

Abstract
The binding of GnRH to its receptor initiates signaling cascades in gonadotropes, which result in enhanced LH and FSH biosynthesis and secretion. This process is necessary for follicular maturation and ovulation. Calcium influx activates MAPKs, which lead to increased transcription of LH and FSH genes. Previous research suggests that two MAPK signaling pathways, ERK and jun-N-terminal kinase, are activated by either calcium influx through L-type calcium channels or by global calcium signals originating from intracellular stores, respectively. Here we continued this investigation to further elucidate molecular mechanisms transducing GnRH receptor stimulation to ERK activation. Although it is known that GnRH activation of ERK requires calcium influx through L-type calcium channels, direct evidence supporting an underlying local calcium signaling mechanism was lacking. Here we used a combination of electrophysiology and total internal reflection fluorescence microscopy to visualize discrete sites of calcium influx (calcium sparklets) in gonadotrope-derived αT3–1 cells in real time. GnRH increased localized calcium influx and promoted ERK activation. The L-type calcium channel agonist FPL 64176 enhanced calcium sparklets and ERK activation in a manner indistinguishable from GnRH. Conversely, the L-type calcium channel antagonist nicardipine inhibited not only localized calcium sparklets but also ERK activation in response to GnRH. GnRH-dependent stimulation of L-type calcium channels was found to require protein kinase C and a dynamic actin cytoskeleton. Taken together, we provide the first direct evidence for localized L-type calcium channel signaling in αT3–1 cells and demonstrate the utility of our approach for investigating signaling mechanisms and cellular organization in gonadotropes.
The hypothalamic neuropeptide GnRH is secreted into the hypophyseal portal circulation and binds to receptors on a subpopulation of anterior pituitary cells termed gonadotropes. The binding of GnRH to its cognate receptor elicits multiple transcriptional and biosynthetic events, leading to increased synthesis and secretion of LH and FSH. Most dramatic is the sharp rise in LH secretion (the LH surge) that precedes and is necessary for final follicular maturation and ovulation (1).
After the GnRH activation of the G protein-coupled GnRH receptor, the Gαq/11 subunit stimulates phospholipase C, leading to the cleavage of plasma membrane-bound phosphatidylinositol-4–5-bisphosphate and the generation of the classical second messengers, inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) (2). Although IP3 promotes calcium (Ca2+) release from the endoplasmic reticulum via activation of IP3 receptors, DAG stimulates various protein kinase C (PKC) isoforms including conventional isoforms (PKC-α, -β, and –γ), which are activated by DAG and Ca2+ and novel isoforms (PKC-δ, -ϵ, -θ, and -η/λ) which are activated by DAG but are Ca2+ independent. Increased PKC activity ultimately stimulates Ca2+ influx through voltage-dependent L-type Ca2+ channels (3).
Increased intracellular Ca2+ contributes to the activation of MAPK signaling in gonadotropes. In general, Ca2+-dependent MAPK initiates transcriptional changes, ultimately leading to increased production of LH and FSH (4). Prior work suggests that these two distinct Ca2+ signals (ie, Ca2+ influx and Ca2+ release from the endoplasmic reticulum) activate two distinct MAPK signaling cascades: IP3-mediated Ca2+ release from the endoplasmic reticulum promotes jun-N-terminal kinase activation, whereas the Ca2+ influx through L-type Ca2+ channels activates ERK (5, 6). Importantly, ERK is the key signal required for the enhanced LH synthesis and the preovulatory LH surge (4, 7).
Previously Roberson and colleagues (5, 6) used a pharmacological approach to frame the hypothesis that a local L-type Ca2+ channel signal insensitive to intracellular chelation was necessary for ERK activation; however, technical limitations precluded direct experimental confirmation of this intriguing hypothesis. Herein we used a powerful Ca2+ imaging approach to directly test in real time the hypothesis that GnRH receptor activation leads to a local L-type Ca2+ channel-mediated signal coupled to ERK activation.
Our approach, based on a combination of voltage-clamp electrophysiology and total internal reflection fluorescence (TIRF) microscopy, has allowed, for the first time, to unambiguously visualize localized Ca2+ influx through L-type Ca2+ channels [ie, Ca2+ sparklets (8–10)] in single αT3–1 gonadotropes. Using this approach, we found that GnRH increases local L-type Ca2+ channel sparklet activity. Furthermore, pharmacological manipulations demonstrate that the L-type Ca2+ channel sparklets are coupled to ERK activation and that the GnRH-dependent activation of local L-type Ca2+ channel function requires PKC and a functional actin cytoskeleton. Our findings are not only consistent with the hypothesis that GnRH induces the local L-type Ca2+ channel signal that is critical for ERK activation, but they also demonstrate directly the existence of a biologically relevant GnRH-induced Ca2+ microdomain in αT3–1 gonadotropes. Importantly, these findings open new and heretofore unavailable opportunities to further uncover the spatial and temporal events underlying the distinct subcellular biochemical and biophysical processes underlying GnRH stimulated gonadotropin synthesis. Finally, because the Ca2+ sparklets have been identified predominantly in cardiovascular tissue, these data suggest a broader regulatory role for this unique Ca2+ signal in mediating a diverse array of cellular events and biological processes.
Materials and Methods
Materials
DMEM was from HyClone, fetal bovine serum was from Atlas Biologicals, and L-glutamine and the antibiotic-antimycotic solution were from Mediatech. Rabbit polyclonal antibodies to ERK were from Santa Cruz Biotechnology, Matrigel was from BD Biosciences, and fluo-5F (pentapotassium salt) was from Invitrogen. All other chemicals were from Sigma.
Cell culture
αT3–1 cells (11), a generous gift from Pam Mellon (University of California, San Diego, San Diego, California), were incubated in high-glucose DMEM supplemented with fetal bovine serum and horse serum (5% each), L-glutamine (2 mM), and antibiotic-antimycotic solution (1%). Cells were maintained at 37°C in 5% CO2 humidified air.
