

Abstract
Mitochondrial dysfunction and oxidative stress are implicated in many neurodegenerative diseases. Mitochondria-targeted drugs that effectively decrease oxidative stress, protect mitochondrial energetics, and prevent neuronal loss may therefore lend therapeutic benefit to these currently incurable diseases. To investigate the efficacy of such drugs, we examined the effects of mitochondria-targeted antioxidants MitoQ10 and MitoE2 on neuronal death induced by neurotrophin deficiency. Our results indicate that MitoQ10 blocked apoptosis by preventing increased mitochondria-derived reactive oxygen species (ROS) and subsequent cytochrome c release, caspase activation, and mitochondrial damage in nerve growth factor (NGF)-deprived sympathetic neurons, while MitoE2 was largely ineffective. In this paradigm, the most proximal point of divergence was the ability of MitoQ10 to scavenge mitochondrial superoxide (O2•−). MitoQ10 also prevented caspase-independent neuronal death in these cells demonstrating that the mitochondrial redox state significantly influences both apoptotic and nonapoptotic pathways leading to neuronal death. We suggest that mitochondria-targeted antioxidants may provide tools for delineating the role and significance of mitochondrial ROS in neuronal death and provide a new therapeutic approach for neurodegenerative conditions involving trophic factor deficits and multiple modes of cell death.
Keywords: MitoQ, apoptosis, caspase, ROS, mitochondria, cytochrome c
Introduction
Mitochondrial dysfunction and oxidative stress are prominent features in both acute neuropathologies and progressive neurodegenerative conditions (Green and Kroemer, 2004). When mitochondrial energetics are impaired, electron leakage from respiratory complexes increases resulting in elevated production of the free radical superoxide (O2•−) via a one electron reduction of O2 (Murphy, 2009). Mitochondria are the primary source of O2•− within mammalian cells and, thus, are also the most likely organelle to suffer damage from O2•− or downstream reactive oxygen species (ROS) resulting from dismutation of O2•− (Turrens, 2003). Protection against oxidative damage is achieved by a series of interdependent mitochondrial antioxidant systems (Kaplowitz et al., 1985; Kowaltowski et al., 2001; Sheeran et al., 2010). A significant imbalance in either system will eventually lead to mitochondrial dysfunction, oxidative stress, and cellular demise (Wallace, 2010). If these events significantly contribute to the etiology of neurodegenerative diseases, therapeutics that protect and preserve mitochondrial energetics may prevent the progression of neuronal death and its consequential symptoms.
Although the factors that trigger programmed cell death (PCD) in neurons differ between pathological settings, most of the subsequent signaling events involve two major pathways and appear highly conserved. Perhaps the most extensively characterized model of PCD in neurons is that of sympathetic neurons deprived of nerve growth factor (NGF) (Davies, 1996; Putcha and Johnson, 2004). Similar to vulnerable neurons in the aging brain, decreased neurotrophic supply in cultured sympathetic neurons reduces their size and number, suppresses neurite outgrowth and decreases cellular glucose uptake (Weissmeler and Wu, 2012; Chang et al., 2003). Within 6 h of deprivation, the proapoptotic protein Bax associates with the outer mitochondrial membrane (OMM) and orchestrates the release of cytochrome c and production of mitochondrial-derived O2•− (Kirkland et al., 2002a, b, 2007, 2010). In the first pathway to cell death, the apoptogenic cytochrome c molecules released into the cytosol activate caspases via the formation of the apoptosome (Vaughn and Deshmukh, 2008; Wright et al., 2006). If the executioner caspases 3/7 are inhibited, cell death is delayed until mitochondrial integrity and membrane potential are lost (Deshmukh et al., 2000; Chang et al., 2002; Chang and Johnson, 2002), at which point cell death occurs through a second, caspase-independent pathway. The established temporal relationship of these events in sympathetic cultures provided an ideal model to evaluate the mechanistic basis of neuroprotective therapeutics.
Mitochondria-targeted antioxidants are a new class of drugs designed to protect the cell from ROS damage at the source. The high potential across the inner mitochondrial membrane (IMM) allows the lipophilic cation, triphenylphosphonium (TPP+) to selectively accumulate in the mitochondrial matrix (James et al., 2007). TPP+ can be covalently conjugated to antioxidants, such as ubiquinone or vitamin E (Fig. 1), enabling these compounds to easily traverse the plasma membrane and the outer mitochondrial membrane (OMM) and selectively accumulate within the matrix and adsorb to the IMM of treated cells (James et al., 2007). In the current study, we examined the effects of two such compounds, MitoQ10 and MitoE2 (Fig. 1) on the events that dictate the commitment to neuronal death (Chang et al., 2002; Chang and Johnson, 2002). Our findings indicate that MitoQ10 but not MitoE2 prevented the progression of both caspase- dependent and-independent pathways to death in cultured sympathetic neurons by protecting the organelle from excessive mitochondrial O2•− and downstream ROS.
Fig. 1.
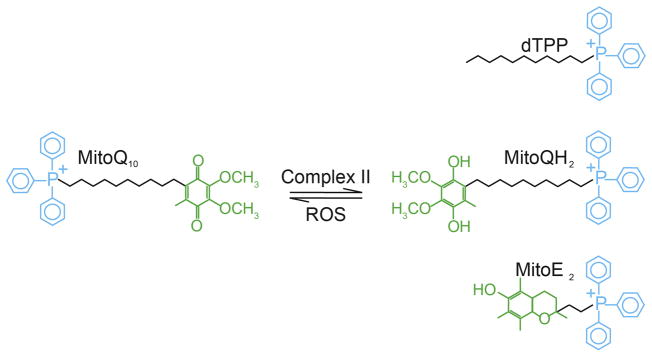
Mitochondria-targeted compounds. These compounds contain a lipophilic triphenylphosphonium cation (TPP+) and can pass directly through lipid membranes to accumulate within the mitochondria driven by the high Δψm of the across the IMM. DecylTPP (dTPP) is similar to MitoQ10 (Asin-Cayuela et al., 2004), but lacks the antioxidant moiety; ubiquinone is attached to the ten-carbon chain to produce MitoQ10 (James et al., 2007), while a two-carbon bridge connects the α-tocopherol moiety of vitamin E to TPP+ in MitoE2 (Smith et al., 1999). In these compounds, the length of the hydrophobic alkyl bridge determines the extent of accumulation and penetration in the mitochondrial inner membrane (Asin-Cayuela et al., 2004) and therefore influences their potential to suppress ROS derived from the mitochondrial electron transport chain. Within the mitochondria, MitoQ10 is continuously reduced to the ubiquinol form (MitoQH2) by respiratory complex II after detoxifying a ROS.
Results
MitoQ10 prevented death of NGF-deprived sympathetic neurons
NGF deprivation induces apoptosis in mouse sympathetic neurons. After 24 h of deprivation, approximately half of the neurons are unable to be rescued by NGF re-addition and are thus committed to die (Deshmukh and Johnson, 1997). To assess the ability of MitoQ10 and MitoE2 to prevent neuronal apoptosis, mouse sympathetic neurons in cell culture were deprived of NGF for 48 h alone or in the presence of MitoQ10 or MitoE2 (250 nM-1 μM), or a combination of both (250 nM each). Approximately 70% of neurons treated with MitoQ10 (250 nM-1 μM) from the time of deprivation were capable of rescue by NGF-readdition 48 h later. Concentrations of MitoQ10 much lower than 250 nM produced no protective effect (e.g., 100 nM from visual screening experiments, not shown) and those above 1 μM were toxic, as demonstrated by the sharp decline in survival with 5 μM MitoQ10 (Fig. 2a). dTPP consists of the mitochondria- targeting moiety (TPP+) attached to a ten-carbon chain and was used to control for nonspecific effects of the lipophilic cation. dTPP produced no detectable changes in the survival rate of NGF- deprived neurons, thus the ubiquinone moiety of MitoQ10 was responsible for its antiapoptotic effect (Fig. 2a, c).
Fig. 2.

