

Abstract
The Toll-like receptors (TLRs) are critical components of the innate immune system that regulate immune recognition in part through NF-κB activation. A human cell-based high throughput screen (HTS) revealed substituted 4-aminoquinazolines to be small molecular weight activators of NF-κB. The most potent hit compound predominantly stimulated through the human TLR4/MD2 complex, and had less activity with the mouse TLR4/MD2. There was no activity with other TLRs and the TLR4 activation was MD-2 dependent and CD14 independent. Synthetic modifications of the quinazoline scaffold at the 2 and 4 positions revealed trends in structure-activity relationships with respect to TLR dependent production of the NF-κB associated cytokine IL-8 in human peripheral blood mononuclear cells, as well as IL-6 in mouse antigen presenting cells. Furthermore, the hit compound in this series also activated the interferon signaling pathway resulting in type I interferon production. Substitution at the O-phenyl moiety with groups such as bromine, chlorine and methyl resulted in enhanced immunological activity. Computational studies indicated that the 4-aminoquinazoline compounds bind primarily to human MD-2 in the TLR4/MD-2 complex. These small molecules, which preferentially stimulate human rather than mouse innate immune cells, may be useful as adjuvants or immunotherapeutic agents.
Keywords: Toll-like receptor 4, MD-2, 4-aminoquinazolines, NF-κB activation, innate immune response
The first line of defense against microbial pathogens such as viruses, bacteria, fungi and protozoa is governed by the innate immune response, which includes barrier tissues and leukocytes. Many innate signaling pathways converge on the NF-κB family of transcription factors,1, 2 which are critical in the regulation of these responses. A small molecular weight molecule that is an effective NF-κB activator could be incorporated into vaccine adjuvants as an innate immune activator. Adjuvants are added to antigens in vaccines to augment adaptive immune responses to poorly immunogenic antigens, and to increase protective antibody titers in at risk populations due to age (infants and the elderly), or disease (diabetes, liver failure). Adjuvants also facilitate the use of smaller doses of antigen, and enable effective immunization with fewer booster immunizations.3 Development of small molecular weight non-lipid ligands that are reactive in human cells might have several advantages to address different immunological requirements for vaccines or immune therapeutics.
As part of our studies on small molecules that can activate NF-κB signaling, we conducted a high through-put screening (HTS) campaign in which a commercially available library of over 170,000 compounds was screened in a human monocyte cell-based NF-κB activation assay. By utilizing a cell based assay we theorized that we could identify small molecules that activated NF-κB through a broad range of mechanisms. Following the cluster enrichment analysis,4 225 compounds were selected for further in vitro biological evaluation involving cytokine induction assays in primary cells, including human peripheral blood mononuclear cells (hPBMC), mouse splenocytes, mouse bone marrow derived dendritic cells (mBMDC) and mouse bone marrow derived macrophages (mBMDM).5 These cells were incubated in triplicate with each of the 225 compounds at a single concentration (1 μM for splenocytes and BMDC, 5 μM for all other mouse cells, and 5 μM for human cells) and the supernatants were tested for the presence of NF-κB dependent cytokines, IL-8 or IL-6, released from the human or mouse cells respectively. Thirty-nine of the 225 compounds stimulated the human and mouse cells to secrete IL-8 or IL-6 above the detectable limit. To further confirm activity, these compounds were repurchased and retested by stimulating hPBMC and mBMDC with titrated doses and assaying for IL-8 and IL-6 respectively.