Electrophysiology and total internal reflection fluorescence microscopy
αT3–1 cells were plated onto Matrigel-coated glass-bottomed Mattek dishes 24 hours prior to experimentation. Simultaneous electrophysiology and Ca2+ imaging experiments were carried out using the conventional dialyzed whole-cell patch clamp technique as described previously (8, 10, 12, 13). Briefly, Ca2+ influx through L-type Ca2+ channels was visualized with a TILL Photonics through-the-lens TIRF system built around an inverted Olympus IX-71 microscope with a ×100 TIRF oil-immersion objective (numerical aperture 1.45) and an Andor iXON EMCCD camera (Andor Technology). To monitor Ca2+ influx, gonadotropes were loaded with the Ca2+ indicator fluo-5F (200 μM) and an excess of EGTA [10 mM; to lower background noise while minimally interfering with fluo-5F (8–10, 13)] via the patch pipette. The membrane potential was controlled with an Axopatch 200B amplifier (Molecular Devices); fluo-5F excitation was achieved with a 491-nm laser with excitation and emission light being separated with appropriate filters. Ca2+ influx was recorded with 2 mM external Ca2+ at a frame rate of 50 Hz and holding potential of −70 mV to increase the driving force for Ca2+ entry. To preclude potential contaminating Ca2+ release events from the endoplasmic reticulum, the Ca2+-ATPase inhibitor thapsigargin (1 μM) was present during all experiments. Cells were imaged for 2 minutes before acute treatment with GnRH (3 nM), the L-type Ca2+ channel agonist FPL 64176 (500 nM), the L-type Ca2+ channel antagonist nicardipine (10 μM), the PKC agonist phorbol 12, 13-dibutyrate (PDBu; 50 nM), the broad-spectrum PKC inhibitor GF109203X (GFX; 1 μM), the PKCα and PKCβ inhibitor Gö6976 (100 nM) or the actin stabilizer jasplakinolide (100 nM) and imaged for an additional 10 minutes. Vehicle controls were performed as appropriate. All experiments were performed at room temperature (22–25°C).
L-type Ca2+ channel sparklet analysis
Background-subtracted fluo-5F fluorescence signals were converted to intracellular Ca2+ concentrations ([Ca2+]i), as described previously (8, 12–14). Briefly, fluo-5F fluorescence images were analyzed with custom software kindly supplied by L. Fernando Santana (University of Washington, Seattle, Washington), and L-type Ca2+ channel sparklet activity was determined by calculating the nPs of each site, where n is the number of quantal levels detected, and Ps is the probability that the site is active. nPs values were obtained using pCLAMP version 10.0 (Molecular Devices) on imported [Ca2+]i time-course records using an initial unitary [Ca2+]i elevation of approximately 20 nM as determined empirically. Active L-type Ca2+ channel sparklet site densities (Ca2+ sparklet sites per square micrometer) were calculated by dividing the number of active sites by the area of cell membrane visible in the TIRF images. Image stacks selected for analysis were obtained between 5 and 10 minutes of pharmacological manipulation.
Normally distributed data are presented as means ± SEM. Two-sample comparisons of these data were performed using either a paired or unpaired (as appropriate) two-tailed Student's t test, and comparisons between more than 2 groups were performed using a one-way ANOVA with Tukey's multiple comparison posttest. L-type Ca2+ channel sparklet activity (ie, nPs) data sets were bimodally distributed (8, 10, 13); thus, two-sample comparisons of nPs data were examined with the nonparametric Wilcoxon matched pairs test (two tailed), and comparisons between more than 2 groups were performed using the nonparametric Friedman test with Dunn's multiple comparison posttest. Arithmetic means of nPs data sets are indicated in the figures (solid gray horizontal lines) for nonstatistical visual purposes, and dashed gray lines mark the threshold for high-activity Ca2+ sparklet sites (nPs ≥ 0.2) (8, 12, 13, 15). Values of P < .05 were considered significant and asterisks used in the figures indicate a significant difference between groups.
Western blot analysis/ERK activation assay
A monolayer of αT3–1 cells (∼8 × 105 cells) in 60-mm tissue culture dishes was washed twice with PBS and incubated in serum-free DMEM for 2 hours. After serum starvation, cells were exposed to either vehicle (0.1% dimethylsulfoxide) or GnRH (3 nM) with or without additional treatments for 5 or 10 minutes (at 37°C) on the same gel. Cells were washed in ice-cold PBS and lysed in radioimmunoprecipitation assay buffer containing a protease inhibitor cocktail. Total cell lysates (10–15 μg) were separated on 10% SDS-PAGE gels and transferred onto nitrocellulose membranes. Phosphorylated ERK, which we used as a surrogate measure of ERK activation, was detected with phosphorylated ERK antibody (1:2000) using the enhanced chemiluminescence prime reagent. After stripping, total ERK was probed with an anti-ERK1 antibody (1:2000) that recognizes ERK-1 and ERK-2 independent of the phosphorylation state on the same blot. Data are presented as mean ± SEM phosphorylated ERK normalized to total ERK; comparisons were performed using ANOVA with Newman-Keuls multiple comparison posttest.
Results
To test the hypothesis that acute application of GnRH induces local Ca2+ influx through L-type Ca2+ channels and ERK activation in pituitary gonadotropes, we proposed five experimental criteria: 1) L-type Ca2+ channels must produce observable sites of localized Ca2+ influx (ie, Ca2+ sparklets) in response to GnRH; 2) L-type Ca2+ channel activators must increase Ca2+ sparklets and ERK signaling; 3) conversely, inhibition of L-type Ca2+ channels must prevent Ca2+ sparklets and decrease ERK signaling; 4) signaling modalities known to regulate localized L-type Ca2+ channel function (eg, PKC) should regulate ERK signaling in a concurrent manner, and 5) changes in actin cytoskeletal organization should disrupt GnRH-induced Ca2+ influx and ERK activation.
TIRF microscopy reveals localized Ca2+ influx in αT3–1 cells after GnRH exposure
Conventional experimental approaches used to investigate intracellular Ca2+ dynamics (eg, wide field fluorescence microscopy) generally provide poor signal discrimination and limited temporal and spatial resolution. To overcome these technical inadequacies, we used a combination of electrophysiology and TIRF microscopy to visualize local Ca2+ influx in αT3–1 cells. This approach provides high temporal and spatial resolution, which is ideal for visualizing small Ca2+ influx events in the plasma membrane (9, 10, 16) due to the high signal to noise ratio achieved with the inherently limited depth of penetration of the TIRF evanescent wave (∼100 nm; Figure 1A).
Figure 1.