MitoQ10 prevented caspase-dependent and independent death induced by NGF deprivation. (a) To determine the effects of the mitochondria-targeted agents on apoptosis in sympathetic neurons, cultures were deprived of NGF alone or in the presence of the indicated concentrations of the broad spectrum caspase inhibitor BAF, dTPP, MitoQ10, MitoE2, or MitoE2 + MitoQ10 (E + Q, 250 nM of each). Neurons that were not committed to die by caspase- dependent death (Commitment 1) were rescued by NGF replacement at 48 h; five days later the cells were fixed, counted, and quantified as the percent of viable neurons in sibling cultures that were maintained in NGF from the time of plating. MitoQ10 treatment (0.25–1 μM) and BAF inhibited caspase-dependent death of NGF-deprived neurons (*p < 0.001). (b) To determine the effect of MitoQ10 (500 nM) and MitoE2 (500 nM) on caspase-independent death, cultures were deprived of NGF alone or with MitoQ10 or MitoE2 from the time of deprivation. These cultures were rescued with NGF 72 h later and quantified as described above. Cultures treated with BAF (50 μM) continued to die after 72 h, despite NGF readdition, indicating that the neurons were committed to die by caspase-independent death (Commitment 2). Cultures receiving MitoQ10 alone and MitoQ10 plus BAF treatment escaped impending death to a similar degree (p = 0.76), indicating that MitoQ10 treatment was neuroprotective against both caspase-dependent and-independent death after 72 h of deprivation (*p < 0.001 for MitoQ10 and MitoQ10 + BAF versus –NGF + BAF). MitoE2 had no effect on caspase- independent death. Results are presented as ±SEM from at least four independent experiments (n = 12–34 cultures). Statistical analysis was performed by ANOVA on ranks, followed by Dunns or Holm-Sidak post hoc tests for multiple comparisons between groups. (c) Representative micrographs of cultures treated as indicated, rescued with NGF after 48 or 72 h and fixed. Easily distinguishable cellular outlines of plump, crystal violet-stained neurons highlight viable neurons over the dead cells that show little or no staining.
MitoE2 was ineffective in preventing neuronal death at all concentrations studied. Ubiquinol enhances the efficacy of vitamin E in membranes and cells by reducing the tocopheroxyl radical back to the active antioxidant after its oxidation by a reactive species (Burton and Traber, 1990; Kagan and Tyurina, 1998; James et al., 2004). We hypothesized that the mitochondria-targeted analogs may perform similarly. To investigate the ability of MitoQ10 to increase the efficacy of MitoE2, cultures were treated with a combination of MitoE2 and MitoQ10 (250 nM each) from the time of deprivation and the effect on the cell death was determined. There was a significant increase in survival over that of untreated NGF-deprived neurons (p < 0.001) but not as great as that observed with 250 nm MitoQ alone (p < 0.001, MitoQ10 250 nM versus MitoE2 + MitoQ10) (Fig. 2a, c).
To determine the effect of MitoQ10 and MitoE2 on caspase-independent death, neuronal cultures were deprived of NGF for 72 h in the presence of the caspase inhibitor, BAF, alone or with MitoQ10 or MitoE2. In this rescue paradigm, ~60% of BAF-treated neurons can be rescued by NGF readdition up to 48 h after NGF-withdrawal, but by 72 h death prevails by a caspase- independent mechanism (Deshmukh et al., 2000). Accordingly, BAF-treatment alone was not sufficient to prevent death after this length of deprivation (Fig. 2b, c). MitoQ10-treated neurons escaped impending death and recovered upon NGF readdition after 72 h of deprivation. Although MitoQ10 treatment did not prevent the commitment to die at later time points (96 h, data not shown), these results indicate that MitoQ10 significantly delayed the commitment to die beyond that of caspase inhibition by BAF.
MitoQ10 prevented cytochrome c redistribution and caspase activation after NGF deprivation (Commitment 1)
The two most critical check-points that dictate a sympathetic neuron’s commitment to die involve the redistribution of mitochondrial cytochrome c into the cytosol and the terminal loss of Δψm (Neame et al., 1998; Deshmukh et al., 2000; Chang et al., 2002; Chang and Johnson, 2002). Because cytochrome c release begins around 18 h after NGF deprivation, approximately half of the deprived sympathetic neurons become committed to die and substantially degraded due to caspase activation by 24 h of NGF withdrawal, which confounds quantification of the effect additives may have on further cytochrome c release and the subsequent terminal loss of Δψm (Deshmukh and Johnson, 1997; Putcha et al., 1999). The ultimate loss of both cytochrome c and Δψm in this paradigm is not dependent on caspase activity (Deshmukh and Johnson, 1998; Desagher et al., 1999; Putcha et al., 1999; Deshmukh et al., 2000; Kirkland and Franklin, 2001; Chang et al., 2002; Chang and Johnson, 2002). Therefore, in order to isolate the ability of mito-antioxidants to modulate these endpoints independent of caspases, degeneration was prevented by inhibiting caspase activity with BAF. Sympathetic neurons were deprived of NGF in the presence of BAF, MitoQ10 + BAF, or BAF plus dTPP, MitoQ10, or MitoE2 for 48 h and the effect on cytochrome c redistribution was quantified by immunocytochemistry (Fig 3a). Only MitoQ10-treated, NGF- deprived neurons maintained mitochondrial cytochrome c staining throughout NGF deprivation (Fig. 3b). Because MitoQ10 inhibited death up to 72 h after NGF deprivation, this treatment did not require BAF addition. The results indicate no difference between MitoQ10 alone and Mito + BAF treatment in this paradigm.
Fig. 3.

MitoQ10 prevented release of cytochrome c from mitochondria and caspase 3 activation during apoptosis induced by NGF deprivation. (a) Micrographs demonstrating the criteria used to score cytochrome c localization. The left panels are differential interference contrast images of three neurons that had been deprived of NGF for 18 h. The right panels are fluorescent micrographs of the same cells showing immunostaining for cytochrome c. The cultures were maintained continuously in the absence of NGF in medium containing BAF (50 mM). BAF does not alter the rate of cytochrome c release (Kirkland and Franklin, 2001; Kirkland et al., 2002). Note the intense punctate staining in one cell and the faint staining in the other two. The intensely-stained neuron is representative of cells that were scored as having retained cytochrome c in mitochondria. The other neurons are representative of cells scored as having released cytochrome c into the cytoplasm where it was rapidly degraded. (b) Sympathetic neurons were deprived of NGF in the presence of MitoQ10, or BAF (50 μM) alone, or with MitoE2, dTPP, or MitoQ10. The concentration of all mito-compounds was 500 nM in each of the experiments here and below. The cultures were fixed after 48 h of treatment and processed for cytochrome c immunocytochemistry. Neurons that lost mitochondrial cytochrome c staining were counted and are shown as the percent of total neurons per treatment. Only MitoQ10 treatment prevented cytochrome c redistribution (*p < 0.001, for –NGF + MitoQ10 + BAF and –NGF + MitoQ10 versus –NGF + BAF). (c) To determine if MitoQ10 could prevent release of mitochondrial cytochrome c when added at later time points, sympathetic neurons were deprived of NGF and maintained in BAF (50 μM), dTPP or MitoQ10 (500 nM each) was added to parallel cultures 18 and 24 h from the time of deprivation, fixed and quantified after 48 h total deprivation. –NGF + BAF neurons were also fixed at 24 h after deprivation to quantify the effect of dTPP and MitoQ10 addition. dTPP failed to attenuate cytochrome c release (p > 0.05 for +dTPP and –NGF + BAF at 24 h). MitoQ10 prevented further release of cytochrome c when added at 18 h or 24 h (*p < 0.001 for cytochrome c retention in –NGF + BAF 24 h and –NGF + MitoQ10 cultures at 48 h) (n ~ 500 neurons). (d) Caspase 3 activity was measured with the Caspase Glo assay. Sympathetic neurons were deprived of NGF for 24 h alone, or with BAF (50 μM), MitoQ10, or MitoE2 (500 nM each). Caspase activity was normalized to NGF-maintained, sibling cultures of equal density. NGF deprivation alone significantly increased caspase 3 activity. MitoQ10 and BAF prevented this increase, while MitoE2 had no effect (*p < 0.001 for –NGF and MitoE2 versus + NGF). Data are from four independent experiments (n = 12–20 cultures).
To determine the time course by which MitoQ10 prevented loss of mitochondrial cytochrome c after NGF deprivation, sympathetic neurons were deprived of NGF in the presence of BAF, and MitoQ10 or dTPP was added to parallel cultures 18 h and 24 h from the initial time of deprivation. The effect on cytochrome c redistribution was assessed at 48 h after deprivation. MitoQ10 addition significantly blocked any further release of cytochrome c from mitochondria of NGF- deprived neurons (Fig. 3c). In contrast, dTPP failed to abort mitochondrial cytochrome c release, as neurons lost a significant amount of cytochrome c after the time of dTTP addition. The results indicate that the antioxidant properties of MitoQ10 acutely prevented mitochondrial cytochrome c depletion.