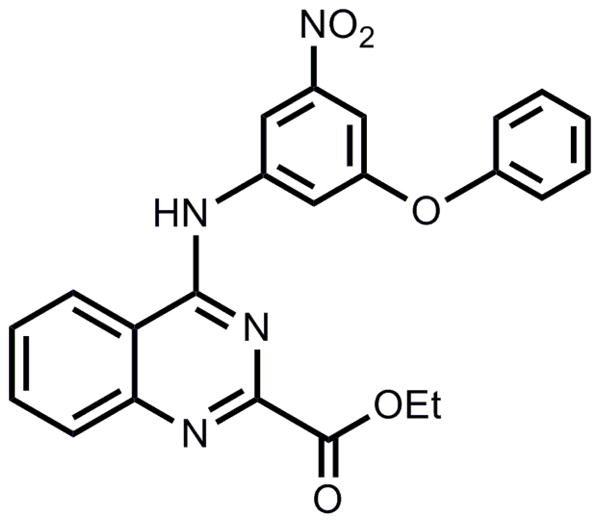
Four structurally diverse library scaffolds were identified in these cytokine assays as having reproducible responses. Among these scaffolds, the substituted 4-aminoquinazolines emerged as the most potent class of compounds in activating human cells. As the Toll-like receptors (TLRs) are the most well understood and studied family of innate immune receptors these receptors were considered as likely targets for the HTS hit compounds. A few examples of pathogen-associated molecular patterns that are natural TLR ligands and their corresponding TLRs include: lipopeptides (TLR2), double-stranded RNA (TLR3), lipopolysaccharide (LPS, TLR4),6 bacterial flagellin (TLR5), guanine and uridine-rich single-stranded RNA (TLR7, 8), and hypo-methylated CpG rich DNA (TLR9).7 The specific innate receptor for the leading hit compound of the 4-aminoquinazolines was determined using primary cells from genetically modified mice, and mouse and human TLR reporter cells. Here we report our discovery and initial structure-activity relationship (SAR) studies of the substituted 4-aminoquinazolines class of ligands and their characterization as TLR4/MD-2 agonists. Within this scaffold cluster, the leading hit from the initial primary and secondary screens was a nitrophenyl-containing 4-aminoquinazoline bearing a phenoxy group on the nitrophenyl moiety (Figure 1).
Figure 1. Structure of hit compound 1a.
To identify the target receptor, we used HEK293 cells stably transfected with the following human TLRs (TLR2, TLR3, TLR4/MD-2/CD14, TLR5, TLR7, TLR8 or TLR9) along with an NF-κB activation reporter producing Secreted alkaline phosphatase (SEAP, Figure 2A). Among the tested TLR transfected cells, only those expressing TLR4/MD-2/CD14 responded to hit compound 1a, as shown in Figure 2A. To further confirm that TLR4 was indeed the receptor, compound 1a was assayed for IL-6 production in mBMDCs from wild type and TLR4 deficient mice (Figure 2B). Genetic disruption of TLR4 completely abrogated IL-6 secretion induced by compound 1a.
Figure 2. Compound 1a is a TLR4 specific ligand that activates NFκB in a TLR4/MD-2-dependent, but CD14-independent manner.

(A) Human TLR2, TLR3, TLR4/MD-2/CD14, TLR5, TLR7, TLR8, and TLR9 HEK 293 Blue cells or NFκB/SEAPorter cells were incubated with 1a (10 μM) for 20–24 h, and activation was evaluated by SEAP secretion in the culture supernatants using SEAPorter assay kit. Data shown are mean ± SEM of triplicates and representative of two to three independent experiments showing similar results. * denotes p < 0.05 was considered significant compared to the vehicle control using Student’s t test. (B–D) mBMDC prepared from wild type mice or mice genetically deficient for TLR4 (B), MD-2 or CD14 (C) were stimulated with 1a (10 μM). IL-6 levels in the culture supernatant were determined by ELISA. (D) Human TLR4 transfectoma cells were incubated with 10 μM 1a or 10 ng/mL LPS in the presence or absence of TLR4 antagonist LPS-RS (12, 111, 1000 ng/mL). Activation of the TLR4/NFκB pathway was evaluated by SEAP secretion in the culture supernatants. * denotes p < 0.05 considered as significant compared to vehicle using one way ANOVA with Dunnett’s post hoc testing. (E) Human PBMC were incubated with 1a alone or 1a with polymyxin B (10 μg/mL) overnight. IL-8 in the culture supernatants was measured by ELISA. Data shown are mean ± SEM of triplicates and representative of two independent experiments showing similar results.
