and S191A (Figure 2A). It should be noted that the Ser190 phosphorylation site in human PR is not responsible for the P4-dependent upshift because it has been shown that other sites are responsible for the upshift (12).
Figure 2.
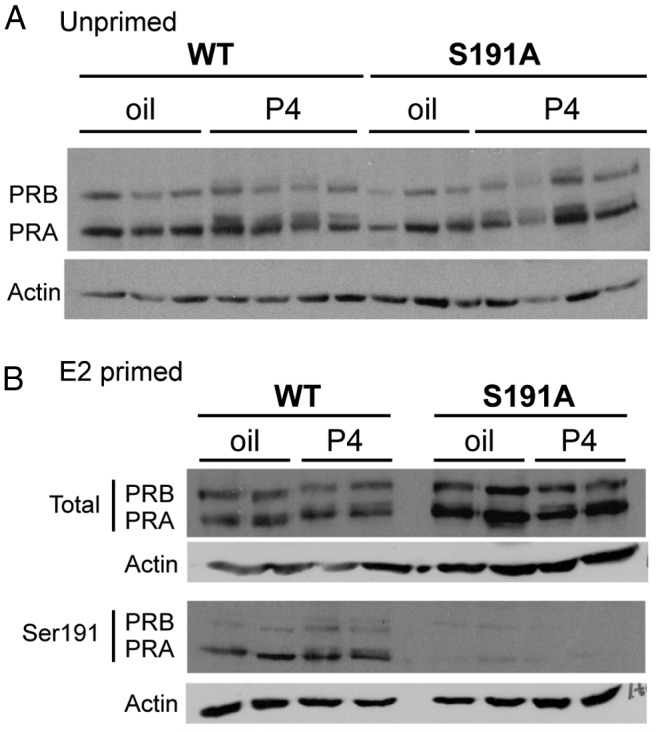
Expression of PR WT and PR S191A protein in mouse uteri is indistinguishable. A, Immunoblotting for total PR in uteri of WT and PR S191A mice pretreated for 6 hours with vehicle (oil) or P4 (1 mg) in non-E2-primed mice. B, Immunoblotting for total (top panel) and phosphorylated (bottom panel) PR (with pS191 specific monoclonal antibody) in uteri of WT and PR S191A mice pretreated for 6 hours with vehicle (oil) or P4 (1 mg) in E2-primed mice.
PR is an estrogen target gene and pretreatment with estradiol increases the expression level of PR protein. Therefore, to maximize PR expression in mouse tissues for subsequent analysis of Ser191 phosphorylation with the phosphospecific antibody, mice were pre-treated for 48 hours with estradiol prior to a 6-hour treatment with vehicle or P4. Total PR protein was expressed at similar levels in WT and S191A mice as expected, but phosphorylation of Ser191 was detected only in the WT mice as indicated by immunoblotting with the phospho-PR (S191) specific antibody (Figure 2B). In human breast cancer cells, serine 190 is basally phosphorylated in the absence of hormone and treatment with P4 increases phosphorylation (11). Thus, Ser190 is not a hormone-dependent phosphorylation site, but its phosphorylation is increased subsequent to P4 binding to PR. Interestingly, there was little change in Ser191 phosphorylation in response to P4 in vivo (Figure 2B), although there is a partial reduction in mobility consistent with phosphorylation of other sites. This difference in hormone dependence was also observed in the HEK 293 cells, in which Ser190 phosphorylation of human PR-B increased in response to the synthetic progestin, R5020, but phosphorylation of Ser191 in mouse showed little or no change (Figure 1B).
P4-dependent responses in the uterus are unaffected by mutation of PR S191
The phenotype of PR S191A mice was compared with that of wild type and PR-null (PRKO) mice because the expectation was that a single point mutation in PR was not likely to result in as severe a phenotype as PRKO mice, and may fall somewhere between that of WT and PRKO. OVX adult virgin mice were treated with P4 as described in Materials and Methods, RNA isolated from the uteri, and PR target genes measured by quantitative PCR. There was no P4-dependent regulation of target genes in PRKO uteri, as expected, and WT and PR S191A mice showed comparable levels of induction of amphiregulin (AREG) and Indian hedgehog and repression of Mucin 1 (Figure 3A). Down-regulation of PR by hormone treatment was also similar for the WT and PR S191A (Figure 3A). Consistent with the lack of changes in the gene expression, no differences in the gross uterine appearance were noted.
Figure 3.