To investigate the apoptogenic activity of cytochrome c that had been released from the mitochondria of NGF-deprived neurons treated with MitoQ10 or MitoE2, sibling cultures of equal density were treated accordingly and induction of caspase 3 activity was determined 24 h later. Treatment with the broad spectrum caspase inhibitor BAF completely blocked the increase in caspase 3 activity in NGF-deprived cultures (Fig. 3d). Likewise, MitoQ10 prevented the increase in caspase activation during NGF deprivation (p > 0.05), indicating that MitoQ10 effectively prevented apoptotic induction of caspase 3, while MitoE2 treatment produced no detectable effect.
ROS scavenging divergence between MitoQ10 and MitoE2
Considering the antioxidant properties of ubiquinol and vitamin E (Burton and Traber, 1990; Mordente et al., 1998; James et al., 2004), we anticipated that any benefit seen from the mitochondria-targeted compounds would likely correspond to their effects on mitochondrial ROS. To explore this relationship, the effect of dTPP, MitoQ10, and MitoE2 on relative mitochondrial O2•− levels in NGF-deprived sympathetic neurons was assessed with the mitochondria-specific, redox-sensitive dye, MitoSOX. Withdrawal of NGF from mouse sympathetic neurons increases mitochondria-derived ROS within 6 h of deprivation, which then peaks after 18–24 h of NGF deprivation (Kirkland et al., 2002b, 2010). In our cultures, NGF deprivation significantly increased MitoSOX fluorescence, which was prevented by MitoQ10 treatment (Fig. 4a, b).
Fig. 4.

Effect of mito-antioxidants on ROS levels in NGF-deprived sympathetic neurons. (a) Representative micrographs comparing MitoSOX fluorescence in NGF-maintained sympathetic neurons to those deprived of NGF for 24 h alone or with the indicated treatments. The cultures were exposed to MitoSOX (2 μM) for 10 min, washed, and analyzed by confocal microscopy. MitoSOX staining is cytoplasmic and punctate, consistent with its demonstrated localization to the mitochondria (Kirkland et al., 2010). (b) Mitochondrial O2•− levels were estimated by the fluorescent intensity of MitoSOX in neurons treated with dTPP, MitoQ10, or MitoE2 (500 nM each) from the time of NGF- withdrawal (24 h), or with MitoQ10 for 1 h prior to analysis at 24 h. MitoSOX dye intensities are normalized to the intensity average of sibling cultures maintained in NGF from the time of plating. Both chronic (24 h) and acute (1 h) MitoQ10 treatments prevented the increase in O2•−sensitive MitoSOX intensity (p < 0.001 versus-NGF). Conversely, neither dTPP nor MitoE2 attenuated mitochondrial O2•−detected by MitoSOX (n = 91–331). (c) Representative micrographs comparing CM-H2DCFDA fluorescence in NGF-maintained sympathetic neurons to those deprived of NGF for 24 h alone or with the indicated treatments. (d) The effect of MitoQ10 and MitoE2 on cellular ROS levels was measured by the dye intensity of CM-H2DCFDA (10 μM for 20 min) and quantified similarly to the method described above. Both MitoQ10 (24 h and 1 h) and MitoE2 significantly prevented the increased CM-H2DCFDA intensity in NGF-deprived neurons (p < 0.001 for MitoQ and MitotE versus-NGF), but only MitoQ10 decreased CM-H2DCFDA to the level of NGF-maintained neurons (p > 0.05 for –NGF + MitoQ10 versus + NGF) (n = 166–254). MitoE was significantly different from both the 1 h and 24 h MitoQ exposures (p < 0.01). Stars indicate statistical difference from neurons deprived of NGF for 24 h (p < 0.001).
To establish the efficiency of mitochondrial O2•− suppression by MitoQ10, MitoQ10 was added after various time points of NGF deprivation up to 24 h, at which time MitoSOX intensity was again quantified and normalized to NGF-maintained controls. Consistent with its rapid uptake into cells and accumulation in the mitochondria (Smith et al., 2003; Ross et al., 2008), the results show that MitoQ10 acutely (1 h treatment starting at 23 h after deprivation) attenuated O2•− as detected by MitoSOX levels in NGF-deprived neurons. A similar suppression was seen at 24 h regardless of when MitoQ was added after NGF withdrawal (e.g., 12 h, not shown). dTTP had no effect on mitochondrial O2•− levels compared to NGF-deprived alone (p > 0.05; Fig. 4b), excluding the possibility that nonspecific effects of lipophilic cation accumulation could account for the decreased MitoSOX intensity. In contrast, MitoE2 had no effect on mitochondrial ROS detected by MitoSOX during NGF deprivation.
To investigate the effects of MitoQ10 and MitoE2 on the intracellular redox state, CM- H2DCFDA fluorescence was determined in sympathetic neurons deprived of NGF alone and in the presence of MitoQ10 or MitoE2, for 24 h. Previous studies with sympathetic neurons in culture have determined that CM-H2DCFDA is relatively insensitive to oxidation by O2•− and the fluorescence it produces in response to NGF deprivation is primarily due to H2O2-associated ROS or reactive nitrogen species (Kirkland et al., 2001, 2002a, b, 2007, McManus et al., 2011). However, this probe does respond to a range of other redox alterations. NGF deprivation resulted in a >5 fold increase in CM-H2DCFDA intensity over that of NGF-maintained controls (Fig. 4c, d). Both MitoQ10 and MitoE2 significantly attenuated the increased intracellular ROS detected by CM-H2DCFDA, but only MitoQ10 decreased these ROS to the level of NGF-maintained neurons (p > 0.05 compared to –NGF+MitoQ10 and +NGF). MitoQ10 also suppressed CM-H2DCFDA-fluorescence within an hour of treatment. Taken together, the data indicate that while MitoE2 has antioxidant activity in these neurons, it was not sufficient to completely eliminate the pro-oxidant state occurring during NGF deprivation.
MitoQ10 prevented loss of Δψm independent of caspase activation (Commitment 2)
NGF-deprived neurons are able to maintain a reduced Δψm after cytochrome c release, and in some cases, for a surprisingly extended period (Neame et al., 1998; Deshmukh et al., 2000; Chang et al., 2002; Chang and Johnson, 2002). Although transient decreases in Δψm occur in healthy and degenerating neurons, permanent loss of Δψm is intimately associated with cell death (Kroemer et al., 2007). The majority of mitochondrial functions that are important for neuronal vitality, such as ATP generation, cytosolic Ca2+ buffering, protein import, and antioxidant regeneration, are dependent on Δψm (von Ahsen et al., 2000; Duchen, 2004; Wallace, 2005). Sustained ATP production is necessary to prevent caspase-independent death. However, sympathetic neurons rely principally on glycolysis for ATP during NGF deprivation (Chang et al., 2003; Vaughn and Deshmukh, 2008). Therefore, maintenance of mitochondrial inner membrane integrity is required for prolonged neuronal survival aside from its role in ATP production, as less than 15% of NGF-deprived, BAF-maintained sympathetic neurons that have lost Δψm are capable of rescue from death (Deshmukh et al., 2000).
MitoQ10 and MitoE2 have structural differences that allow MitoQ10 to adsorb to the IMM to a greater degree than MitoE. Previous studies have shown that the length of the alkyl chain connecting these antioxidants to the TPP+ moiety is directly correlated with the extent of IMM adsorption (Asin-Cayuela et al., 2004; James et al., 2004, 2007). Because MitoQ10 has a much longer hydrophobic linking chain than MitoE2 (see Fig. 1), we sought to determine if improved protection of the IMM by MitoQ10 would correlate with its enhanced efficacy over MitoE2. The ability of MitoQ10 and MitoE2 to preserve the functional integrity of the IMM was estimated by assessing Δψm with the potential-dependent fluorescent dye, TMRM+. By 24 h after NGF withdrawal, a time at which roughly half of the cells are committed to die, TMRM+ intensity had significantly decreased (Fig. 5b; Deshmukh et al., 2000; Kirkland et al., 2010). Treating cells with BAF prevented this decrease suggesting that it was caused, at least in part, by caspase activity, possibly secondary to caspase cleavage of components of the electron transport chain (Ricci et al., 2004). Part of the decline is also likely due to the loss of cytochrome c from the chain. In accordance with the time-line previously established (Deshmukh et al., 2000), our results reveal that the majority of BAF-treated sympathetic neurons had greatly decreased Δψm after 48 h of NGF deprivation, suggesting the involvement of caspase-independent mechanisms such as loss of cytochrome c from the electron transport chain (Fig. 5a, b). MitoQ10-treated, NGF-deprived neurons retained TMRM+ staining at the level of NGF-maintained neurons, while neither MitoE2 nor dTPP produced any detectable protection. The ability of NGF-deprived neurons treated with MitoQ10 alone to retain TMRM+ intensity for 48 h indistinguishable from that of cultures supported in NGF indicates that MitoQ10 prevented both caspase-dependent and-independent influences on IMM integrity.