TLR4 signals through two distinct pathways, leading respectively to NF-κB dependent cytokines and type I interferon (IFN) production. Several naturally occurring TLR4 ligands and MPLA have been reported to require CD14 to activate the IFN regulatory pathway.8 The hTLR4 reporter cell line (Figure 2A) also overexpresses hMD-2 and hCD14, which are TLR4 accessory proteins.9, 10 Compound 1a, however, was not dependent on CD14, but dependent on MD-2 for IL-6 production, as demonstrated using mBMDC genetically deficient for CD14 or MD-2 (Figure 2C).
The binding of compound 1a to the TLR4/MD-2/CD14 complex was further confirmed using a competitive antagonist for the TLR4 binding complex, LPS-RS (LPS from Rhodobacter sphaeroides).11 LPS-RS inhibited NF-κB activation in hTLR4/MD-2/CD14 transfectoma cells induced by compound 1a in a dose dependent manner, indicating that compound 1a bound to the TLR4 complex (Figure 2D). Since TLR4/MD-2/CD14 was the receptor complex for the active compounds in this series, it was important to rule out the possibility that activity might have been caused by LPS contamination. Therefore, compound 1a (and all active derivatives) were assayed for LPS (endotoxin) levels using a commercially available detection system and found to contain less than 2 EU/μmol of endotoxin. To further exclude contamination, compound 1a was re-synthesized according to Scheme 1. Both samples of compound 1a displayed indistinguishable physicochemical and biological properties, indicating that the positive biological activity was not due to LPS or another contaminant. Furthermore, the IL-8 inducing ability of compound 1a was not reduced when assayed in the presence of polymyxin B, an LPS binding agent (Figure 2E) although the same concentration of polymixin B significantly suppressed IL-8 release by LPS stimulation (Figure 2F).
Scheme 1.

The engagement by active compounds, such as 1a, of the TLR4/MD-2/CD14 complex may result in signaling through the two pathways mentioned above. To evaluate whether the type I IFN pathway was activated, hPBMC were incubated with compound 1a or with LPS or vehicle overnight. The release of type I IFN in the culture supernatants was then determined in a type I IFN reporter cell assay. Compound 1a was found to induce the release of type I IFN (Figure 3A).
Figure 3. Compound 1a induces type I IFN and expression of co-stimulatory molecules.

(A) Human PBMC were incubated with 1a (10 μM), LPS (10 ng/mL), or vehicle overnight. The release of type I IFN in the culture supernatants was measured by luciferase release in L929-ISRE reporter cells. Data shown are mean ± SEM of triplicates. (B) 1a- or vehicle- treated wild type or Tlr4 −/− mBMDC were stained for CD11c and either CD40, CD80, or CD86. The expression levels of CD40, CD80, or CD86 were evaluated in the gated CD11c+ population. DMSO 0.5% served as the vehicle control. (C) Human PBMC were incubated with compound 1a (5 μM) overnight. CD80 and CD83 expression in gated CD11c+HLA-DR+ population was analyzed by flow cytometer. Data are representative of two independent experiments showing similar results.
To evaluate whether treatment of DCs with this compound can induce maturation in these cells, mBMDC prepared from wild type or TLR4 deficient mice were exposed to compound 1a and then assayed for expression of co-stimulatory molecules CD40, CD80, and CD86 in the CD11c+ population (Figure 3B). The expression of all three co-stimulatory molecules was increased in the wild type cells treated with compound 1a, but not in the TLR4 deficient cells. Induction of co-stimulatory molecules by compound 1a was also confirmed in human PBMC. After incubation of PBMC with compound 1a overnight, increased expression of CD80 and CD83 was observed in the DC-enriched (CD11c+HLA-DR+) gated population (Figure 3C).