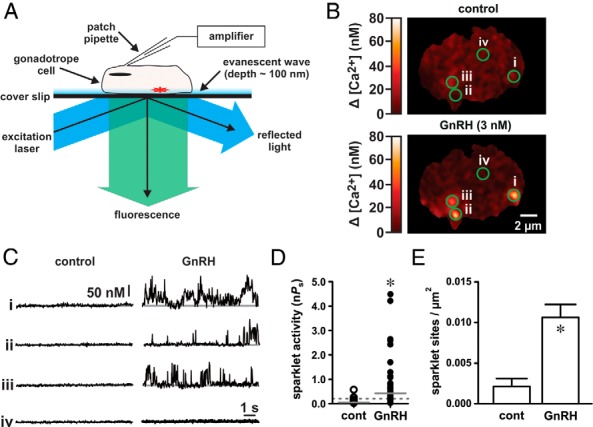
GnRH induces localized Ca2+ influx in αT3–1 cells. A, Schematic illustrating the electrophysiological and TIRF imaging method used to visualize Ca2+ influx in αT3–1 cells. B, Representative TIRF images showing localized Ca2+ influx in αT3–1 cells before and after GnRH (3 nM). C, Traces showing the time course of localized Ca2+ influx at the four circled sites before and after GnRH. The scale bar provides reference for changes in [Ca2+]i during the traces (see Materials and Methods). D, Plot of Ca2+ sparklet site activities (nPs; see Materials and Methods) before and after GnRH (n = 32 cells); solid gray lines are the arithmetic means of each group, and dashed lines mark the threshold for high-activity Ca2+ sparklet sites [nPs ≥ .2; (10)]. E, Plot of mean ± SEM Ca2+ sparklet site densities (Ca2+ sparklet sites/μm2) before and after GnRH (n = 32 cells). *, P < .05. cont, control.
To begin, we applied GnRH (3 nM) to αT3–1 cells and monitored for changes in the Ca2+ influx across the plasma membrane. To preclude potential contaminating Ca2+ release events from the endoplasmic reticulum, the Ca2+-ATPase inhibitor thapsigargin (1 μM) was present during all experiments. In addition, cells were dialyzed with an excess of the Ca2+ chelator EGTA (10 mM) via the patch pipette to buffer global changes in intracellular Ca2+. Consistent with our hypothesis that GnRH induces a local Ca2+ signal, GnRH exposure promoted discrete sites of repetitive Ca2+ influx (ie, Ca2+ sparklets; Figure 1, B and C, and Supplemental Movie 1; n = 32 cells). The average area of these Ca2+ sparklets was 2.65 ± 0.10 μm2. Note that many areas of the plasma membrane visible in the TIRF image were optically silent (ie, showed no evidence of Ca2+ influx) as illustrated by region iv in Figure 1, B and C.
To quantify Ca2+ sparklet activity, we determined the nPs value for each site where n is the number of quantal levels observed and Ps is the probability that the Ca2+ sparklet site is active (8–10, 12). Similar to arterial smooth muscle cells (10, 12), Ca2+ sparklet site activity (nPs) in αT3–1 cells was bimodal with low activity sites (nPs < .2) and high activity sites (nPs ≥ .2). Qualitatively, low nPs sites are characterized as having short Ca2+ sparklet events resulting in minimal Ca2+ influx, whereas high nPs sites have longer, more continuous Ca2+ sparklet events, resulting in substantial Ca2+ influx (9). In αT3–1 cells, GnRH increased Ca2+ sparklet site activity (nPs) compared with vehicle control (Figure 1D; P < .05, n = 32 cells). The density of detected Ca2+ sparklet sites (number of Ca2+ sparklet sites per square micrometer) also increased after GnRH exposure (Figure 1E; P < .05, n = 32 cells). These data demonstrate that GnRH induces localized Ca2+ influx in αT3–1 cells by increasing the number of active Ca2+ sparklet sites and by increasing the activity at each of those sites.
GnRH stimulates L-type Ca2+ channel sparklets in αT3–1 cells which contribute to ERK activation
Next, we used a pharmacological approach to test the hypothesis that Ca2+ sparklets observed in αT3–1 cells are produced by L-type Ca2+ channels. Consistent with this hypothesis, the nondihydropyridine (17) L-type Ca2+ channel agonist FPL 64176 (500 nM) increased Ca2+ sparklet activity and density (Figure 2, A and B; P < .05, n = 12 cells). Interestingly, the Ca2+ sparklet site activity and density after the FPL 64176 exposure was not different from that induced by GnRH (P > .05). To more directly establish that Ca2+ sparklets induced by GnRH are produced through L-type Ca2+ channels, we applied GnRH in the presence of the dihydropyridine L-type Ca2+ channel antagonist nicardipine (10 μM). In contrast to GnRH alone (see Figure 1), GnRH did not significantly increase Ca2+ sparklet activity or density in the presence of nicardipine (Figure 2, C and D; P > .05, n = 7 cells). These data provide strong evidence that the subplasmalemmal Ca2+ influx in αT3–1 cells as a result of GnRH is due to L-type Ca2+ channel sparklets.
Figure 2.

GnRH-induced Ca2+ sparklets are mediated by L-type Ca2+ channels and promote ERK activation. A and C, Representative traces showing time courses of Ca2+ influx in an αT3–1 cell before and after application of the L-type Ca2+ channel agonist FPL 64176 (500 nM) (A) and GnRH (3 nM) (C) in the presence of the L-type Ca2+ channel antagonist nicardipine (10 μM). B and D, Plots of Ca2+ sparklet site activities (nPs) and mean ± SEM Ca2+ sparklet site densities (Ca2+ sparklet sites per square micrometers) before and after FPL 64176 (n = 12 cells) (B) and GnRH in the presence of nicardipine (n = 7 cells) (D). E, Western blot analysis of ERK activation (as measured by ERK phosphorylation) in αT3–1 cells exposed to GnRH (3 nM) or FPL 64176 (500 nM) in the presence of nicardipine (10 μM) for 5 or 10 minutes (n ≥ 3 independent experiments). *, P < .05 vs nicardipine; †, P < .05 vs nicardipine and P < .05 vs GnRH. cont, control; nic, nicardipine.
To further establish the importance of L-type Ca2+ channels, we examined the effects of FPL 64176 (500 nM) on ERK activation in αT3–1 cells. For these and related experiments, we used ERK phosphorylation as a surrogate measure of ERK activation. Similar to GnRH, FPL 64176 (which promoted localized Ca2+ influx; Figure 2A) also promoted ERK activation (Figure 2E; P < .05, n ≥ 3 independent experiments). Conversely, inhibition of L-type Ca2+ channels with nicardipine (10 μM) abolished FPL 64176-induced ERK activation (Figure 2E; P > .05, n ≥ 3 independent experiments). Inhibition of local L-type Ca2+ channel signaling with nicardipine (eg, Figure 2C) attenuated but did not eliminate GnRH-dependent ERK activation (Figure 2E; P > .05, n ≥ 3 independent experiments). The observed loss or attenuation of ERK activation after the inhibition of localized L-type Ca2+ channel activity in these experiments supports the hypothesis that localized L-type Ca2+ channel function regulates ERK activation in αT3–1 cells.