Fig. 5.

MitoQ10 prevented loss of Δψm in NGF-deprived sympathetic neurons. Sympathetic neurons were deprived of NGF in the presence of BAF (50 μM) alone, or with 500 nM of MitoE2, MitoQ10, or dTPP. The effects of mitochondria-targeted compounds on Δψm were measured by the fluorescent intensity produced by the membrane potential–dependent dye TMRM+. (a) Representative TMRM+-stained neurons. (b) The average TMRM+ intensity of NGF-deprived, or NGF-deprived and BAF-maintained neurons within each treatment group was normalized to that of sibling cultures maintained in NGF from the time of plating. TMRM+ fluorescence was Δψm-specific, as 1 h treatment of NGF-maintained neurons with the mitochondrial uncoupling agent FCCP (1.6 μM) completely abolished TMRM+ staining (**p < 0.001, for FCCP versus +NGF and-NGF). All NGF-deprived or NGF-deprived + BAF-treated cultures exhibited significant loss of Δψm by 24 or 48 h (*p < 0.001 versus + NGF), except those also treated with MitoQ10 (p > 0.05 for MitoQ10 + BAF versus + NGF). MitoQ10 treatment prevented Δψm loss in both caspase-dependent (without BAF) and caspase-independent conditions (p < 0.001 for MitoQ10 and MitoQ10 + BAF versus –NGF + BAF) (n = 30–220).
Discussion
This study evaluated the ability of mitochondria-targeted compounds, MitoQ10 and MitoE2, to prevent caspase-dependent and-independent neuronal death. In cultured neurons deprived of trophic factors, MitoQ10 but not MitoE2 prevented all critical checkpoints that dictate a neuron’s commitment to death (Fig. 6) (Deshmukh et al., 2000; Chang et al., 2002; Chang and Johnson, 2002) and proved a more potent mitochondrial antioxidant than MitoE2. MitoQ10 treatment restored mitochondrial redox balance, prevented cytochrome c release, activation of caspase 3, and delayed the commitment to die for 72 h. The mitochondrial protection afforded by MitoQ10 also inhibited the progression of death in BAF-treated cultures, suggesting that mitochondria- derived ROS are also actively involved in caspase-independent death.
Fig. 6.
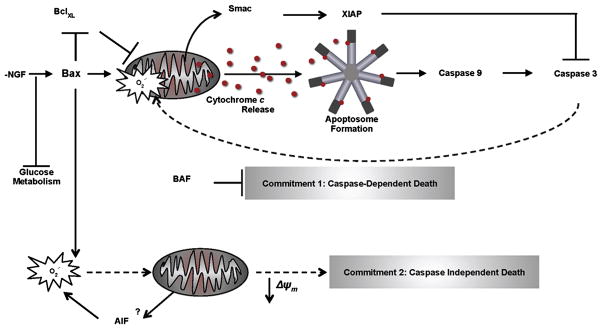
Proposed role of mitochondrial ROS in the commitment to neuronal death: implications from mitochondria-targeted antioxidants. In the absence of NGF, neurons shift to a pro-oxidant state as metabolism slows and mitochondria produce more ROS, thus compromising endogenous antioxidant defenses. Proapoptotic Bcl-2 proteins, such as Bax or Bid, may directly permeabilize the OMM, mobilizing proteins from the intermembrane space, such as Smac and cytochrome c. Released Smac may bind the X-linked inhibitor of apoptosis proteins (XIAPs), removing the block and allowing caspase activation. A potentially moderate level of caspase-3 activation may then provoke mitochondrial O2•− production as caspases now have access to the electron transport chain due to OMM compromise. The induction of mitochondrial ROS supports further cytochrome c release by oxidizing cardiolipin. Cytochrome c causes formation of the apoptosome and the inevitable onset of apoptotic death (Commitment 1). In BAF-treated neurons, Bax-dependent events lead to mitochondrial O2•−accumulation by unknown mechanisms, but likely involving metabolic decline and possibly apoptosis inducing factor (AIF)(Kondo et al., 2010). The mitochondrial ROS that escape detoxification may damage metabolic enzymes and lipids of the IMM, both of which become deleterious for sympathetic neuron vitality as mitochondrial dysfunction ensues with the collapse of Δψm (Commitment 2). Dashed arrows represent pathways that were blocked by MitoQ10 treatment in NGF-deprived sympathetic neurons.
Increased generation of O2•− by the mitochondrial respiratory chain is one of the earliest apoptotic events that occurs following NGF withdrawal from sympathetic neurons (Greenlund et al., 1995; Dugan et al., 1997; Kirkland et al., 2007). Accordingly, it was the most proximal point of divergence between MitoQ10 and MitoE2 in this study. MitoQ10 acutely attenuated increased mitochondrial O2•−, while MitoE2 only affected downstream cytoplasmic ROS or other reactive species detected by CM-H2DCFDA, suggesting that the antioxidant activity of MitoE2 does not involve direct modification of mitochondrial O2•− production. These results suggest that efficient suppression of mitochondrial ROS was of paramount importance in the cascade of apoptotic events that follow.
These results also suggest that the mitochondrial redox state may be a more influential mediator of apoptotic progression than the total cellular redox state (Martensson et al., 1990; Kim, 2007; Circu et al., 2008), in part by influencing cytochrome c release and caspase activation. Here, MitoQ10 addition was sufficient to restore both the mitochondrial and cytosolic redox states and prevent cytochrome c redistribution in NGF-deprived neurons. Previous studies show that MitoQ10 directly detoxifies O2•−/HOO and lipid-derived radicals, and effectively prevents peroxidation of the IMM within isolated mitochondria and cells (Kelso et al., 2001; James et al., 2005, 2007, Maroz et al., 2009). Our results suggest that MitoQ10 may act similarly in neurons, as oxidation of the major IMM phospholipid, cardiolipin, disrupts its association with cytochrome c, and may be necessary to release mitochondrial cytochrome c into the cytosol of NGF-deprived cells (Kirkland et al., 2002a; Iverson and Orrenius, 2004).
The divergent effects of MitoQ10 and MitoE2 on mitochondrial ROS and neuronal death offer other important implications. First, selective accumulation within the IMM and effective recycling of radical scavengers appear as a critical determinant of long-term antioxidant activity. Both MitoQ10 and MitoE2 are lipophilic, radical scavengers that are targeted to the mitochondria, however the antioxidant moiety of MitoQ10 is recycled after it detoxifies reactive species, whereas there is no available evidence of this for MitoE2 (Kelso et al., 2001; James et al., 2007). Although oxidative phosphorylation does not produce significant ATP in deprived neurons, electron transport maintains Δψm after NGF deprivation (Chang et al., 2003), enabling the regeneration of MitoQ10 until the point of terminal Δψm loss. Thus the ability to be continuously regenerated by respiratory complex II may have sustained the protective effect of MitoQ10 in both pathways to neuronal death. The effectual disparity between these compounds also reflects structural differences that allow improved adsorption onto and penetration into the IMM by MitoQ (Kelso et al., 2001; Asin-Cayuela et al., 2004; James et al., 2007).
Second, previous studies have shown that antiapoptotic agents, such as GSH and NGF, restore the cytosolic redox balance (as assessed by CM-H2DCFDA levels) and not that of the mitochondria (as assessed by MitoSOX levels) when added to NGF-deprived sympathetic neurons (Kirkland et al., 2007). However, the antioxidant action of MitoE2 in this study was not sufficient to prevent the same events. Therefore, our results may signify the importance of other signaling pathways that are regulated by GSH and NGF (Hall, 1999; Vaughn and Deshmukh, 2008; Franco and Cidlowski, 2009; Wallace, 2010), but not by vitamin E, that may be necessary to prevent death during stressful events, such as loss of trophic support (Yan et al., 1995). MitoQ10 acts solely as an antioxidant within mitochondria (James et al., 2005), yet it was sufficient to restore cytosolic redox balance, consistent with mitochondria as the major producer of cellular ROS in this situation. Taken together, these results suggest that the mitochondrial redox balance and ROS production is an important determinant of the cytosolic redox state (Franco and Cidlowski, 2009), but the reverse may not be true when the bioenergetic balance is challenged by apoptotic stimuli.