The original HTS of the compound library was conducted using THP-1 cells, a human monocytic leukemia cell line, wherein four scaffolds of active compounds were identified. For further in vivo immunological studies, mouse models are commonly used. It was therefore of interest to determine the relative level of immunoactivity in both mouse and human cells for the 4-aminoquinazolines. To examine this, RAW264.7 cells (a mouse macrophage cell line) or THP-1 cells, both containing a Fluorescence Resonance Energy Transfer (FRET) β-lactamase reporter gene construct downstream of an NF-κB promoter sequence, were incubated with graded concentrations of compound 1a for 18 h. The levels of NF-κB activation were determined, normalized to the level induced by LPS and expressed as percent activation (Figure 4A). The hit compound 1a is significantly more potent in human THP-1 versus mouse RAW264.7 cells. Cytokine induction was evaluated by exposing mBMDC or hPBMC to graded concentrations of compound 1a and levels of IL-6 or IL-8, respectively, were determined by ELISA (Figure 4B and C). Results show that even at concentrations below 1 μM, compound 1a was able to induce detectable levels of IL-8 by hPBMC whereas higher concentrations were required to induce detectable levels of IL-6 by mBMDC, thus supporting the notion that the active hit compound in this class is more potent in human cells than in mouse cells. The viability of mouse BMDC and human PBMC following 18 h incubation with compound 1a (10 μM) was 117 ± 2.6% and 112 ± 2.1% by MTT assay, respectively, indicating that the lower activity of compound 1a was not due to its cytotoxicity. In fact, in vitro studies using BMDM prepared from transgenic mice12 that were genetically altered to express human TLR4/MD-2 and deficient for murine TLR4/MD-2 showed that compounds 1a and 1g in this series do indeed activate the transgenic human TLR4/MD-2 complex to a greater extent relative to mouse TLR4/MD-2 (Figure 4C and 4D).
Figure 4. Compound 1a is more potent in human cells.

(A) CellSensor® RAW264.7 or CellSensor® THP-1 cells were incubated with graded concentrations of 1a. The levels of NFκB activation were normalized with LPS and expressed as percent activation. The average response ratio of LPS (5 ng/mL) in RAW and THP-1 CellSensorκ cells was 6.5 ± 0.17 and 3.27 ± 0.67, respectively. (B and C) Murine BMDC (B) or hPBMC (C) were stimulated with graded concentrations of 1a overnight. Murine IL-6 or hIL-8 in the culture supernatant was determined by ELISA. Data shown are mean ± SEM of triplicates and representative of two independent experiments showing similar results.
The innate immune receptor that is responsible for the physiological recognition of LPS is TLR4, in association with MD-2.13, 14 The structural basis of receptor specificity and of the mechanism of activation by LPS have recently been elucidated by determining the crystal structure of the TLR4/MD-2-LPS complex at 3.1 A resolution.15 Binding of agonistic ligands, such as LPS, cause dimerization of the extracellular domains to form a TLR4/MD-2-LPS complex. Like the extracellular domains of other TLRs, TLR4 contains leucine-rich repeats and adopts a characteristic horseshoe-like shape. MD-2 is non-covalently bound to the side of the horseshoe ring and also directly interfaces with the ligand. MD-2 has a β-cup fold structure composed of two antiparallel-β sheets forming a large hydrophobic pocket for ligand binding. LPS binds to this pocket and directly mediates dimerization of the two TLR4/MD-2 complexes. TLR4 can be activated by structurally diverse LPS molecules, which have been predicted to occupy this pocket in MD-2.