Progesterone-dependent growth and decidualization in uteri of WT and PR S191A C57BL/6 mice do not differ. A, P4-regulated gene expression in uteri from C57BL/6 WT and PR S191A mice. Mice were OVX at 6 weeks of age, rested for 2 weeks, and injected with vehicle (oil) or P4 (1 mg) at 6 hours prior to harvest. RNA was extracted and quantitative PCR was used to analyze expression of selected genes including PR, Areg, Indian hedgehog (Ihh), and mucin-1 (Muc1). Mouse numbers include the following: WT oil, n = 3; WT P4, n = 8; PR S191A oil, n = 7; PR S191A P4, n = 8; PRKO oil, n = 5; and PRKO P4, n = 5. Bars represent SEM. Quantitative PCR values are calculated in relation to PCR standards and normalized to 18S. B, Gross morphology of the uterine decidual response in WT (left panel) and PR S191A (right panel) mice. Graph of the ratio of the traumatized uterine horn weight to the control horn weight of WT (n = 4) vs PR S191A (n = 4) mice, 5 days after the decidual stimulus. Scale bars, 1 cm. C, Expression of select P4-dependent target genes in decidual tissues (FKBP5, BMP2, and Wnt4) in WT and PR S191A mice. WT control (n = 2) and decidual (n = 4) vs PR S191A control (n = 2) and decidual (n = 4) is shown. Control horn, white bars; decidualized horn, black bars. Bars represent SEM. Quantitative PCR values are calculated in relation to PCR standards and normalized to 18S.
We also performed an artificial P4-dependent decidual response assay, as described in Materials and Methods, to determine whether PR S191A mice display compromised uterine function. After hormone treatment, the decidualization response was initiated in the right uterine horn by scratching with a burred needle. The left horn was left unstimulated as a control. Gross morphology of the stimulated and unstimulated horns was examined as a measure of decidualization. PR S191A mice exhibited a robust response to the trauma and quantification of this response by measuring uterine wet weight reveals that the decidual response is comparable with that of the WT mice (Figure 3B, right panel). This contrasts with the lack of P4-dependent decidualization in PRKO mice, a process important for embryo implantation and fertility (9, 13). Expression of FKBP5, BMP2, and Wnt4, three P4-dependent target genes in decidualized tissue, was analyzed by quantitative PCR; the regulation of these genes was similar in WT and PR S191A mice (Figure 3C).
PR Ser191 site-specific phosphorylation modulates fertility and the estrous cycle
Previous studies have shown that PRKO females are infertile (13). Thus, we compared the fertility of WT and S191A mutant mice over a period of 6 months (Figure 4). We did not find a difference in the number of litters per female or in days between litters, but the average litter size was significantly decreased, by 19% (Figure 4). We also examined the estrous cycle length of these mice to determine whether the estrous cycles were normal. Daily vaginal smears were taken over a period of 60 days to determine estrous staging by cell cytology. The PR S191A mice had fewer normal cycles of less than 5 days, and more longer cycles of 5 days or more relative to the WT mice (Figure 4). Thus, phosphorylation at Ser191 is required for optimal fertility.
Figure 4.
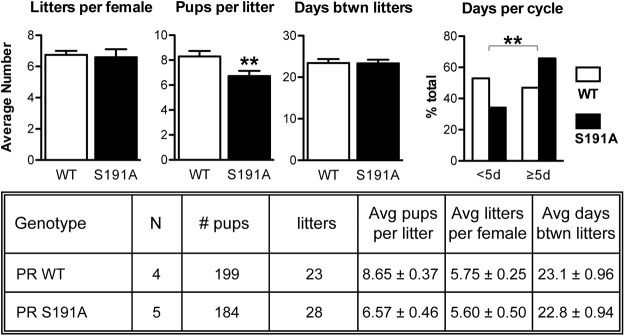
PR S191A mice have significantly smaller litter sizes and an extended estrous cycle. A 6-month fertility study carried out for the C57BL/6 mice showing the averages (left panel) for the number of litters, pups per litter, and days between litters. Average days between litters for the individual mice between WT (n = 4) and PR S191A mice (n = 5) are shown. A Student's t test was used to determine statistical significance. **, P < .005. A 60-day cyclicity study carried out for the C57BL/6 mice shows the average estrous cycle length for the group (WT, n = 12; PR S191A, n = 11). A Fisher's test was used to determine statistical significance. **, P < .005.