Sympathetic neurons normally maintain a highly reduced intracellular environment by NGF-stimulation of the pentose phosphate pathway, which generates the reducing equivalents (NADPH) necessary to prevent excessive oxidation of molecules (Ho et al., 2007). Sympathetic neurons require this reduced environment to ensure the release of cytochrome c alone will not lead to caspase-dependent death, as cytochrome c must be oxidized to induce apoptosome formation (Wright, et al., 2007; Vaughn and Deshmukh, 2008). Our results more specifically support a functional role of the mitochondrial redox state in regulating apoptotic activation of cytochrome c as MitoQ10 addition to NGF-deprived cultures promptly decreased mitochondrial free radicals and derailed this check-point (Commitment 1; Fig. 6), while MitoE2 treatment appeared to only affect general, cytosolic ROS levels and did not prevent progression of this commitment to death (Chang et al., 2002; Chang and Johnson, 2002; Kirkland et al., 2002b).
As depicted in Fig. 6, unchecked mitochondrial ROS production commits cells to caspase-dependent death in NGF-deprived sympathetic neurons. However, the mechanisms leading to the collapse of Δψm that dictates caspase-independent death (Commitment 2) thus far remain unknown (Deshmukh et al., 2000). Available evidence suggests that mitochondrial processes occuring downstream of Bax and upstream of caspase activation are required for caspase-independent death (Chang et al., 2003; Kirkland et al., 2002a, b, 2007a, b, 2010). The ability of MitoQ10 to prevent Bax-dependent events and extend Commitment 2 in these neurons may indicate that mitochondrial O2•− mediates Bax-dependent events in caspase-independent death.
In many cell types, the ultimate loss of Δψm occurs by activation of the mitochondrial permeability transition pore (mPTP) due to increased levels of mitochondrial ROS, calcium or other events, but the effect of mPTP inhibition on caspase-independent Δψm loss in mouse sympathetic neurons appears inconsequential (Kroemer et al., 2007; Papa et al., 2007; Juhaszova et al., 2008; Chang et al., 2002; Chang and Johnson, 2002). Our results establish a role for mitochondrial ROS generation in orchestrating the ultimate loss of Δψm, independent of the known effects on caspase activation and mPTP opening (Juhaszova et al., 2008). Mitochondrial ROS may lead to loss of Δψm by i) directly damaging the respiratory complexes that are responsible for generating Δψm, ii) by initiating lipid peroxidation events that ultimately dismantle the IMM and drive the cell to death, and/or iii) assembly of the mPTP, permeabilization and swelling of the IMM (Ricci et al., 2004; Kroemer et al., 2007; Kondo et al., 2010).
Most cell types can undergo a caspase-mediated apoptotic type of cell death. In many cases, ROS is an important component of this death (Franklin, 2011). ROS-mediated, caspase-independent cell death has been reported in macrophages (Xu et al., 2006), retinal ganglion cells (Tezel and Yang, 2004), human neutrophils (Maianski et al., 2003), and many other cell types (Lang-Rollin et al., 2003; Galluzzi et al., 2012). Therefore, the findings reported here for the block by a mitochondria-targeted antioxidant of both caspase-mediated and caspase-independent cell death in sympathetic neurons may be broadly applicable multiple forms of cell death in many other types of cells. The potential therapeutic benefits of MitoQ have been explored in both animal models and human clinical trials. We demonstrated that MitoQ can block the development of Alzheimer’s disease-like behavioral and neuronal pathologies in the 3xTG-AD mouse model of the disease (McManus et al., 2011). Other animal studies show that MitoQ can prevent diabetic neuropathy in a mouse model of the disease (Snow et al., 2010), that it can protect against organ damage in a mouse model of sepsis (Lowes et al., 2008), and that it can delay disease progression in a mouse model of multiple sclerosis (Peizhong, 2013). MitoQ decreased liver damage in phase II clinical trials of patients with hepatitis C (Gane et al., 2010) but failed to slow the progression of Parkinsonism, perhaps because treatment was started too late in the course of the disease (Snow et al., 2010). Many other studies investigating the potential therapeutic value of MitoQ or other mitochondria-l-targeted antioxidants have been done or are in progress (Smith and Murphy, 2010; Subramanian et al., 2010; Skulachev, 2012).
In summary, we report that the ability of MitoQ10 to suppress redox processes within mitochondria was both necessary and sufficient to protect the functional integrity of the IMM in caspase-dependent and caspase-independent pathways to neuronal death. These findings suggest that mitochondria-ROS production may orchestrate the execution of cell death independently from their influence on caspase activity. As such, mitochondria-targeted antioxidant therapeutics may enhance treatment options for neurodegenerative and other conditions that involve multiple modes of death (Friedlander, 2003; Stefanis, 2005; Spires-Jones et al., 2009; Weissmeler and Wu, 2012).
Experimental methods
All animal work was performed according to the AVMA guidelines and University of Georgia IACUC approved protocols.
Materials
[10-(4,5-dimethoxy-2-methyl-3,6-dioxo-1,4-cycloheexadienl-yl)decyl] triphenylphosphonium methanesulfonate (MitoQ10) and [2-(3,4-dihydro-6-hydroxy-2,5,7,8- tetramethyl-2H-1-benzopyran-2-yl) ethyl] triphenylphosphonium chloride (MitoE2) (Kelso et al., 2001; Smith et al., 1999) were synthesized in one of our laboratories (Murphy). MitoSOX Red and 5-(and-6)- chloromethyl-2′, 7′-dicholorodihydrofluorescein diacetate (CM-H2DCFDA) were purchased from Invitrogen (Eugene, OR). NGF 2.5S was purchased from Harlan Bioproducts (Indianapolis, IN). All other chemicals were purchased from Sigma (St. Louis, MO) unless otherwise noted.
Neuronal culture and treatment
Cultures of sympathetic neurons were prepared from the superior cervical ganglia (SCG) of neonatal male and female C57BL/6 mice as described (Johnson and Argiro, 1983). ARRIVE guidelines were followed. Breeders were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). The SCG were enzymatically dissociated by successive 30-min treatments in 1 mg/ml collagenase and 2.5 mg/ml trypsin at 35° C, washed with Leibovitz’s 15 medium (L-15), then mechanically dissociated by trituration into growth media, plated onto collagen-coated 24-well Costar plates (Corning, Inc., Corning, NY, USA) for survival assays, or #1 glass coverslips for microscopy experiments, and maintained at 35°C in a humidified incubator containing 5% CO2/95% air. To assure equal plating densities within an experiment, all ganglia from a dissection were dissociated together and resuspended in growth media. Equal volumes of the suspended cells were plated onto the tissue culture plates or coverslips. Plating density was identical for all cultures plated at the same time. The growth medium (Eagle’s minimum essential medium with Earle’s salts, Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 20 μM fluorodeoxyuridine, 20 μM uridine, 1.4mM L-glutamine, and 50 ng/ml 2.5S NGF) was replaced after 3–4 days in vitro (DIV). After 6 DIV, NGF deprivation was achieved by rinsing cultures twice with medium lacking NGF, followed by addition of medium containing goat anti-mouse 2.5S NGF neutralizing antiserum (anti-NGF, Cedarlane Labs, Burlington, Ontario, Canada). Mitochondria-targeted compounds were included in media at the time of deprivation unless otherwise stated. The concentrations (0.1–1 μM) of these compounds were chosen based on results from initial toxicity screens, which revealed that long-term exposure (7 days) to concentrations in this range were nontoxic to NGF-maintained neurons.