The 4-aminoquinazoline hit compound discovered in the present study was also thought to bind to the TLR4/MD-2 complex. Accordingly, we examined the predicted binding mode(s) of compound 1a to the crystal structure of the human TLR4/MD-2 complex (PDB: 3fxi) using the docking program HEX,16 and AMPAC17 for optimization of the compound conformation. We selected the best configurations of 1a bound to this complex based on molecular surface shape complementarity and the most favorable intermolecular energy of interactions. Interestingly, the best docking position for 1a was within the LPS pocket.15
Figure 5 shows the predicted binding mode of compound 1a in the TLR4/MD-2 model, while the set of binding interactions that may keep the compound in the MD-2 pocket bound to both TLR4 and MD-2 is depicted in Figure 6. There is a set of possible hydrogen bonds formed by the residues Gln436 and Glu439 of TLR4 and Arg90 of MD-2. Noteworthy is the interaction of the two polar nitro oxygens of the compound with the backbone nitrogens of Ile124 and Lys122 of the MD-2 protein. The positive charge of the Lys122 provides a significant attractive force for a nitro oxygen in the human TLR4/MD-2 complex, whereas in the mouse complex, this residue is replaced by glutamic acid. Thus, the negative charge of the Glu122 in the mouse MD-2 causes an increase in repulsion energy as it encounters the negative oxygens of the nitro function in the compound and is likely at least partially responsible for the decreased activity of the compound in mouse cells relative to human cells. Also, there are several hydrophobic interactions with MD-2, involving residues Leu87, Phe126, Ile124, Phe121, and Phe119. Taken together, such interactions of the compound with two proteins can improve the free energy of complex formation by approximately 10 kcal/mole.
Figure 5. Predicted binding mode of compound 1a to human TLR4/MD-2 complex.

The compound has attractive interactions with the seven residues of MD-2, and three residues of TLR-4 which significantly increase the probability and stability of binding.
Figure 6. Predicted binding interactions of compound 1a with hTLR4/MD-2 complex (PDB ID: 3fxi).

The set of H-bond and hydrophobic interactions supports the prediction of the compound position in the pocket. Purple—TLR4, yellow—MD-2.
The above data indicate that compound 1a activates NF-κB preferentially through the human TLR4/MD-2 complex rather than through the murine TLR4/MD-2 complex, in a CD14 independent manner. Considering this selective activity, it was thought that compound 1a might serve as a useful chemical probe to investigate TLR4/MD-2 receptor-ligand specificity. Hence, compound 1a provided a starting point for structure-activity studies.
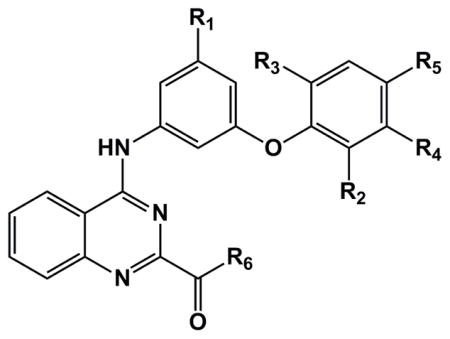
The primary HTS library contained a very small family of compounds in the 4-aminoquinazoline class and therefore provided only a limited indication of structural features important for activation of NF-κB. Substitutions at three sites were first undertaken to probe structural requirements and limitations with respect to optimization of cytokine induction in hPBMC and induction of NF-κB activation in TLR4 reporter cells (Table 1). While keeping all other structural features of hit compound 1a constant, we prepared a series of derivatives with variations at 1) the phenoxy moiety; 2) the nitro group; and 3) the ester function. The synthesis began with construction of the appropriately substituted nitroaniline as shown in Scheme 1. Trinitrobenzene was carefully reacted with appropriately substituted phenols according to the general method of Shevelev et al.18 to provide the corresponding dinitrophenoxy compounds 2 a–j, and k. The dinitro intermediates were then partially reduced to the corresponding nitroanilines (3 a–j, and k) using ammonium sulfide. These nitroanilines were condensed with the chloroquinazoline compound 419 to provide the corresponding substituted 4-aminoquinazolines 1 a–j and k. Thus, we re-synthesized the original hit compound 1a by this method along with a variety of phenoxy derivatives substituted with alkyl and halogen groups. As indicated in Table 1, the results from assays using the TLR4 reporter cells and hPBMC suggest that when the phenoxy group in 1a is substituted in the ortho position by groups such as chloro (1f), bromo (1g), iodo (1h), or methyl (1b), the activity is enhanced relative to hydrogen (values in the Table are normalized to LPS positive control=100). However, if the alkyl group is larger than methyl, such as ethyl (1e), a substantial loss of activity is observed. Likewise, if the aryloxy group is larger than phenoxy, such as naphthyloxy (1k), a loss of activity is also encountered. The importance of the nitro group in compound 1a for cytokine induction was investigated by reduction of the nitro to the primary amine compound (5a) using Raney nickel which resulted in complete loss of activity. Next the ester function was modified to include conversion to the free carboxylic acid (6a, 7a, 7g) and then to the carboxamide (9a) and the N-ethylcarboxamide (8a). Other esters were formed including the methyl (10a), isopropyl (11a) and isoamyl (12a) esters. The compound that combines the features of the ortho-bromophenoxy moiety and the isopropyl ester (13g) was also prepared.