P4-dependent responses in the mammary gland are not overtly affected by mutation of the Ser191 phosphorylation site
In the developing mammary gland, PR is required for terminal end bud formation, proliferation, and ductal side branching. During pregnancy, PR is essential for the proliferation and expansion of the epithelium followed by subsequent morphogenesis to form lobuloalveoli. PRKO mammary glands exhibit a reduction in ductal epithelium proliferation and ductal branching, with the absence of lobuloalveolar structures (9, 13). To mimic a pregnancy-like state, intact 9-week-old female adult PR S191A mutant mice and WT littermates were exposed to chronic, 4-week hormone stimulation with EP (see Materials and Methods). PRKO mice were included in this study as a negative control because these mammary glands are unresponsive to hormone treatment (13). Examination of inguinal mammary gland whole mounts showed that WT mammary glands display increased ductal side branching and lobuloalveolar development compared with untreated controls (compare Figure 5, D and J, with Figure 5, A and G). As expected, the PRKO mice displayed a lack of ductal side branching and failed to respond to EP treatment (Figure 5, C, F, I, and L). However, the PR S191A mutant displayed a normal mammary gland morphology in the untreated animals and responded to EP in a way similar to WT littermates (Figure 5, B, E, H, and K).
Figure 5.

Response of mammary gland to prolonged EP treatment. Adult female mice (9–12 wk) were treated with beeswax pellets containing E2 and P4 for a period of 4 weeks as described in Materials and Methods. A–L, Whole-mount staining with Carmine Red at ×10 magnification (A–F) and ×50 magnification (G–L) of inguinal mammary glands from adult (13–14 wk) 129/C57BL/6 mixed WT treated (D and J) and untreated (A and G), PR S191A treated (E and K) and untreated (B and H), and PRKO treated (F and L) and untreated (C and I). Treatment was carried out for 4 weeks. UN, untreated. Animal numbers include the following WT, n = 3 treated, n = 1 untreated; PR S191A, n = 4 treated, n = 2 untreated; PRKO, n = 3 treated, n = 2 untreated; WT on same strain background as PRKO, n = 3 treated, n = 2 untreated. Scale bars, 1 mm. Representative images are shown.
Expression of select PR target genes, known to be induced by P4 in the mammary gland, was also evaluated in WT and PR S191A mice by quantitative PCR using total RNA extracted from whole inguinal mammary glands from 8-week-old OVX mice treated with vehicle or EP for 76 hours. We were unable to detect any significant difference in basal or hormone-induced expression of Wnt4, RANKL, or amphiregulin between WT and PR S191A mice under these conditions (data not shown). These genes have all been shown to have important roles in mammary gland function, display hormone-dependent induction, and exhibit differential induction between normal and PRKO mice (9, 14–17).
P4 induction of selected target genes is impaired in the mammary epithelial compartment of PR S191A mice
Unlike the uterus, in which PR is expressed in most cell types, PR is expressed in only a subset of ductal epithelial cells in the mammary gland and not in the stroma or fat pad, which makes up most of the tissue. Therefore, mRNA isolated from whole mammary glands is diluted with an abundance of mRNA from PR-negative cells. To analyze responsiveness of the mammary epithelium only, we have taken advantage of a previously described MEC three-dimensional (3D) primary culture system that closely recapitulates the mammary gland ducts in vivo. Enriched populations of MECs isolated from mammary glands of adult mice are embedded in Matrigel, as a source of basement membrane, and are grown in a defined medium that maintains expression of PR and functional responses to progestins. After 14 days in culture, the acini formed a single layer of polarized myoepithelial and luminal epithelial cells surrounding a hollow lumen (18).
PR is expressed heterogeneously in luminal epithelial cells, which are segregated from proliferative cells. This expression pattern is also observed in the mammary gland in vivo and is characteristic of proliferation mediated by paracrine signaling pathways. These primary 3D MEC cultures derived from WT or PR S191A mice formed similar normal-like 3D acinar structures with a heterogeneous pattern of PR expression and approximately 20% of PR-positive cells as detected by IHC (Figure 6, B and C). PR was down-regulated similarly in WT and S191A MECs after chronic (13 d) exposure to R5020, a synthetic progestin, at both the mRNA and protein levels (Figure 6, A–C). We confirmed the results from 3D acini by analyzing mammary gland tissue and primary MECs isolated from WT or S191A mice treated with vehicle or P4 for 76 hours. Pieces of mammary glands were sectioned and stained for PR, showing similar levels of PR-positive MECs in WT and S191A and equivalent reduction in the number of PR-positive MECs in response to P4 (Figure 6B). RNA isolated from the primary MECs showed comparable levels of PR mRNA in the two genotypes and both were down-regulated in response to hormone (Figure 6A, right panel). Proliferation in response to progestin was also evaluated by staining acini for Ki67 or mammary gland pieces with BrdU (Figure 6, D and E). In each case, proliferation was induced in response to hormone with no difference between WT and PR S191A mutant. These results indicate that culturing primary MECs in 3D culture, in the presence of extracellular matrix, recapitulates the environment of the whole mammary gland tissue.