PKC promotes localized L-type Ca2+ channel signaling and subsequent ERK activation in response to GnRH
In arterial smooth muscle cells, PKC promotes localized L-type Ca2+ channel sparklet activity similar to what we describe after GnRH exposure in αT3–1 cells (9, 10). In gonadotropes PKC is known to induce global Ca2+ signals via the dihydropyridine-sensitive Ca2+ channels in response to GnRH (5). Accordingly, we hypothesized that exposing αT3–1 cells to GnRH induces GnRH receptor-dependent PKC activation, which in turn stimulates local L-type Ca2+ channel sparklets. Consistent with this hypothesis, the PKC agonist PDBu (50 nM) increased the L-type Ca2+ channel sparklet site activity and density (Figure 3, A and B; P < .05, n = 11 cells). The increase in L-type Ca2+ channel sparklet activity and density induced by PDBu was not different from that observed with GnRH or FPL 64176 (see Figures 1 and 2; P > .05). Suggesting that the population of L-type Ca2+ channels stimulated by GnRH and PDBu is one in the same, the effects of GnRH and PDBu were not additive with respect to the Ca2+ sparklet activity (median GnRH nPs = .19, interquartile range 0.41; median GnRH + PDBu nPs = .24, interquartile range 0.71; P > .05, n = 16 cells) or density (GnRH density 0.010 ± 0.002 sparklet sites/μm2; GnRH + PDBu density = 0.007 ± 0.001 Ca2+ sparklet sites/μm2; P > .05, n = 16 cells).
Figure 3.

GnRH-induced L-type Ca2+ channel sparklets and ERK activation require PKC. A and C, Representative traces showing time courses of Ca2+ influx in an αT3–1 cell before and after application of the PKC activator PDBu (50 nM) (A) and GnRH (3 nM) (C) in the presence of the broad-spectrum PKC inhibitor GFX (1 μM). B and D, Plots of Ca2+ sparklet site activities (nPs) and mean ± SEM Ca2+ sparklet site densities (Ca2+ sparklet sites per square micrometer) before and after PDBu (n = 11 cells) (B) and GnRH in the presence of GFX (n = 9 cells) (D). E, Western blot analysis of ERK activation (as measured by ERK phosphorylation) in αT3–1 cells exposed to PDBu (50 nM) or PDBu with nicardipine (1 μM) for 5 minutes and αT3–1 cells exposed to GnRH (3 nM) or FPL 64176 (500 nM) for 5 minutes in the presence or absence of GFX (1 μM; n ≥ 3 independent experiments). *, P < .05. cont, control; nic, nicardipine.
We then used the broad-spectrum PKC inhibitor GF109203X (18, 19) to link GnRH-dependent activation of PKC to stimulation of L-type Ca2+ channel sparklets. Indicative of the necessity of PKC, inhibition with GFX (1 μM) abolished the GnRH-dependent stimulation of L-type Ca2+ channel sparklets (Figure 3, C and D; P > .05, n = 9 cells). At a concentration of 1 μM, GFX inhibits the following PKC isoforms known to be expressed in αT3–1 cells: α, β, δ, and ϵ (20). To narrow our investigation of the isoforms involved with L-type Ca2+ channel sparklets in αT3–1 cells, we used the PKC inhibitor Gö6976, which is selective for PKCα and PKCβ (21). Application of Gö6976 (100 nM) had no significant effect on GnRH-dependent stimulation of L-type Ca2+ channel sparklet activity (median GnRH nPs = .19, interquartile range 0.41; median GnRH + Gö6976 nPs = .06, interquartile range 0.43; P > .05, n = 7 cells) or density (GnRH density = 0.010 ± 0.002 sparklet sites/μm2; GnRH + Gö6976 density = 0.011 ± 0.002 Ca2+ sparklet sites/μm2; P > .05, n = 7 cells). These data indicate that PKCα and PKCβ are not essential for GnRH-dependent activation of L-type Ca2+ channels in αT3–1 cells. This observation is consistent with previous work, suggesting that PKCδ and PKCϵ play a greater role in GnRH-dependent ERK activation than PKCα and PKCβ (20).
In agreement with our Ca2+ sparklet data, PKC stimulation with PDBu (50 nM) increased ERK activation (Figure 3E; P < .05, n ≥ 3 independent experiments). Demonstrating the necessity of Ca2+ influx through L-type Ca2+ channels, αT3–1 cells preincubated with nicardipine (10 μM) showed no significant increase in ERK signaling after the PDBu exposure (Figure 3E; P > .05, n ≥ 3 independent experiments). Once again in agreement with the effect on Ca2+ sparklets, PKC inhibition with GFX (1 μM) reduced GnRH-dependent ERK activation (Figure 3E; P < .05, n ≥ 3 independent experiments), whereas Gö6976 (1 μM) was without effect (P > .05, n ≥ 3 independent experiments; data not shown). Note that PKC inhibition with GFX had no effect on ERK phosphorylation induced by direct stimulation of L-type Ca2+ channels with FPL 64176 (Figure 3E; P > .05, n ≥ 3 independent experiments). This suggests that PKC activity occurs upstream of L-type Ca2+ channels in the signaling cascade linking GnRH receptor stimulation to ERK activation. Taken together, our data indicate that GnRH induces localized PKC-dependent L-type Ca2+ channel sparklets and that this mechanism plays a fundamental role in GnRH-dependent activation of ERK signaling in αT3–1 cells.
A dynamic actin cytoskeleton is necessary for transducing GnRH receptor activation to localized Ca2+ influx and ERK activation
Our data support the concept that precise structural and molecular elements create a microenvironment suitable for localized subplasmalemmal L-type Ca2+ channel signaling necessary for gonadotrope function. Thus, it is logical to suggest that cellular structural features such as the cortical actin cytoskeleton could be involved in the organization of molecular components responsible for transducing GnRH receptor activation to the L-type Ca2+ channel. In accordance with this hypothesis, previous work has demonstrated that rapid remodeling of the actin cytoskeleton occurs after GnRH treatment and that an intact actin cytoskeleton is necessary for GnRH signaling to ERK (22–24). However, the point(s) in this pathway in which actin dynamics participate in ERK activation is (are) unclear.