Survival assays
The commitment to die was defined as the portion of neurons that could be rescued from death by readdition of NGF to deprived cultures (Deshmukh et al., 2000). Briefly, sympathetic neurons were equally distributed among 24-well plates and allowed to establish for 6 DIV. Cultures were deprived of NGF for 48 or 72 h. They were then rinsed to remove residual anti-NGF and incubated in NGF-containing media for at least 5 days. In this rescue paradigm, neurons not committed to die hypertrophy and regain phenotypic characteristics of NGF-maintained neurons in the days following NGF-readdition, while those that have made the decision to die continue to atrophy and lyse. After rescue, cultures were fixed with 4% paraformaldehyde in phosphate buffered saline (PBS, pH 7.2) for > 1 h, washed with PBS, and stained by a brief rinse with 0.1% crystal violet. The cultures were coded and counted by a blinded observer. Survival within each experimental group is presented as a percent of the average of the total number of neurons in sibling cultures that were maintained in NGF from the time of plating. These control cultures consisted of 3–4 cultures maintained for the entire period in medium containing NGF. The number of cells in the NGF-rescued experimental cultures was normalized to the average number counted in these cultures. When the number of neurons counted in the 3–4 control dishes from each of seventeen randomly chosen platings was subtracted from the normalized average of each, the average % difference from the control mean was 0.07 % ± 0.24 SD. Treatments were run in triplicate or quadruplicate within each of at least three independent experiments.
Microscopy
Confocal microscopy was performed with a Nikon C1 laser scanning confocal microscope mounted on a Nikon Eclipse TE 300 inverted microscope (Melville, NY, USA). The confocal microscope was controlled by EZC1 software running on a Dell computer. Neurons were observed with a 60x plan-apo oil immersion lens, chosen at random and scanned. Laser power, pinhole size, and photo- multiplier gain were maintained at constant levels during each experiment. Wide field fluorescence microscopy was conducted using a 20x objective with a Xenon lamp light source. Images were collected by a cooled CCD camera. (Micro-Max, Princeton Instruments, Trenton, NJ, USA). Filters were operated by a Lambda 10–2 optical filter changer (Sutter Instruments, Novato, CA, USA). Experiments were replicated at least three times.
All images were quantified by measuring the raw pixel intensities in the cytoplasm of neuronal somas with the region tool of MetaMorph software (Universal Imaging, West Chester, PA, USA). The area quantified covered a 60 μm2 area of cytoplasm in confocal images. The light intensity for each neuron was normalized to that of control neurons receiving the same concentration of dye for the same time as the experimental cells. Normalized data are shown as fold change from the intensity of the dye measured in sibling cultures of sympathetic neurons maintained in NGF-containing medium from the time of plating. All microscopy was done at room temperature.
ROS measurement
Relative mitochondrial O2•− levels were determined by MitoSOX Red. MitoSOX is a redox- sensitive dye composed of hydroethidine linked by a hexyl carbon chain to a TPP+ group that targets the dye to the mitochondria. The 408 nm laser line of the confocal microscope was used to select for the O2•−/MitoSOX product (Robinson et al., 2006; Johnson-Cadwell, et al., 2007), and the red photomultiplier channel was used for image acquisition. Our lab previously characterized the use of this dye to detect O2•− in sympathetic neurons. Selectivity of MitoSOX for mitochondria-derived O2•− in these neurons is supported by its co-localization with MitoTracker Green, increased intensity upon treatment with the O2•−- generator menadione, and lack of fluorescent induction in response to H2O2 (Kirkland et al., 2007; 2010). Cultures were incubated for 10 min at 35°C in the appropriate experimental medium containing MitoSOX (2 μM). After incubation, cultures were washed twice with L-15 and kept in the second wash for microscopy.
ROS were also detected using the redox-sensitive dye CM-H2DCFDA. The reduced form of CM-H2DCFDA becomes fluorescent after oxidation by multiple reactive species, thus providing a general indicator for the intracellular redox state (Kirkland et al., 2001, 2002b). Cultures were incubated in the appropriate experimental medium containing CM-H2DCFDA (10 μM) for 20 min at 35°C. They were then washed twice with L-15 medium and left in the last wash for confocal microscopy. CM- H2DCFDA was excited with a 488 nm laser and imaged using the green channel.
Mitochondrial membrane potential (Δψm) measurement
The cationic fluorescent probe, tetramethylrhodamine methyl ester (TMRM+), accumulates in cellular compartments in response to local membrane potential differences. Neurons were incubated with TMRM+ (10 nM) for 30 min in culture medium containing the experimental treatments. They were then rinsed once with L-15 medium containing TMRM+ (10 nM) and the experimental treatments to remove culture medium. They were bathed in the same L-15 medium for image acquisition. At equilibrium, this low concentration of TMRM+ provides a quantitative estimate of Δψm such that the fluorescence produced is a direct function of Δψm, and complications due to self-quenching of the dye in the mitochondrial matrix are eliminated (Nicholls and Ward, 2000; Duchen et al., 2003). Using this technique, we measured TMRM+ fluorescence within individual neurons and normalized these values to those of control cultures maintained in NGF. Specificity of TMRM+ fluorescence for Δψm was confirmed by the complete loss (p < 0.001) of staining upon treatment with the mitochondrial uncoupling agent, carbonyl cyanide p-trifluromethoxy-phenylhydrazone (FCCP), which dissipates Δψm (Duchen, 1999).
Immunocytochemistry
Neurons were immunostained for cytochrome c as described (Kirkland et al., 2007). Cultures were fixed for 60 min at 4°C with 4% paraformaldehyde in PBS, washed with Tris-buffered saline (TBS, pH 7.2), incubated for 60 min at room temperature in blocking buffer (TBS containing 0.3% Triton X-100 and 5% normal goat serum), rinsed, and incubated overnight at 4°C in blocking buffer containing the anti-cytochrome c antibody (200 ng/ml, PharMingen, San Diego, CA, USA). The next day, cultures were washed 3X with cold TBS and incubated for 2–4 h at room temperature in blocking buffer containing a FITC-conjugated anti-mouse secondary antibody (1.7 μg/ml, PharMingen), washed, and then viewed by fluorescence microscopy. Most cytochrome c in these neurons is released from mitochondria into the cytoplasm over a short period. This release is followed by rapid degradation of cytochrome c in the cytoplasm resulting in two populations of cells, those that have released cytochrome c and those that have not. Neurons with a bright, mitochondrial pattern of staining were counted positive for cytochrome c retention, whereas those with faint or no staining were counted negative (Neame et al., 1998; Putcha et al., 2000; Kirkland et al., 2007).
Caspase 3/7 activity assay
To assess caspase 3/7 activity, the Caspase-Glo™ Assay (Promega, Madison, WI, USA) was used according to the manufacturer’s protocol. Briefly, sympathetic neurons were grown in a 96- well plate for 6 DIV and then deprived of NGF for 24 h in the presence or absence of MitoQ10 or MitoE2 (500 nM). Additional NGF-deprived cultures were treated with the pan-caspase inhibitor, boc-aspartyl fluoromethyl ketone (BAF, 50 μM), for comparison. After the treatment period, an equal amount of Caspase-Glo 3/7 Reagent was added to each well. The magnitude of the luminescence generated represents both caspase 3 and 7 activity. However, only caspase 3 is present in these cells (Wright et al., 2006). Luminescence intensity was recorded with a Spectramax M2 microplate reader (Molecular Devices, Sunnyvale, CA, USA). Results represent the average of four independent experiments in which the treatments were run in duplicate or triplicate and are shown as fold change from the respective NGF-maintained sibling cultures.
Statistical analysis
Statistical comparisons were made by Kruskal-Wallis one-way ANOVA, followed by Dunn’s post hoc test unless otherwise stated. Data analysis and statistical comparisons were performed using SigmaPlot 11.0 (Systat Software, Inc. San Jose, CA, USA). All data is presented as mean ± standard error of the mean (SEM).
Acknowledgments
This study was supported by National Institutes of Health Grant RO1NS37110. We thank Dr. Martin Picard and Grace Truong for scientific and editorial review of the manuscript. MPM holds patents and consults for a company (Antipodean Pharmaceutical, Inc.) in the area of commercialization of mitochondria-targeted antioxidants.