Figure 6.

Phosphorylation of Ser191 does not alter the pattern of PR expression or P4-induced proliferation of mammary epithelial cells. Primary MECs from WT or PR S191A mice, either acini cultured and treated for 13 days with ethanol vehicle (EtOH) or 100 nM R5020 or glands from mice treated for 76 hours with oil or 1 mg P4, were used to isolate total RNA or were fixed and sectioned to perform IHC. A, Quantitative PCR was performed from triplicate experiments to determine the expression of PR, relative to GAPDH. B, Sections of acini or mammary glands were stained with an anti-PR antibody, and positive cells were quantified relative to total MECs. C, Representative images of PR staining of sectioned WT acini treated with either EtOH or 100 nM R5020 for 13 days. Scale bars, 50 μm. D, Sections of acini or mammary glands were stained with anti-Ki67 or anti-BrdU antibodies, and positive cells were quantified relative to total MECs. E, Representative images of Ki67 staining of sectioned WT acini treated with either ethanol or 100 nM R5020 for 13 days. Scale bars, 50 μm. Statistical significance, determined by a two-way ANOVA, is indicated by one to three asterisks corresponding to P ≤ .05 or P ≤ .01 or P ≤ .001, respectively, or P = ns (not significant).
We further applied the 3D culture system to examine the effects of the PR S191A mutation on P4-induced expression of several known target genes. Quantitative PCR was performed using RNA isolated from cultures after treatment with vehicle or the synthetic progestin R5020 (Figure 7). The R5020-induced expression of RANKL and calcitonin was significantly reduced in the S191A mutant MECs as compared with WT MECs (Figure 7A), whereas other gene targets, such as Wnt4, Defensin-β1 (Defb1), and AP-(AP-2β), were not affected by mutation of the S191 phosphorylation site (Figure 7C). To determine whether this difference was also observed in vivo, the expression of RANKL and calcitonin was analyzed in MECs enriched from mammary glands after treatment of mice in vivo for 76 hours with vehicle or P4. A similar reduction of P4-stimulated expression of RANKL and calcitonin was observed in MECs derived from S191A mice (Figure 7B), confirming a role for Ser191 phosphorylation in regulating the expression of select genes in the mouse mammary gland.
Figure 7.

P4 induction of selected target genes is impaired in the mammary epithelial compartment of PR S191A mice. A, Primary MECs from WT or PR S191A mice cultured and treated for 13 days with EtOH or 100 nM R5020 were used to isolate total RNA. Quantitative RT-PCR was performed from triplicate experiments to determine the expression of RANKL, calcitonin (Calca), Wnt4, Defensin-β1, and AP-2β, relative to GAPDH (n = 3 samples in triplicate). B, WT or PR S191A mice were treated in vivo for 76 hours with oil or 1 mg P4. MECs were then harvested, enriched, and used to isolate total RNA. Quantitative RT-PCR was performed to determine the expression of RANKL and calcitonin (Calca), relative to GAPDH. Statistical significance, determined by a two-way ANOVA, is indicated by one to three asterisks corresponding to P ≤ .05 or P ≤ 0.01 or P ≤ 0.001, respectively, or P = ns (not significant).
A ChIP assay was used to determine whether the lack of Ser191 phosphorylation altered binding of PR to enhancers in the RANKL or calcitonin genes. We previously reported that RANKL contains multiple P4-responsive distal enhancers (D1-D6), located between 16 and 88 kb upstream of the transcription start site, that bind PR in a P4-dependent manner (18). Moreover, PR binding was shown to be important for P4-induced expression of RANKL (18–20). A full progesterone response element (PRE) binding site was identified 10 kb upstream of the calcitonin transcription start site in a recent ChIP-sequence screen performed from mouse mammary gland tissue treated with EP (21). Primary MECs isolated from WT or PR S191A mice were treated acutely with vehicle or P4, and ChIP assays were performed with an anti-PR antibody. Immunoprecipitated DNA fragments bound by PR were amplified and quantified by quantitative PCR using primers spanning a range of 150–650 bp within each of the enhancers (Supplemental Table 1). As previously reported for WT MECs, P4-stimulated binding of PR to multiple RANKL enhancers, including D1, D3, D4, D5, and D6. As compared with WT MECs, a significant reduction of P4-dependent binding of PR to enhancers D3 and D4 was observed with MECs from S191A mice (Figure 8). Whereas WT PR bound to D5, the hormone-dependent increase in S191A binding to this element was not statistically significant. P4-dependent PR binding was either the same (D1) or increased (D2 and D6) at the other RANKL enhancers in the PR S191A MECs compared with WT. Why P4 did not stimulate binding of PR with enhancer D2 in WT MECs is not known because this was observed previously. However, this may be due to differences in mouse strains (18).