Therefore, we investigated whether actin dynamics are involved with GnRH induction of localized Ca2+ sparklets upstream of ERK activation to narrow down where actin reorganization may be necessary in this signaling pathway. To begin, we examined the effect of jasplakinolide (100 nM for 5 min), which stabilizes filamentous actin, on localized L-type Ca2+ channel function. By itself, jasplakinolide had no effect on basal L-type Ca2+ channel activity (Figure 4, B and C; P > .05, n = 9 cells). However, jasplakinolide reduced the stimulatory effect of GnRH (3 nM) on L-type Ca2+ channel sparklet activity (Figure 4, A and B; P < .05, n = 9 cells). Consistent with previous work (22), ERK activation in response to GnRH was also reduced by jasplakinolide (Figure 4D; P < .05; n ≥ 3 independent experiments).
Figure 4.

Actin stabilization disrupts GnRH-dependent Ca2+ sparklet stimulation. A, Representative traces showing time courses of Ca2+ influx in αT3–1 cells exposed to GnRH (3 nM) in the presence or absence of jasplakinolide (100 nM for 5 min). B and C, Plots of Ca2+ sparklet site activities (nPs) and mean ± SEM Ca2+ sparklet site densities (Ca2+ sparklet sites per square micrometer) for GnRH (3 nM; n = 9 cells in each group) with or without jasplakinolide (100 nM for 5 min). D, Western blot analysis of ERK activation (as measured by ERK phosphorylation) in αT3–1 cells exposed to GnRH (3 nM) with or without jasplakinolide (100 nM for 5 min; n ≥ 3 independent experiments). *, P < .05; †, P < .05 vs jasplakinolide and P < .05 vs GnRH. cont, control; jas, jasplakinolide.
Unlike Ca2+ sparklet site activity (ie, nPs), Ca2+ sparklet site density (ie, the number of Ca2+ sparklet sites/μm2) in response to GnRH was not reduced by jasplakinolide (Figure 4C; P > .05; n = 9 cells). These observations suggest that actin stabilization does not perturb the number of functional L-type Ca2+ channels available for stimulation after GnRH application. Rather, actin stabilization with jasplakinolide disrupts the ability of GnRH signaling to evoke high-activity Ca2+ sparklet sites. To test this hypothesis, we examined the effect of the direct L-type Ca2+ channel activator FPL 64176 (500 nM) on Ca2+ sparklets in the presence and absence of jasplakinolide. In contrast to GnRH, jasplakinolide had no inhibitory effect on Ca2+ sparklet activation by FPL 64176 (Figure 5, A and C; P > .05; n = 8). Jasplakinolide also had no effect on Ca2+ sparklet density (ie, the number of Ca2+ sparklet sites/μm2; Figure 5D; P > .05; n = 8) induced by FPL 64176 and had no effect on subsequent ERK activation (Figure 5E; P > .05; n ≥ 3 independent experiments). These observations are consistent with the hypothesis that stabilization of filamentous actin disrupts communication between the GnRH receptor and the L-type Ca2+ channel rather than interfering with the L-type Ca2+ channel itself or subsequent ERK activation.
Figure 5.

Actin stabilization does not disrupt Ca2+ sparklet stimulation by direct activation of L-type Ca2+ channels or PKC. A and B, Representative traces showing time courses of Ca2+ influx in αT3–1 cells exposed to the L-type Ca2+ channel agonist FPL 64176 (500 nM) (A) or the PKC activator PDBu (50 nM) (B) with or without jasplakinolide (100 nM for 5 min). C and D, Plots of Ca2+ sparklet site activities (nPs) and mean ± SEM Ca2+ sparklet site densities (Ca2+ sparklet sites per square micrometer) for FPL 64176 (500 nM; n = 8 cells in each group) and PDBu (50 nM; n = 11 cells in each group) with or without jasplakinolide (100 nM for 5 min). E, Western blot analysis of ERK activation (as measured by ERK phosphorylation) in αT3–1 cells exposed to FPL 64176 (500 nM) and PDBu (50 nM) with or without jasplakinolide (100 nM for 5 min; n ≥ 3 independent experiments). *, P < .05. cont, control; jas, jasplakinolide.
The observed effects of jasplakinolide indicate that actin dynamics are important for the transduction of GnRH receptor activation to the stimulation of the L-type Ca2+ channel. Given the importance of PKC in this transduction pathway (6) and the data presented above (Figure 3), we examined the effect of the PKC activator PDBu (50 nM) on the cells pretreated with jasplakinolide. In contrast to GnRH, but similar to FPL 64176, jasplakinolide had no effect on the PDBu-dependent stimulation of Ca2+ sparklets (Figure 5, B–D; P > .05, n = 11 cells) and ERK activation (Figure 5E; P > .05; n ≥ 3 independent experiments). These data suggest that the stabilization of filamentous actin does not preclude the PKC-dependent activation of local L-type Ca2+ channel function and ERK activation. Taken together, our data support the concept that precise structural and molecular elements create a microenvironment suitable for localized subplasmalemmal Ca2+ signaling necessary for gonadotrope function.
Discussion
Consistent with the hypothesis that GnRH binding to the GnRH receptor evokes a rapid increase in localized subplasmalemmal Ca2+ signals that lead to ERK activation in αT3–1 gonadotropes, we find the following: 1) GnRH application promoted experimentally evident localized sites of Ca2+ influx across the plasma membrane (ie, Ca2+ sparklets); 2) direct pharmacological activation of L-type Ca2+ channels induced localized Ca2+ influx and increased ERK signaling; 3) pharmacological inhibition of L-type Ca2+ channels decreased observable Ca2+ influx events and decreased ERK signaling; 4) PKC activation, which is associated with increased ERK signaling in gonadotropes and stimulation of L-type Ca2+ channel sparklets in arterial myocytes (10, 12, 13), increased localized L-type Ca2+ channel function in αT3–1 cells; 5) PKC inhibition decreased GnRH-dependent stimulation of L-type Ca2+ channel sparklets and ERK activation; and 6) actin cytoskeletal rearrangement, which is involved with GnRH receptor signaling (22–24), is necessary for GnRH-induced stimulation of localized Ca2+ influx through L-type Ca2+ channels and ERK activation. From these observations we conclude that GnRH-dependent activation of ERK signaling in αT3–1 gonadotropes requires localized PKC-dependent L-type Ca2+ channel activity.