Abbreviations
- BAF
boc-aspartyl fluoromethyl ketone
- FCCP
p-trifluromethoxy-phenylhydrazone
- IMM
inner mitochondrial membrane
- NGF
nerve growth factor
- OMM
outer mitochondrial membrane
- ROS
reactive oxygen species
- O2•−
superoxide
- TPP+
triphenylphosphonium,
- MitoQ10
[10-(4,5-dimethoxy-2-methyl-3,6-dioxo-1,4-cycloheexadienl-yl)decyl] triphenylphosphonium methanesulfonate
- MitoE2
[2-(3,4-dihydro-6-hydroxy-2,5,7,8-tetramethyl-2H-1-benzopyran-2-yl) ethyl] triphenylphosphonium chloride
- CM-H2DCFDA
chloromethyl-2′, 7′-dicholorodihydrofluorescein diacetate
- L-15
Leibovitz’s 15 medium
- DIV
days in vitro
- PBS
phosphate buffered saline
- Δψm
mitochondrial membrane potential
- TMRM+
tetramethylrhodamine methyl ester
- TBS
tris-buffered saline
- SEM
standard error of the mean
- dTPP
n-decyl triphenylphosphonium
- mPTP
mitochondrial permeability transition pore
Footnotes
MJM and JLF declare that they have no conflicts of interest.
Contributor Information
James L. Franklin, Email: jfrankli@rx.uga.edu.
Meagan J. McManus, Email: megangm@gmail.com.
Michael P. Murphy, Email: michael.murphy@mrc-mbu.cam.ac.uk.
References
- Asin-Cayuela J, Manas AB, James AM, Smith RAJ, Murphy MP. Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett. 2004;571:9–16. doi: 10.1016/j.febslet.2004.06.045. [DOI] [PubMed] [Google Scholar]
- Burton GW, Traber MG. Vitamin E: antioxidant activity, biokinetics, and bioavailability. Ann Rev Nutr. 1990;10:357–382. doi: 10.1146/annurev.nu.10.070190.002041. [DOI] [PubMed] [Google Scholar]
- Chacko BK, Reily C, Srivastava A, Johnson MS, Ye Y, Ulasova E, Agarwal A, Zinn KR, Murphy MP, Kalyanaraman B, Darley-Usmar V. Biochem J. 2010;432:9–19. doi: 10.1042/BJ20100308. Prevention of diabetic nephropathy in Ins2(+/)−(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LK, Putcha GV, Deshmukh M, Johnson EM., Jr Mitochondrial involvement in the point of no return in neuronal apoptosis. Biochimie. 2002;84:223–231. doi: 10.1016/s0300-9084(02)01372-x. [DOI] [PubMed] [Google Scholar]
- Chang LK, Johnson EM., Jr Cyclosporin A inhibits caspase-independent death of NGF-deprived sympathetic neurons: a potential role for mitochondrial permeability transition. J Cell Biol. 2002;157:771–781. doi: 10.1083/jcb.200112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LK, Schmidt RE, Johnson EM. Alternating metabolic pathways in NGF-deprived sympathetic neurons affect caspase-independent death. J Cell Biol. 2003;162:245–256. doi: 10.1083/jcb.200302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radical Bio Med. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AM. The neurotrophic hypothesis: where does it stand? Philos T Roy Soc B. 1996;351:389–394. doi: 10.1098/rstb.1996.0033. [DOI] [PubMed] [Google Scholar]
- Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh M, Johnson EM., Jr Programmed Cell Death in Neurons: Focus on the Pathway of Nerve Growth Factor Deprivation-Induced Death of Sympathetic Neurons. Mol Pharmacol. 1997;51:897–906. doi: 10.1124/mol.51.6.897. [DOI] [PubMed] [Google Scholar]
- Deshmukh M, Johnson EM., Jr Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- Deshmukh M, Kuida K, Johnson E. Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J Cell Biol. 2000;150:131–143. doi: 10.1083/jcb.150.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol Aspects Med. 2004;25:365–451. doi: 10.1016/j.mam.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signaling and cell death. J Physiol. 1999;516:1–17. doi: 10.1111/j.1469-7793.1999.001aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Surin A, Jacobson J. Imaging mitochondrial function in intact cells. In: Method Enzymol. 2003;361:353–389. doi: 10.1016/s0076-6879(03)61019-0. [DOI] [PubMed] [Google Scholar]
- Dugan L, Creedon D, Johnson E, Holtzman D. Rapid suppression of free radical formation by nerve growth factor involves the mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA. 1997;94:4086–4091. doi: 10.1073/pnas.94.8.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Cidlowski JA. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ. 2009;16:1303–1314. doi: 10.1038/cdd.2009.107. [DOI] [PubMed] [Google Scholar]
- Franklin JL. Redox regulation of the intrinsic pathway in neuronal apoptosis. Antioxid Redox Signal. 2011;14:1437–1448. doi: 10.1089/ars.2010.3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander R. Apoptosis and caspases in neurodegenerative diseases. New Engl J Med. 2003;348:1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nuňez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RAJ, Murphy MP. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver. 2010;30:1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Greenlund LJ, Deckwerth TL, Johnson EM., Jr Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- Hall AG. The role of glutathione in the regulation of apoptosis. Eur J Clin Invest. 1999;29:238–245. doi: 10.1046/j.1365-2362.1999.00447.x. [DOI] [PubMed] [Google Scholar]
- Ho HY, Cheng ML, Chiu DT. Glucose-6-phosphate dehydrogenase from oxidative stress to cellular functions and degenerative diseases. Redox Rep. 2007;12:109–118. doi: 10.1179/135100007X200209. [DOI] [PubMed] [Google Scholar]
- Iverson SL, Orrenius S. The cardiolipin-cytochrome c interaction and the mitochondrial regulation of apoptosis. Arch Biochem Biophys. 2004;423:37–46. doi: 10.1016/j.abb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- James AM, Smith RA, Murphy MP. Antioxidant and prooxidant properties of mitochondrial coenzyme Q. Arch Biochem Biophys. 2004;423:47–56. doi: 10.1016/j.abb.2003.12.025. [DOI] [PubMed] [Google Scholar]
- James AM, Cochemé HM, Smith RAJ, Murphy MP. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. J Biol Chem. 2005;280:21295–21312. doi: 10.1074/jbc.M501527200. [DOI] [PubMed] [Google Scholar]
- James AM, Sharpley MS, Manas AR, Frerman FE, Hirst J, Smith RAJ, Murphy MP. Interaction of the Mitochondria-targeted Antioxidant MitoQ with Phospholipid Bilayers and Ubiquinone Oxidoreductases. J Biol Chem. 2007;282:14708–14718. doi: 10.1074/jbc.M611463200. [DOI] [PubMed] [Google Scholar]
- Johnson MI, Argiro V. Techniques in the tissue culture of rat sympathetic neurons. Method Enzymol. 1983;103:334–347. doi: 10.1016/s0076-6879(83)03022-0. [DOI] [PubMed] [Google Scholar]
- Johnson-Cadwell LI, Jekabsons MB, Wang A, Polster BM, Nicholls DG. Mild uncoupling does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J Neurochem. 2007;101:1619–31. doi: 10.1111/j.1471-4159.2007.04516.x. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Wang S, Zorov DB, Nuss HB, Gleichmann M, Mattson MP, Sollott SJ. The identity and regulation of the mitochondrial permeability transition pore. Ann NY Acad Sci. 2008;1123:197–212. doi: 10.1196/annals.1420.023. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Tyurina YY. Recycling and redox cycling of phenolic antioxidants. Ann NY Acad Sci. 1998;854:425–434. doi: 10.1111/j.1749-6632.1998.tb09921.x. [DOI] [PubMed] [Google Scholar]
- Kaplowitz N, Aw TY, Ookhtens M. The regulation of hepatic glutathione. Ann Rev Pharmacol Tox. 1985;25:715–744. doi: 10.1146/annurev.pa.25.040185.003435. [DOI] [PubMed] [Google Scholar]
- Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EL, Smith RA, Murphy MP. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Yune TY, Han CT, Kim YC, Oh YJ, Markelonis GJ, Oh TH. Mitochondrial isocitrate dehydrogenase protects human neuroblastoma sh-sy5y cells against oxidative stress. J Neurosci Res. 2007;85:139–152. doi: 10.1002/jnr.21106. [DOI] [PubMed] [Google Scholar]
- Kirkland RA, Adibhatla RM, Hatcher JF, Franklin JL. Loss of cardiolipin and mitochondria during programmed neuronal death: evidence of a role for lipid peroxidation and autophagy. Neuroscience. 2002a;115:587–602. doi: 10.1016/s0306-4522(02)00512-2. [DOI] [PubMed] [Google Scholar]