Figure 8.

PR S191A mutation alters P4-dependent binding of PR with RANKL or calcitonin enhancers. Recruitment of PR to six distal RANKL enhancers (A) or a calcitonin enhancer (B) was analyzed by a ChIP assay of primary MECs derived from WT or PR S191A mice. Isolated, enriched MECs were treated in vitro for 4.5 hours with vehicle (EtOH) or P4 (100 nM). Immunoprecipitated DNA was amplified by quantitative PCR using primers flanking potential PREs described in the schematic. Amplified ChIP DNA was normalized to the input and displayed as progesterone-induced fold PR recruitment over vehicle, setting the value of vehicle treated primary MECs to 1. Results are mean ± SEM from three independent experiments. Statistical significance, determined by a two-way ANOVA, is indicated by one to three asterisks corresponding to P ≤ .05 or P ≤ .01 or P ≤ .001, respectively, or P = ns (not significant).
Binding to the calcitonin enhancer was strongly induced by P4 in the WT MECs but was abrogated completely in the S191A MECs. Although binding to this site is lost in the PR S191A mammary gland, there is some P4-induced gene expression of calcitonin in the mutant, suggesting there may be other unidentified PRE half-sites that contribute to the hormone-induced expression of the gene. These data, showing an alteration in the pattern of PR binding to multiple enhancers, suggest that phosphorylation of S191 plays a role in regulating PR binding to specific DNA sites, presumably through protein-protein interactions, as a contributing factor to P4-induced expression and RANKL or calcitonin.
Discussion
Studies to define a role for PR phosphorylation have been performed previously by exogenously expressing WT and mutant PRs in human breast cancer cell lines that no longer express endogenous PR and measuring the activity of the exogenous receptors. To examine the role of phosphorylation in normal cells and tissues at physiological levels of PR, we generated a mouse model with an alanine substitution for a serine at phosphorylation site 191 (S191A) to mimic unphosphorylated receptor. Mice lacking PR (PRKO) are infertile, fail to undergo full differentiation of mammary glands in response to EP treatment, and exhibit a number of uterine defects including a reduced uterine weight and failure to undergo decidualization (13). The S191A mouse exhibited no overt morphological differences compared with WT in response to treatment with P4 or EP. For example, decidualization was normal and there were no modifications in P4-stimulated side branching and alveologenesis (Figure 5). However, a fertility study showed a decrease in litter size with no changes in the numbers of litters. In addition, the length of the estrous cycle was significantly increased in the S191A mice.
We also took advantage of our recently described 3D primary MEC culture system to compare the characteristics of WT and PR S191A MECs. The proliferative response to P4 and the P4-mediated down-regulation of PR was similar in the WT and PR S191A MECs, regardless of the assay (isolated primary MECs or 3D culture). In contrast, elimination of the Ser 191 phosphorylation site substantially limited the ability of PR to induce expression of RANKL and calcitonin, whereas having no effect on several other target genes such as Areg and Wnt4. RANKL is known to be a key paracrine mediator of progesterone-dependent proliferation in adjacent steroid receptor-negative mammary epithelium in the mouse, and more recently the P4-RANKL signaling axis for proliferation was shown to be conserved in human breast epithelium (22). Calcitonin, a small peptide hormone involved in calcium homeostasis, is induced by progesterone and expressed in the luminal epithelial cells of the pregnant mammary gland (23). Even with a decreased expression of RANKL, proliferation of S191A mutant MECs was unaffected. This is most likely due to the redundancy of these paracrine factors because other known factors, Wnt4 and amphiregulin, were not affected in the mutant (Figure 7). It is not known what role calcitonin plays in mammary epithelial cell function because the receptor for calcitonin is expressed in myoepithelial cells (23).