Our data provide strong evidence that Ca2+ influx in αT3–1 gonadotropes is highly localized across the plasma membrane (see Supplemental Movie 1). To quantify the spatial confinement of the observed Ca2+ influx, we compared the average Ca2+ sparklet surface area (2.65 ± 0.10 μm2) to the estimated total surface area of the αT3–1 cells studied (2096 ± 56.04 μm2) as determined by cell capacitance measurements obtained with voltage clamp (25). Individual Ca2+ sparklet sites therefore corresponded to approximately 0.13% (2.65 per 2096) of the αT3–1 cell surface area; each cell had approximately 20.96 ± 0.56 active sites (2096 μm2 × 0.01 Ca2+ sparklet sites/μm2). Thus, in αT3–1 cells stimulated with GnRH, Ca2+ influx was highly localized and limited to approximately 2.72% (0.13% × 20.96) of the total surface area. In addition, high activity (nPs ≥ .2) Ca2+ sparklet sites with values greater than 1 were occasionally observed. This is of interest because it indicates clustering and functional coupling of L-type Ca2+ channels (16, 26). Future experiments to characterize the biochemical nature of these clusters in gonadotropes in greater detail are needed.
Previous efforts using conventional experimental approaches have established the importance of PKC-mediated Ca2+ influx through L-type Ca2+ channels with respect to ERK activation in gonadotropes (5, 6). On the basis of substantial indirect evidence, a local Ca2+ signaling mechanism was hypothesized. Using our experimental approach, we are able to visualize Ca2+ influx through L-type Ca2+ channels. This allows us not only to test directly and confirm a local Ca2+ signaling hypothesis but also permits investigation of participating signaling components (eg, PKC and actin cytoskeletal dynamics) with unprecedented spatial and temporal resolution. Our data demonstrate that GnRH and PDBu stimulate Ca2+ sparklet activity within 5 minutes (at 22–25°C). This time course is similar to that observed by direct channel activation with the L-type Ca2+ channel activator FPL 64176. Consistent with a highly localized signaling process, GnRH receptor stimulation and (subsequent) PKC stimulation therefore appears to involve a relatively rapid mechanism of L-type Ca2+ channel activation.
Consistent with previous work (20) and our Ca2+ sparklet data, we found that the inhibition of PKC with the broad-spectrum PKC inhibitor GFX (but not the PKCα/β selective inhibitor Gö6976) reduced GnRH-dependent activation of ERK signaling. These observations suggest that other PKC isoforms such as PKCδ and/or PKCϵ are involved in ERK activation in αT3–1 cells. Future experiments with isoform-specific PKC inhibitors and isoform-selective knockdown approaches are necessary to further clarify the PKC isoform(s) involved with GnRH-dependent activation of L-type Ca2+ channels. Note that PKC inhibition with GFX produced a modest reduction in ERK activation when compared with L-type Ca2+ channel inhibition with nicardipine. One scenario likely contributing to this observation involves differences in the effect of nicardipine and PKC inhibition on L-type channel Ca2+ sparklets: nicardipine inhibits low (nPs < .2) and high activity (nPs ≥ .2) L-type channel Ca2+ sparklets, whereas the effects of PKC inhibition are limited primarily to high-activity Ca2+ sparklets (9, 10, 12, 27). In addition, when using 2 mM external Ca2+, as in our experiments, many low-activity Ca2+ sparklets are likely below our detection threshold (10). Thus, the substantial low-activity Ca2+ sparklets (not necessarily evident in our recordings) present after the PKC inhibition could result in more Ca2+ influx and ERK activation as compared with L-type Ca2+ channel inhibition with nicardipine.
The findings presented here are consistent with previous research in arterial smooth muscle cells in which localized L-type Ca2+ channel sparklets were previously described (8–10, 12, 16, 28). Similar to the vasoconstrictor angiotensin II (8, 29), which stimulates Gq-coupled angiotensin II type 1 receptors (30), GnRH receptor activation promoted Ca2+ influx in αT3–1 cells by increasing Ca2+ sparklet site activity (nPs) and density (sites per square micrometer). As with Ca2+ sparklet activity in arterial smooth muscle cells, Ca2+ sparklets in αT3–1 cells were stimulated by L-type Ca2+ channel agonists and inhibited by L-type Ca2+ channel antagonists (10, 12). Also similar to arterial smooth muscle (8, 10, 12, 27, 31), L-type channel Ca2+ sparklets in αT3–1 cells are subject to PKC regulation (see Figure 3). Interestingly, and in contrast to arterial smooth muscle in which PKCα is the isoform responsible for the stimulation of L-type channel Ca2+ sparklets (12), GnRH-dependent activation of Ca2+ sparklets in αT3–1 cells does not appear to be dependent on PKCα but rather seems to require other isoforms such as PKCδ and/or PKCϵ. Future studies should focus on clarifying this important issue.
Although PKC is known to activate ERK, the underling mechanism remains unclear (4, 32, 33). Direct activation of Raf-1 kinase by PKC has been proposed, with little supporting evidence, and Raf-1 involvement in GnRH-dependent activation of ERK activation is not apparent (34). Here we demonstrate that GnRH-dependent activation of PKC stimulates Ca2+ influx through L-type Ca2+ channels. Further emphasizing the temporal relationship between GnRH receptor and L-type Ca2+ channel activation is our observation of enhanced ERK phosphorylation (via Western blot analysis) within 5 minutes of GnRH application (at 37°C). Thus, the time course of L-type Ca2+ channel sparklet activation (recordings made between 5 and 10 min after applying GnRH) approximately parallels that of ERK activation. Furthermore, our Western blot data (at 5 and 10 min) supports the previous work in which ERK was shown to be maximally phosphorylated within 5–15 minutes (20). Future efforts are necessary to clarify the intermediate molecular mechanisms by which PKC stimulation of L-type Ca2+ channels leads to ERK activation in gonadotropes.
Our data demonstrate the importance of spatially restricted subplasmalemmal Ca2+ microdomain signaling in mediating ERK activation in response to GnRH receptor stimulation. These findings suggest that a local Ca2+ signal (ie, L-type Ca2+ channel sparklets) could promote discrete sites of ERK activation at or near the sites of Ca2+ influx. Consistent with this hypothesis, work in pancreatic β-cells has shown that localized Ca2+ influx through L-type Ca2+ channels was sufficient for activating ERK in response to glucagon-like peptide-1 (35). In gonadotropes, evidence supporting the concept of localized ERK activation includes the observation that phosphorylated ERK not only associates with membrane fractions containing the GnRH receptor, but also the two proteins coimmunoprecipitate with one another (36). Given these observations, we hypothesize that the localized L-type Ca2+ channel sparklet signaling described in this study could lead to localized ERK activation at the sites of Ca2+ influx. Future studies are required to examine this important concept.