Table 1.
Activity of substituted 4-aminoquinazolines in hTLR4 transfectoma and primary human PBMC.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| cmpd | R1 | R2 (o) | R3 (o2) | R4 (m) | R5 (p) | R6 | hTLR41 | PBMC2 | HepG2 |
| 0.5 μM | 0.5 μM | MTT3 | |||||||
| LPS | 100 | 100 | 100.9 | ||||||
| 1a | NO2 | H | H | H | H | OC2H5 | 49.1 | 22.2 | 93.5 |
| 1b | NO2 | CH3 | H | H | H | OC2H5 | 45.5 | 20.5 | 96.7 |
| 1c | NO2 | H | H | CH3 | H | OC2H5 | 14.5 | 5.8 | 109.9 |
| 1d | NO2 | H | H | H | CH3 | OC2H5 | 0.5 | 0.4 | 108.8 |
| 1e | NO2 | Et | H | H | H | OC2H5 | 4.2 | 3.2 | 98.9 |
| 1f | NO2 | Cl | H | H | H | OC2H5 | 73.9 | 27.3 | 107.4 |
| 1g | NO2 | Br | H | H | H | OC2H5 | 79.0 | 35.6 | 90.8 |
| 1h | NO2 | I | H | H | H | OC2H5 | 65.5 | 34.2 | 93.8 |
| 1i | NO2 | CF3 | H | H | H | OC2H5 | 58.7 | 26.5 | 67.1 |
| 1j | NO2 | CH3 | CH3 | H | H | OC2H5 | 0.6 | 0.0 | 106.3 |
| 1k | NO2 | H | H | * | * | OC2H5 | 0.8 | 0.0 | 111.4 |
| 5a | NH2 | H | H | H | H | OC2H5 | 0.6 | 0.3 | 106.1 |
| 6a | NH2 | H | H | H | H | OH | 1.1 | 0.3 | 105.0 |
| 7a | NO2 | H | H | H | H | OH | 0.6 | 0.5 | 84.5 |
| 7g | NO2 | Br | H | H | H | OH | 1.6 | 0.0 | 96.3 |
| 8a | NO2 | H | H | H | H | NHC2H5 | 2.2 | 1.5 | 107.4 |
| 9a | NO2 | H | H | H | H | NH2 | 0.0 | 0.3 | 78.4 |
| 10a | NO2 | H | H | H | H | OCH3 | 0.4 | 0.2 | 116.4 |
| 11a | NO2 | H | H | H | H | O-isopropyl | 7.4 | 2.1 | 65.6 |
| 12a | NO2 | H | H | H | H | O-isoamyl | 8.4 | 2.2 | 63.4 |
| 13g | NO2 | Br | H | H | H | O-isopropyl | 67.8 | 19.2 | 106.5 |
2-Naphthol instead of Phenol
Average Quantiblue OD630 of hTLR4 HEK Blue cells incubated with LPS (100 ng/mL) was 1.22± 0.17.
Average IL-8 release by 100 ng/mL LPS was 25.9 ± 5.6 ng/mL.