Previous studies of exogenously expressed PR and PR mutants in breast cancer cells have used the addition of progestins under basal conditions depleted of steroids and other growth factors (serum free or charcoal stripped serum). Under these limiting conditions, mutation of other PR phosphorylation sites has been reported to alter gene expression (24). Interestingly, in the mouse, our analyses that did not include exogenous administration of P4 (fertility and estrous cycle) both showed modest, but significant, differences between the WT and S191A mice. When P4 is exogenously administered, differences are detected only at the level of gene regulation.
Although P4-mediated induction of RANKL and calcitonin was reduced by the S191A substitution, induction of other genes was unaffected. Mechanisms for the regulation of RANKL by PR have been reported recently (18). P4 induces the recruitment of signal transducer and activator of transcription (STAT)-5a to a subset of the RANKL enhancers (D1, D3, D4, D5, and D6) (18). Conversely, PR is not recruited to the D2, D4, D5, or D6 elements in MECs derived from STAT5a knockout mice, and P4 induction of RANKL was also lost. These data indicate that PR and STAT5a act cooperatively to bind and function at multiple RANKL enhancers. These data raise the possibility that the phosphorylation of Ser 191 plays a role in PR-protein interactions, such as with STAT5a, which are required for maximal binding of PR to select target gene enhancers.
We have described some of the consequences of eliminating a single phosphorylation site in PR. PR has numerous phosphorylation sites in its amino terminus (Figure 1), and most reside in Ser-Pro motifs, suggesting that several may be coordinately regulated by a single kinase. Human Ser 190, which corresponds to mouse Ser 191, is one of several sites that are substrates for cyclin A/Cdk2 in vitro (25), but hormone-dependent phosphorylation in breast cancer cells is not blocked by a cyclin-dependent kinase (CDK)-1/CDK2 inhibitor and the extent of Ser190 phosphorylation in G2/M phase is comparable with its phosphorylation in the S phase, in which cyclin A/CDK2 is active (26). Thus, a direct test of this hypothesis awaits the identification of the kinase(s) that phosphorylate the site.
Reductions in the phosphorylation of multiple sites likely will have more profound effects on the activity of PR. Our results with the serine to alanine substitution mutation alone do not provide definitive proof that the phenotype observed is due to the elimination of phosphorylation of S191 as opposed to an effect of the alanine substitution on conformation of PR, independent of phosphorylation. Additional studies with other amino acid substitutions such as phosphorylation mimics (glutamic acid) and elimination of a kinase signaling pathway in vivo that phosphorylates S191 would be required to make a more definitive conclusion. It should be noted that most phosphorylation sites of steroid receptors including PR are located in the NTD, and the NTD is largely composed of intrinsically disordered (ID) protein. ID regions of proteins lack stable secondary and tertiary structure and exist as dynamic ensembles of interconverting conformers capable of undergoing a disorder-to-order transition upon interacting with other proteins or DNA. There is also a preference for post-translational modifications, such as phosphorylation, to occur in ID regions due to their structural flexibility (27). It has also been shown by Kumar and colleagues (28) that site-specific phosphorylation of Ser 211 in the NTD of glucocorticoid receptors causes the ID activation function-1 region to adopt a more stable, folded conformation that, in turn, facilitates binding of coregulatory proteins. Kumar and colleagues used both site-directed mutations and phosphorylation and dephosphorylation of Ser 211 with kinases and phosphatases in vitro to come to this conclusion. Because the NTD of PR is also intrinsically disordered and is able to undergo a disorder-to-order transition upon binding coregulatory proteins (29), it is formally possible that phosphorylation of Ser 191 directly affects conformation of the NTD. Additional experiments will be needed to sort out these mechanistic possibilities.
These mice were maintained under optimal laboratory conditions and were not subjected to any form of stress. Under less optimal conditions, including a natural environment, the loss of even a single phosphorylation may have more profound effects. Although a 20% reduction in litter size is a modest change, the selective advantage for WT mice over multiple generations would be very substantial. In conclusion, these studies provide the first evidence for a physiological role for PR phosphorylation in normal tissues and suggests that one of the roles of S191 phosphorylation is to facilitate binding of PR to complex regulatory elements, which selectively regulate transcription of a subset of PR target genes.
Additional material
Supplementary data supplied by authors.
Acknowledgments
We thank the Baylor College of Medicine Breast Center Pathology Core and Laboratory for their excellent technical assistance in processing and staining the three-dimensional culture samples. We also thank the Mouse ES Cell Core Facility and Genetically Engineered Mouse Core at Baylor College of Medicine for their help in creating the PR S191A mice. In addition, we thank Cristian Coarfa and Kimal Rajapakshe (Molecular and Cellular Biology Department, Baylor College of Medicine) for assistance with the statistical analyses of data.