Dynamic remodeling of the actin cytoskeleton is necessary for GnRH receptor-dependent ERK activation (22–24). Here we have identified that the necessity of actin cytoskeletal organization occurs downstream of GnRH receptor activation but upstream of PKC-dependent activation of L-type Ca2+ channels as direct stimulation of the channels (with FPL 64176) or PKC (with PDBu) leads to Ca2+ influx in the presence of jasplakinolide. These data suggest that the stabilization of filamentous actin does not preclude PKC-dependent activation of local L-type Ca2+ channel function and ERK activation per se. However, because global phorbol ester-induced activation of PKC is nonphysiological, it is unreasonable to conclude that actin dynamics are not involved with physiologically relevant instances of PKC activation (eg, by GnRH receptor stimulation). Future studies are required to address this important issue and to further dissect the role of actin cytoskeletal dynamics with respect to the subcellular structural organization required for the generation of biologically relevant L-type channel Ca2+ microdomains in gonadotropes.
Although we provide compelling evidence demonstrating the importance of localized Ca2+ influx in mediating ERK activation, our observations do not preclude the involvement of other mechanisms. Close inspection of our data suggests that other mechanisms could be involved. PKC inhibition and actin cytoskeletal disruption with jasplakinolide abolished the stimulatory effect of GnRH on L-type Ca2+ channels, but the inhibitory effects on ERK activation were incomplete. Similarly, nicardipine eliminated L-type Ca2+ channel sparklet activity in response to GnRH but did not fully eliminate changes in ERK phosphorylation. These findings are in contrast to experiments in which we bypassed GnRH receptor signaling altogether and stimulated the L-type Ca2+ channels directly with FPL 64176. With this approach we found that nicardipine not only eliminated Ca2+ sparklet activity but also abolished FPL 64176-mediated ERK activation. Taken together, we conclude that although local L-type Ca2+ channel signaling is a major mechanism underlying ERK activation in response to GnRH, other contributing mechanisms are likely involved.
In summary, we provide the first direct evidence for localized, subplasmalemmal Ca2+ signaling in gonadotropes. Because our approach allows us to detect and quantify GnRH-induced Ca2+ sparklets in real time, the field is now positioned to address an entirely new set of questions as to the identity of proximate cellular and molecular events that convey the extracellular GnRH signal to its most distal and biologically critical intracellular targets. From the therapeutic perspective, this is key because the ability of the gonadotrope to integrate multiple molecular events in response to a GnRH signal resides at the core of the hypothalamic-pituitary-gonadal axis and thus reproductive function in mammals. In light of this, it is not surprising that the gonadotrope is the primary target of the vast majority of pro- and antifertility drugs (37–40). Thus, increasingly refined pharmacological modulation of reproductive and sexual function unequivocally depends on a complete understanding of GnRH signaling in the pituitary. Finally, understanding localized Ca2+ signaling in αT3–1 cells may well have relevance beyond GnRH action. Indeed, the similarities between the L-type Ca2+ channel sparklets in arterial smooth muscle (16) and pituitary endocrine cells suggests a conserved signaling mechanism fundamental to a diverse group of physiological and cellular processes.
Additional material
Supplementary data supplied by authors.
Acknowledgments
This work was supported in whole or in part by the National Institutes of Health Grants R01HD065943 (to C.M.C.), 1R01HL111060 (to G.C.A.), and P20GM103432 (to A.M.N.); the Colorado State University College Research Council (to G.C.A. and C.M.C.); the National Center for Research Resources Grant P20RR016474 (to A.M.N.); and the Pew Charitable Trusts (to G.C.A.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- [Ca2+]i
- intracellular calcium concentration
- DAG
- diacylglycerol
- GFX
- GF109203X
- IP3
- inositol-1,4,5-trisphosphate
- nPs
- number of quantal levels detected and probability that the site is active
- PDBu
- phorbol 12, 13-dibutyrate
- PKC
- protein kinase C
- TIRF
- total internal reflection fluorescence.
References
- 1. Counis R, Laverrière JN, Garrel G, et al. Gonadotropin-releasing hormone and the control of gonadotrope function. Reprod Nutr Dev. 2005;45(3):243–254. [DOI] [PubMed] [Google Scholar]
- 2. Grosse R, Schmid A, Schoneberg T, et al. Gonadotropin-releasing hormone receptor initiates multiple signaling pathways by exclusively coupling to G(q/11) proteins. J Biol Chem. 2000;275(13):9193–9200. [DOI] [PubMed] [Google Scholar]
- 3. Naor Z. Signaling by G-protein-coupled receptor (GPCR): studies on the GnRH receptor. Front Neuroendocrinol. 2009;30(1):10–29. [DOI] [PubMed] [Google Scholar]
- 4. Roberson MS, Misra-Press A, Laurance ME, Stork PJ, Maurer RA. A role for mitogen-activated protein kinase in mediating activation of the glycoprotein hormone alpha-subunit promoter by gonadotropin-releasing hormone. Mol Cell Biol. 1995;15(7):3531–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mulvaney JM, Zhang T, Fewtrell C, Roberson MS. Calcium influx through L-type channels is required for selective activation of extracellular signal-regulated kinase by gonadotropin-releasing hormone. J Biol Chem. 1999;274(42):29796–29804. [DOI] [PubMed] [Google Scholar]
- 6. Mulvaney JM, Roberson MS. Divergent signaling pathways requiring discrete calcium signals mediate concurrent activation of two mitogen-activated protein kinases by gonadotropin-releasing hormone. J Biol Chem. 2000;275(19):14182–14189. [DOI] [PubMed] [Google Scholar]
- 7. White BR, Duval DL, Mulvaney JM, Roberson MS, Clay CM. Homologous regulation of the gonadotropin-releasing hormone receptor gene is partially mediated by protein kinase C activation of an activator protein-1 element. Mol Endocrinol. 1999;13(4):566–577. [DOI] [PubMed] [Google Scholar]