MTT assay data (compounds tested at 10 μM) were normalized with OD570–650 of vehicle treatment in the related experiments. The average MTT reading for HepG2 cells treated with vehicle was 1.326 ± 0.294.
Besides the necessity of a nitro group in the aminophenyl moiety for TLR4 binding activity, a comparison in hTLR4 reporter cells of selected compounds from the SAR studies for modifications at the ortho-position of the phenoxy moiety suggests that lower alkyl or halogen groups at this position are favored for activity (Figure 7A). Likewise, for the ester function at the 2-position of the quinazoline ring, the ethyl ester is preferred over a smaller or larger alkyl group (Figure 7B). In primary hPBMC, the same trends are apparent, particularly at the lower, more physiologically relevant concentration of 0.5 μM (Figure 7C and 7D).
Figure 7. Representative results of substituted 4-aminoquinazolines using hTLR4 transfectoma and primary hPBMC.

(A and B) Human TLR4 transfectoma cells were incubated with graded concentrations of 1a or indicated SAR compounds (A: 1e and 1g, B: 11a, and 13a) for 20–22 h. Release of SEAP was measured by OD630 in a QuantiBlue assay. (C and D) Human PBMC from two independent donors were stimulated with 0.5 or 5 μM1a overnight. IL-8 in the culture supernatant was determined by ELISA. Data shown are mean ± SEM of triplicate and represents two to three experiments showing similar results.
In the course of an HTS designed to identify activators of innate immunity, a series of substituted 4-aminoquinazolines was discovered as selective TLR4 ligands. Small molecules of this class are unique among TLR4 activators in that they are “non-lipid-like”. Structure-activity evaluation in both mouse and human cells revealed that this series of small molecules activates the human TLR4/MD-2 complex more potently than the corresponding mouse complex and this activation was observed to be MD-2-dependent and CD14-independent. Interestingly, a recent report describes a natural product, Euodenine A,20 that activates human TLR4 more potently than the corresponding mouse receptor and shares a similar immunologic profile with the 4-aminoquinazolines described here. The 4-aminoquinazolines species preference phenomenon could be explained, at least in part, by the observation that certain amino acid residues of the MD-2 proteins of the TLR4/MD-2 complex vary in mouse versus human at critical interaction binding sites for the 4-aminoquinazoline compounds. Indeed, a simple change in residue 122 of MD-2 from Lys in human to Glu in mouse is predicted to reverse the electrostatic influence of binding of the nitro oxygen(s) of, for example, the active compound 1a. The result is an attractive force in the human complex for the compound, but a repulsive force at the same site in the mouse complex.
Initial SAR studies revealed several findings. First, the nitro group on the phenylamino moiety is essential for activity since reduction to amino abrogated the activity. Indeed, computational studies showed that interactions at the predicted binding site of MD-2 for the nitro compound were much more favorable (by as much as 6.1 kcal/mol total energy improvement in interaction energy) compared to the corresponding amino compound. Aromatic or heteroaromatic nitro groups are well known in drug design. Examples are the 5-nitrofurans and 2- and 5-nitroimidazoles. These drugs are used as therapeutic agents against a variety of protozoan and anaerobic bacterial infections in humans and animals.21 However, the compounds reported here represent the first known class of nitro-containing small molecules that can activate a human innate immune receptor. Second, a small alkyl or halogen group at the ortho position of the phenoxy moiety enhances activity. Third, a lower alkyl ester at the 2-position of the quinazoline ring appears to be favored for activity, while compounds having the corresponding carboxylic acid form or unsubstituted amide are not active. Interestingly none of the substitutions increased the activity of the compound to preferentially stimulate murine rather than human cells, a finding opposite from that observed with certain other small molecule TLR4 ligands, pyrimido[5,4-b]indoles, that were also discovered in the same HTS process.5 Lead optimization studies to improve the activity of the compounds using computational methods are currently underway in our laboratories. Optimized lead compounds in the 4-aminoquinazoline class that stimulate innate immune cells with minimal toxicity may be useful as adjuvants or immunotherapeutic agents.