This work was supported by Grant CA57539 (to N.L.W., D.P.E., J.P.L.), National Cancer Institute Cancer Center Support Grant P30CA125123 (to Genetically Engineered Mouse core, Monoclonal Antibody/Protein Expression core), Grant DK040930 (to D.P.E.), and Grant HD038129 (to D.P.E.); National Institutes of Health T32 training Grant HD007165 (to A.E.O., H.L.F., J.M.R.); and National Institutes of Health T32 training Grant DK007696 (to J.M.R.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Ala
- alanine
- Areg
- amphiregulin
- BrdU
- bromodeoxyuridine
- CDK
- cyclin-dependent kinase
- ChIP
- chromatin immunoprecipitation
- 3D
- three dimensional
- E2
- estradiol
- EP
- estrogen and progesterone
- ES
- embryonic stem
- EtOH
- ethanol
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- HA
- hemagglutinin
- HEK
- human embryonic kidney
- ID
- intrinsically disordered
- IHC
- immunohistochemistry
- MEC
- mammary epithelial cell
- NTD
- amino-terminal domain of PR
- OVX
- ovariectomized
- P4
- progesterone
- PR
- progesterone receptor
- PRE
- progesterone response element
- PRKO
- progesterone receptor knockout mouse
- R5020
- synthetic progestin
- RANKL
- receptor activator of nuclear factor-κB ligand
- Ser
- serine
- S191A
- Ser to Ala at amino acid 191 of mouse PR
- Thr
- threonine
- STAT
- signal transducer and activator of transcription
- WT
- wild type.
References
- 1. Hill KK, Roemer SC, Churchill ME, Edwards DP. Structural and functional analysis of domains of the progesterone receptor. Mol Cell Endocrinol. 2012;348(2):418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ward RD, Weigel NL. Steroid receptor phosphorylation: Assigning function to site-specific phosphorylation. BioFactors. 2009;35(6):528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Trevino LS, Weigel NL. Phosphorylation: a fundamental regulator of steroid receptor action. Trends Endocrinol Metab. 2013;24(10):515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen W, Dang T, Blind RD, et al. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 2008;22(8):1754–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Breitwieser W, Lyons S, Flenniken AM, et al. Feedback regulation of p38 activity via ATF2 is essential for survival of embryonic liver cells. Genes Dev. 2007;21(16):2069–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. York B, Yu C, Sagen JV, et al. Reprogramming the posttranslational code of SRC-3 confers a switch in mammalian systems biology. Proc Natl Acad Sci USA. 2010;107(24):11122–11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kastner P, Krust A, Turcotte B, et al. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. 1990;9(5):1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mulac-Jericevic B, Mullinax RA, DeMayo FJ, Lydon JP, Conneely OM. Subgroup of reproductive functions of progesterone mediated by progesterone receptor-B isoform. Science. 2000;289(5485):1751–1754. [DOI] [PubMed] [Google Scholar]
- 9. Mulac-Jericevic B, Lydon JP, DeMayo FJ, Conneely OM. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc Natl Acad Sci USA. 2003;100(17):9744–9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takimoto GS, Hovland AR, Tasset DM, Melville MY, Tung L, Horwitz KB. Role of phosphorylation on DNA binding and transcriptional functions of human progesterone receptors. J Biol Chem. 1996;271(23):13308–13316. [DOI] [PubMed] [Google Scholar]
- 11. Clemm DL, Sherman L, Boonyaratanakornkit V, Schrader WT, Weigel NL, Edwards DP. Differential hormone-dependent phosphorylation of progesterone receptor A and B forms revealed by a phosphoserine site-specific monoclonal antibody. Mol Endocrinol. 2000;14(1):52–65. [DOI] [PubMed] [Google Scholar]
- 12. Zhang Y, Beck CA, Poletti A, Edwards DP, Weigel NL. Identification of a group of Ser-Pro motif hormone-inducible phosphorylation sites in the human progesterone receptor. Mol Endocrinol. 1995;9(8):1029–1040. [DOI] [PubMed] [Google Scholar]
- 13. Lydon JP, DeMayo FJ, Funk CR, et al. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9(18):2266–2278. [DOI] [PubMed] [Google Scholar]
- 14. Humphreys RC, Lydon J, O'Malley BW, Rosen JM. Mammary gland development is mediated by both stromal and epithelial progesterone receptors. Mol Endocrinol. 1997;11(6):801–811. [DOI] [PubMed] [Google Scholar]