- 8. Amberg GC, Earley S, Glapa SA. Local regulation of arterial L-type calcium channels by reactive oxygen species. Circ Res. 2010;107(8):1002–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Amberg GC, Navedo MF, Nieves-Cintrón M, Molkentin JD, Santana LF. Calcium sparklets regulate local and global calcium in murine arterial smooth muscle. J Physiol. 2007;579(Pt 1):187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Navedo MF, Amberg GC, Votaw VS, Santana LF. Constitutively active L-type Ca2+ channels. Proc Natl Acad Sci USA. 2005;102(31):11112–11117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Windle JJ, Weiner RI, Mellon PL. Cell lines of the pituitary gonadotrope lineage derived by targeted oncogenesis in transgenic mice. Mol Endocrinol. 1990;4(4):597–603. [DOI] [PubMed] [Google Scholar]
- 12. Navedo MF, Amberg GC, Nieves M, Molkentin JD, Santana LF. Mechanisms underlying heterogeneous Ca2+ sparklet activity in arterial smooth muscle. J Gen Physiol. 2006;127(6):611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chaplin NL, Amberg GC. Hydrogen peroxide mediates oxidant-dependent stimulation of arterial smooth muscle L-type calcium channels. Am J Physiol Cell Physiol. 2012;302(9):C1382–C1393. [DOI] [PubMed] [Google Scholar]
- 14. Maravall M, Mainen ZF, Sabatini BL, Svoboda K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys J. 2000;78(5):2655–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Navedo MF, Cheng EP, Yuan C, et al. Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ Res. 2010;106(4):748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Navedo MF, Amberg GC. Local regulation of L-type Ca2+ channel sparklets in arterial smooth muscle. Microcirculation. 2013;20(4):290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zheng W, Rampe D, Triggle DJ. Pharmacological, radioligand binding, and electrophysiological characteristics of FPL 64176, a novel nondihydropyridine Ca2+ channel activator, in cardiac and vascular preparations. Mol Pharmacol. 1991;40(5):734–741. [PubMed] [Google Scholar]
- 18. Toullec D, Pianetti P, Coste H, et al. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266(24):15771–15781. [PubMed] [Google Scholar]
- 19. Gekeler V, Boer R, Überall F, et al. Effects of the selective bisindolylmaleimide protein kinase C inhibitor GF 109203X on P-glycoprotein-mediated multidrug resistance. Br J Cancer. 1996;74(6):897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dobkin-Bekman M, Rahamin-Ben Navi L, Shterntal B, et al. Differential role of PKC isoforms in GnRH and phorbol 12-myristate 13-acetate activation of extracellular signal-regulated kinase and Jun N-terminal kinase. Endocrinology. 2010;151(10):4894–4907. [DOI] [PubMed] [Google Scholar]
- 21. Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase Cμ by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996;392(2):77–80. [DOI] [PubMed] [Google Scholar]
- 22. Navratil AM, Knoll JG, Whitesell JD, Tobet SA, Clay CM. Neuroendocrine plasticity in the anterior pituitary: gonadotropin-releasing hormone-mediated movement in vitro and in vivo. Endocrinology. 2007;148(4):1736–1744. [DOI] [PubMed] [Google Scholar]
- 23. Navratil AM, Dozier MG, Whitesell JD, Clay CM, Roberson MS. Role of cortactin in dynamic actin remodeling events in gonadotrope cells. Endocrinology. 2014;155(2):548–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davidson L, Pawson AJ, Millar RP, Maudsley S. Cytoskeletal reorganization dependence of signaling by the gonadotropin-releasing hormone receptor. J Biol Chem. 2004;279(3):1980–1993. [DOI] [PubMed] [Google Scholar]
- 25. Hille B. Ion Channels of Excitable Membranes. 3rd ed Suderland, MA: Sinauer Associates, Inc; 2001. [Google Scholar]
- 26. Dixon RE, Yuan C, Cheng EP, Navedo MF, Santana LF. Ca2+ signaling amplification by oligomerization of L-type Cav1.2 channels. Proc Natl Acad Sci USA. 2012;109(5):1749–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Navedo MF, Nieves-Cintrón M, Amberg GC, et al. AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin II-induced hypertension. Circ Res. 2008;102(2):e1–e11. [DOI] [PubMed] [Google Scholar]
- 28. Santana LF, Navedo MF, Amberg GC, Nieves-Cintrón M, Votaw VS, Ufret-Vincenty CA. Calcium sparklets in arterial smooth muscle. Clin Exp Pharmacol Physiol. 2008;35(9):1121–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nieves-Cintrón M, Amberg GC, Navedo MF, Molkentin JD, Santana LF. The control of Ca2+ influx and NFATc3 signaling in arterial smooth muscle during hypertension. Proc Natl Acad Sci USA. 2008;105(40):15623–15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52(4):639–672. [PubMed] [Google Scholar]
- 31. Chaplin NL, Amberg GC. Stimulation of arterial smooth muscle L-type calcium channels by hydrogen peroxide requires protein kinase C. Channels (Austin). 2012;6(5):385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Naor Z, Benard O, Seger R. Activation of MAPK cascades by G-protein-coupled receptors: the case of gonadotropin-releasing hormone receptor. Trends Endocrinol Metab. 2000;11(3):91–99. [DOI] [PubMed] [Google Scholar]
- 33. Sundaresan S, Colin IM, Pestell RG, Jameson JL. Stimulation of mitogen-activated protein kinase by gonadotropin-releasing hormone: evidence for the involvement of protein kinase C. Endocrinology. 1996;137(1):304–311. [DOI] [PubMed] [Google Scholar]
- 34. Bliss SP, Navratil AM, Xie J, Roberson MS. GnRH signaling, the gonadotrope and endocrine control of fertility. Front Neuroendocrinol. 2010;31(3):322–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Selway J, Rigatti R, Storey N, Lu J, Willars GB, Herbert TP. Evidence that Ca2+ within the microdomain of the L-type voltage gated Ca2+ channel activates ERK in MIN6 cells in response to glucagon-like peptide-1. PLoS One. 2012;7(3):e33004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bliss SP, Navratil AM, Breed M, Skinner DC, Clay CM, Roberson MS. Signaling complexes associated with the type I gonadotropin-releasing hormone (GnRH) receptor: colocalization of extracellularly regulated kinase 2 and GnRH receptor within membrane rafts. Mol Endocrinol. 2007;21(2):538–549. [DOI] [PubMed] [Google Scholar]
- 37. Kousta E, White DM, Franks S. Modern use of clomiphene citrate in induction of ovulation. Hum Reprod Update. 1997;3(4):359–365. [DOI] [PubMed] [Google Scholar]
- 38. Hughes E, Collins J, Vandekerckhove P. Clomiphene citrate for ovulation induction in women with oligo-amenorrhoea. Cochrane Database Syst Rev. 2000(2):CD000056. [DOI] [PubMed] [Google Scholar]
- 39. Practice Committee of the American Society for Reproductive Medicine. Use of clomiphene citrate in infertile women: a committee opinion. Fertil Steril. 2013;100(2):341–348. [DOI] [PubMed] [Google Scholar]
- 40. Tomao F, Lo Russo G, Spinelli GP, et al. Fertility drugs, reproductive strategies and ovarian cancer risk. J Ovarian Res. 2014;7:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.