- Kirkland RA, Windelborn JA, Kasprzak JM, Franklin JL. A Bax-induced pro oxidant state is critical for cytochrome c release during programmed neuronal death. J Neurosci. 2002b;22:6480–6490. doi: 10.1523/JNEUROSCI.22-15-06480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland RA, Saavedra GM, Cummings BS, Franklin JL. Bax Regulates production of superoxide in both apoptotic and nonapoptotic neurons: role of caspases. J Neurosci. 2010;30:16114–16127. doi: 10.1523/JNEUROSCI.2862-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland RA, Franklin JL. Evidence for redox regulation of cytochrome c release during programmed neuronal death: antioxidant effects of protein synthesis and caspase inhibition. J Neurosci. 2001;21:1949–1963. doi: 10.1523/JNEUROSCI.21-06-01949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland RA, Saavedra GM, Franklin JL. Rapid activation of antioxidant defenses by nerve growth factor suppresses reactive oxygen species during neuronal apoptosis: evidence for a role in cytochrome c redistribution. J Neurosci. 2007;27:11315–11326. doi: 10.1523/JNEUROSCI.3590-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K, Obitsu S, Ohta S, Matsunami K, Otsuka H, Teshima R. Poly (ADP-ribose) polymerase (PARP)-1-independent apoptosis-inducing factor (AIF) release and cell death are induced by eleostearic acid and blocked by alpha-tocopherol and MEK inhibition. J Biol Chem. 2010;285:13079–13091. doi: 10.1074/jbc.M109.044206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaltowski AJ, Castilho RF, Vercesi AE. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001;495:12–15. doi: 10.1016/s0014-5793(01)02316-x. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Lang-Rollin ICJ, Rideout HJ, Noticewala M, Stefanis L. Mechanisms of caspase-independent neuronal death: energy depletion and free radical generation. J Neurosci. 2003;23:11015–11025. doi: 10.1523/JNEUROSCI.23-35-11015.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowes DA, Thottakam BMV, Webster NR, Murphy MP, Galley HF. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide–peptidoglycan model of sepsis. Free Radic Biol Med. 2008;45:1559–1565. doi: 10.1016/j.freeradbiomed.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Maianski NA, Roos D, Kuijpers TW. Tumor necrosis factor α induces a caspase-independent death pathway in human neutrophils. Blood. 2003;101:1987–1995. doi: 10.1182/blood-2002-02-0522. [DOI] [PubMed] [Google Scholar]
- Maroz A, Anderson RF, Smith RA, Murphy MP. Reactivity of ubiquinone and ubiquinol with superoxide and the hydroperoxyl radical: implications for in vivo antioxidant activity. Free Radical Bio Med. 2009;46:105–109. doi: 10.1016/j.freeradbiomed.2008.09.033. [DOI] [PubMed] [Google Scholar]
- Martensson J, Lai JC, Meister A. High-affinity transport of glutathione is part of a multicomponent system essential for mitochondrial function. Proc Natl Acad Sci USA. 1990;87:7185–7189. doi: 10.1073/pnas.87.18.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus MJ, Murphy M, Franklin JL. The mitochondria-targeted antioxidant, MitoQ, prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2011;31:15703–15715. doi: 10.1523/JNEUROSCI.0552-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordente A, Martorana GE, Minotti G, Giardina B. Antioxidant properties of 2,3-dimethoxy-5-methyl- 6-(10-hydroxydecyl)-1,4-benzoquinone (Idebenone) Chem Res Toxicol. 1998;11:54–63. doi: 10.1021/tx970136j. [DOI] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neame SJ, Rubin LL, Philpott KL. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J Cell Biol. 1998;142:1583–1593. doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Ward MW. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- Papa L, Gomes E, Rockwell P. Reactive oxygen species induced by proteasome inhibition in neuronal cells mediate mitochondrial dysfunction and a caspase-independent cell death. Apoptosis. 2007;12:1389–1405. doi: 10.1007/s10495-007-0069-5. [DOI] [PubMed] [Google Scholar]
- Peizhong M, Mancazk M, Shirendeb UP, Reddy PH. MitoQ, a mitochondria-targeted antioxidant, delays disease progression and alleviates pathogenesis in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. BBA Molecular Basis of Disease. 2013;12:2322–2331. doi: 10.1016/j.bbadis.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson EM., Jr BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Johnson EM., Jr Men are but worms: neuronal cell death in C. elegans and vertebrates. Cell Death Differ. 2004;11:38–48. doi: 10.1038/sj.cdd.4401352. [DOI] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson EM., Jr Inhibition of apoptotic signaling cascades causes loss of trophic factor dependence during neuronal maturation. J Cell Biol. 2000;149:1011–1018. doi: 10.1083/jcb.149.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci JE, Muñoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, Scheffler IE, Ellisman MH, Green DR. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell. 2004;117:773–786. doi: 10.1016/j.cell.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci USA. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross MF, Prime TA, Abakumova I, James AM, Porteous CM, Smith RA, Murphy MP. Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochem J. 2008;411:633–645. doi: 10.1042/BJ20080063. [DOI] [PubMed] [Google Scholar]
- Sheeran FL, Rydström J, Shakhparonov MI, Pestov NB, Pepe S. Diminished NADPH transhydrogenase activity and mitochondrial redox regulation in human failing myocardium. BBA-Bioenergetics. 2010;1797:1138–1148. doi: 10.1016/j.bbabio.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Skulachev VP. Mitochondria-targeted antioxidants as promising drugs for treatment of age-related brain diseases. J Alzheimer’s Dis. 2012;28:283–289. doi: 10.3233/JAD-2011-111391. [DOI] [PubMed] [Google Scholar]
- Smith RA, Porteous CM, Coulter CV, Murphy MP. Selective targeting of an antioxidant to mitochondria. Eur J Biochem. 1999;263:709–716. doi: 10.1046/j.1432-1327.1999.00543.x. [DOI] [PubMed] [Google Scholar]
- Smith RAJ, Porteous CM, Gane AM, Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci USA. 2003;100:5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RAJ, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann NY Acad Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O’Sullivan JD, Fung V, Smith RAJ, Murphy MP, Taylor KM. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease Mov. Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- Spires-Jones TL, Stoothoff WH, de Calignon A, Jones PB, Hyman BT. Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci. 2009;32:150–159. doi: 10.1016/j.tins.2008.11.007. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Zhou Y, Martin B, Maudsley S. Pharmacomimetics of exercise: novel approaches for hippocampally-targeted neuroprotective agents. Curr Med Chem. 2009;16:4668–4678. doi: 10.2174/092986709789878292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanis L. Caspase-dependent and-independent neuronal death: two distinct pathways to neuronal injury. Neuroscientist. 2005;11:50–62. doi: 10.1177/1073858404271087. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Kalyanaraman B, Migrino RQ. Mitochondrially Targeted Antioxidants for the Treatment of Cardiovascular Diseases. Recent Pat Cardiovasc Drug Discov. 2010;5:54–65. doi: 10.2174/157489010790192601. [DOI] [PubMed] [Google Scholar]
- Tezel G, Yang X. Caspase-independent component of retinal ganglion cell death, in vitro. Invest Ophthalmol Vis Sci. 2004;45:4049–4059. doi: 10.1167/iovs.04-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol. 2008;10:1477–1483. doi: 10.1038/ncb1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Ahsen O, Renken C, Perkins G, Kluck RM, Bossy-Wetzel E, Newmeyer DD. Preservation of Mitochondrial Structure and Function after Bid- or Bax-Mediated Cytochrome c. J Cell Biol. 2000;150:1027–1036. doi: 10.1083/jcb.150.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. The epigenome and the mitochondrion: bioenergetics and the environment. Gene Dev. 2010;24:1571–1573. doi: 10.1101/gad.1960210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmiller AM, Wu C. Current advances in using neurotrophic factors to treat neurodegenerative disorder. Transl Neurodeg. 2012;1:1–14. doi: 10.1186/2047-9158-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright KM, Vaughn AE, Deshmukh M. Apoptosome dependent caspase-3 activation pathway is non-redundant and necessary for apoptosis in sympathetic neurons. Cell Death Differ. 2006;14:625–633. doi: 10.1038/sj.cdd.4402024. [DOI] [PubMed] [Google Scholar]
- Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem. 2006;281:19179–19187. doi: 10.1074/jbc.M513377200. [DOI] [PubMed] [Google Scholar]
- Yan CY, Ferrari G, Greene LA. N-acetylcysteine-promoted survival of PC12 cells is glutathione-independent but transcription-dependent. J Biol Chem. 1995;270:26827–26832. doi: 10.1074/jbc.270.45.26827. [DOI] [PubMed] [Google Scholar]