- 15. Said TK, Conneely OM, Medina D, O'Malley BW, Lydon JP. Progesterone, in addition to estrogen, induces cyclin D1 expression in the murine mammary epithelial cell, in vivo. Endocrinology. 1997;138(9):3933–3939. [DOI] [PubMed] [Google Scholar]
- 16. Tranguch S, Cheung-Flynn J, Daikoku T, et al. Cochaperone immunophilin FKBP52 is critical to uterine receptivity for embryo implantation. Proc Natl Acad Sci USA. 2005;102(40):14326–14331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang Z, Wolf IM, Chen H, et al. FK506-binding protein 52 is essential to uterine reproductive physiology controlled by the progesterone receptor A isoform. Mol Endocrinol. 2006;20(11):2682–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Obr AE, Grimm SL, Bishop KA, Pike JW, Lydon JP, Edwards DP. Progesterone receptor and Stat5 signaling crosstalk through RANKL in mammary epithelial cells. Mol Endocrinol. 2013;27(11):1808–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bishop KA, Meyer MB, Pike JW. A novel distal enhancer mediates cytokine induction of mouse RANKl gene expression. Mol Endocrinol. 2009;23(12):2095–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim S, Yamazaki M, Zella LA, Shevde NK, Pike JW. Activation of receptor activator of NF-κB ligand gene expression by 1,25-dihydroxyvitamin D3 is mediated through multiple long-range enhancers. Mol Cell Biol. 2006;26(17):6469–6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lain AR, Creighton CJ, Conneely OM. Research resource: progesterone receptor targetome underlying mammary gland branching morphogenesis. Mol Endocrinol. 2013;27(10):1743–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tanos T, Sflomos G, Echeverria PC, et al. Progesterone/RANKL is a major regulatory axis in the human breast. Sci Transl Med. 2013;5(182):182ra155. [DOI] [PubMed] [Google Scholar]
- 23. Ismail PM, DeMayo FJ, Amato P, Lydon JP. Progesterone induction of calcitonin expression in the murine mammary gland. J Endocrinol. 2004;180(2):287–295. [DOI] [PubMed] [Google Scholar]
- 24. Hagan CR, Daniel AR, Dressing GE, Lange CA. Role of phosphorylation in progesterone receptor signaling and specificity. Mol Cell Endocrinol. 2012;357(1–2):43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y, Beck CA, Poletti A, et al. Phosphorylation of human progesterone receptor by cyclin-dependent kinase 2 on three sites that are authentic basal phosphorylation sites in vivo. Mol Endocrinol. 1997;11(6):823–832. [DOI] [PubMed] [Google Scholar]
- 26. Narayanan R, Edwards DP, Weigel NL. Human progesterone receptor displays cell cycle-dependent changes in transcriptional activity. Mol Cell Biol. 2005;25(8):2885–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Blackford JA, Jr, Brimacombe KR, Dougherty EJ, et al. Research resource: modulators of glucocorticoid receptor activity identified by a new high-throughput screening assay. Mol Endocrinol. 2014;28(7):1194–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garza AM, Khan SH, Kumar R. Site-specific phosphorylation induces functionally active conformation in the intrinsically disordered N-terminal activation function (AF1) domain of the glucocorticoid receptor. Mol Cell Biol. 2010;30(1):220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kumar R, Moure CM, Khan SH, et al. Regulation of the structurally dynamic N-terminal domain of progesterone receptor by protein-induced folding. J Biol Chem. 2013;288(42):30285–30299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Finn CA, Martin L. Endocrine control of the timing of endometrial sensitivity to a decidual stimulus. Biol Reprod. 1972;7(1):82–86. [DOI] [PubMed] [Google Scholar]
- 31. Goldman JM, Murr AS, Cooper RL. The rodent estrous cycle: characterization of vaginal cytology and its utility in toxicological studies. Birth Defects Res B Dev Reprod Toxicol. 2007;80(2):84–97. [DOI] [PubMed] [Google Scholar]
- 32. Puchtler H, Waldrop FS, Meloan SN, Terry MS, Conner HM. Methacarn (methanol-Carnoy) fixation. Practical and theoretical considerations. Histochemie. 1970;21(2):97–116. [DOI] [PubMed] [Google Scholar]
- 33. Muller PY, Janovjak H, Miserez AR, Dobbie Z. Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques. 2002;32(6):1372–1374, 1376,, 1378–1379. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.