Supplementary Material
Acknowledgments
We are grateful for the assistance provided by Dr. Yongxuan Su of the Department of Chemistry at UCSD for high resolution mass spectrometry. We acknowledge the services provided by the UCSF SMDC. This project was supported by a contract from the National Institute of Allergy and Infectious Diseases (HHSN272200900034C) from the National Institutes of Health (to DAC).
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supplementary data (experimental procedures, characterization of final compounds and biological assays protocols) associated with this article can be found, in the on-line version, at XXXXXX.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Dev A, Iyer S, Razani B, Cheng G. Curr Top Microbiol Immunol. 2011;349:115. doi: 10.1007/82_2010_102. [DOI] [PubMed] [Google Scholar]
- 2.Li Q, Verma IM. Nat Rev Immunol. 2002;2:725. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 3.Coffman RL, Sher A, Seder RA. Immunity. 2010;33:492. doi: 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pu M, Hayashi T, Cottam H, Mulvaney J, Arkin M, Corr M, Carson D, Messer K. Stat Med. 2012 doi: 10.1002/sim.5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan M, Hayashi T, Mathewson RD, Nour A, Hayashi Y, Yao S, Tawatao RI, Crain B, Tsigelny IF, Kouznetsova VL, Messer K, Pu M, Corr M, Carson DA, Cottam HB. J Med Chem. 2013;56:4206. doi: 10.1021/jm301694x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang H, Wu Y, Huang X, Wang W, Bing A, Cao X, Wan T. J Biol Chem. 2011;286:30393. doi: 10.1074/jbc.M111.266528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. Nature. 2000;408:740. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 8.Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, Freudenberg M, Beutler B. Nat Immunol. 2005;6:565. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 9.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. Science (Washington, DC, United States) 1990;249:1431. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 10.Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, Kitamura T, Kosugi A, Kimoto M, Miyake K. Nat Immunol. 2002;3:667. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- 11.Coats SR, Pham TTT, Bainbridge BW, Reife RA, Darveau RP. J Immunol. 2005;175:4490. doi: 10.4049/jimmunol.175.7.4490. [DOI] [PubMed] [Google Scholar]
- 12.Hajjar AM, Ernst RK, Fortuno ES, 3rd, Brasfield AS, Yam CS, Newlon LA, Kollmann TR, Miller SI, Wilson CB. PLoS Pathog. 2012;8:e1002963. doi: 10.1371/journal.ppat.1002963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. J Exp Med. 1999;189:1777. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr Nature (London) 1997;388:394. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 15.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. Nature (London, U K) 2009;458:1191. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 16.Macindoe G, Mavridis L, Venkatraman V, Devignes MD, Ritchie DW. Nucleic Acids Res. 2010;38:W445. doi: 10.1093/nar/gkq311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dewar MJS. Mod Tech Comput Chem: MOTECC-91. 1991:455. [Google Scholar]
- 18.Shevelev SA, Dutov MD, Vatsadze IA, Serushkina OV, Rusanov AL, Andrievskii AM. Mendeleev Communications. 1995;5:157. [Google Scholar]
- 19.Mekuskiene G, Udrenaite E, Gaidelis P, Vainilavicius P. Chemija. 1999;10:214. [Google Scholar]
- 20.Neve JE, Wijesekera HP, Duffy S, Jenkins ID, Ripper JA, Teague SJ, Campitelli M, Garavelas A, Nikolapoulos G, Le PV, de Leone AP, Pham NB, Shelton P, Fraser N, Carroll AR, Avery VM, McCrae C, Williams N, Quinn RJ. J Med Chem. 2014;57:1252. doi: 10.1021/jm401321v. [DOI] [PubMed] [Google Scholar]
- 21.Raether W, Hanel H. Parasitol Res. 2003;90(Supp 1):S19. doi: 10.1007/s00436-002-0754-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.