

Abstract
Gestational exposure to alcohol can result in long-lasting behavioral deficiencies generally described as fetal alcohol spectrum disorder (FASD). FASD-modeled rodent studies of acute ethanol exposure typically select one developmental window to simulate a specific context equivalent of human embryogenesis, and study consequences of ethanol exposure within that particular developmental epoch. Exposure timing is likely a large determinant in the neurobehavioral consequence of early ethanol exposure, as each brain region is variably susceptible to ethanol cytotoxicity and has unique sensitive periods in their development. We made a parallel comparison of the long-term effects of single-day binge ethanol at either embryonic day 8 (E8) or postnatal day 7 (P7) in male and female mice, and here demonstrate the differential long-term impacts on neuroanatomy, behavior and in vivo electrophysiology of two systems with very different developmental trajectories. The significant long-term differences in odor-evoked activity, local circuit inhibition, and spontaneous coherence between brain regions in the olfacto-hippocampal pathway that were found as a result of developmental ethanol exposure, varied based on insult timing. Long-term effects on cell proliferation and interneuron cell density were also found to vary by insult timing as well as by region. Finally, spatial memory performance was affected in P7-exposed mice, but not E8-exposed mice. Our physiology and behavioral results are conceptually coherent with the neuroanatomical data attained from these same mice. Our results recognize both variable and shared effects of ethanol exposure timing on long-term circuit function and their supported behavior.
Keywords: FASD, neural circuit development, piriform, hippocampus, parvalbumin
I. Introduction
Fetal alcohol spectrum disorder (FASD) describes the range of clinically observed developmental deficits resulting from gestational alcohol exposure. Despite widespread acceptance and awareness of the teratogenic effects of alcohol consumption during pregnancy, it persists as one of the greatest causes of mental deficits, impacting an estimated 1 in 100 children born in the U.S. (Abel, 1998, May and Gossage, 2001), with reports of much greater incidence in certain global regions (Harris and Bucens, 2003, Viljoen et al., 2005, May et al., 2011). There are numerous and multi-modal risk factors for FASD that potentiate gestational alcohol exposure. For instance, a woman’s age, extent and frequency of consumption, as well as the form of alcohol consumed are all implicated in variable FASD risk (Abel, 1998, May and Gossage, 2011). In addition to these factors, the developmental timing of binge drinking is particularly influential toward the type of neurobehavioral pathology expressed in both human FASD (O’Leary et al., 2010) and animal-modeled FASD (Maier et al., 1997). This suggests that specific brain regions have critical sensitive periods to alcohol toxicity, which can influence pathological outcome.
Temporally confined ethanol exposure (binge or extended binge/semi-chronic) in rodents causes multiple deficits in brain anatomy, physiology and behavior, shown both immediately following exposure (Ikonomidou et al., 2000, Galindo et al., 2005), and in the long-term (Wozniak et al., 2004, Izumi et al., 2005, Wilson et al., 2011, Sadrian et al., 2012). The resulting profiles of pathology in each of these studies originate from a specific epoch of ethanol exposure (as brief as a single day), and therefore limit developmental correlates to a snapshot. Furthermore, the comparative impacts on long-term in vivo circuit function from different developmental points of exposure have not been examined. It is of particular importance to examine circuit function outcome in fully developed adults, so that the long-term impacts of developmental ethanol exposure may be further investigated in a coordinated way that has clinical relevance for those affected with FASD.
We and others have previously shown that single day binge ethanol at P7 creates an immediate wave of neurodegeneration in specific brain regions that include the hippocampus and specific cortical regions (Ikonomidou et al., 2000, Saito et al., 2010, Wilson et al., 2011). In the same mice we also observed long-term differences in local and regional neuronal circuit function within the olfacto-hippocampal pathway, thereby establishing a model of long-term ethanol-induced neuronal circuit dysfunction. These effects were prevented when the neuroprotective agent lithium was given on the same day as ethanol (Sadrian et al., 2012). We hypothesize that the immediate wave of neurodegeneration caused by binge ethanol toxicity initiates disruptive cascades in circuit maturation, the effects of which are sustained long after the ethanol has been metabolized. The susceptibility to ethanol-induced cell loss has been shown to vary based on developmental stage of exposure (Ikonomidou et al., 2000) as well as by cell type (Tran and Kelly, 2003). The specific timing of ethanol-derived cytotoxicity is likely of crucial importance with regards to the specific long-term functional outcomes.
FASD-modeled rodent studies examining acute early embryonic exposure (around embryonic day 8 – E8) have been used to simulate alcohol toxicity during human brain development in the first trimester. During this period gastrulation and neurulation occur, and acute ethanol exposure induces excessive apoptotic cell death in many regions, especially in the developing CNS (Dunty et al., 2001). Acute exposure at postnatal day 7 (P7) in rodents on the other hand represents exposure during a sensitive midpoint in the human embryonic third trimester equivalent (Cudd, 2005), which is considered the brain growth spurt period of rampant synaptogenesis and refinement in behaviorally relevant circuits (Dobbing and Sands, 1979, Bonthius and West, 1991). Alcohol insult during different stages of embryogenesis will affect different populations of cells, disrupt different developmental processes, and make different long-term functional impacts, which have not been fully described previously.
Here, we took advantage of an extended functional circuit – the olfacto-hippocampal pathway which includes regions expressing very different neurobehavioral developmental trajectories (Webster et al., 1983, Seress, 2007, Sarma et al., 2011, Burd et al., 2012). For example, the primary olfactory system (including the olfactory bulb and piriform cortex) is functional at birth in rodents (Brunjes, 1994, Miller and Spear, 2009, Sarma et al., 2011), and in fact is necessary for infant survival (Moriceau and Sullivan, 2004). In contrast, the hippocampal formation which receives robust olfactory input via the entorhinal cortex, is a relatively late developing system with hippocampal-dependent behaviors not emerging until near weaning in rodents (Freeman et al., 1994, Rudy, 1994, Raineki et al., 2010). Despite these differences in functional emergence, it is interesting that both the olfactory bulb and hippocampal formation show continued neurogenesis throughout life (Lledo et al., 2006). Given the differences in timing of development between these two components of the olfacto-hippocampal pathway, we hypothesized different temporal sensitivities to developmental ethanol exposure. We utilized the single day acute (binge-like) exposure model in mice, delivering ethanol or saline either intraperitoneally to pregnant mothers at embryonic day 8 (E8) or via direct subcutaneous injection to postnatal day 7 (P7) pups. The binge model was specifically chosen to allow a developmental dissection of when different cell types and circuits are particularly vulnerable to ethanol exposure (Sadrian et al., 2013). Each group was tested as adults at 15 weeks of age for long-term outcomes in cell proliferation, interneuron cell count, spontaneous and odor-evoked in vivo physiology, and spatial memory performance (please see schema in Figure 1).
Figure 1.

Schematic of ethanol and control saline treatment timing for each experimental group, including points for each measure taken. Acute anaesthetized recordings were made immediately prior to tissue fixation for subsequent immunohistochemical analysis.
II. Methods
Subjects
C57BL/6By mice were bred at the Nathan Kline Institute animal facility, and maintained on ad lib food and water at all times. All procedures were approved by the Nathan Kline Institute IACUC and were in accordance with NIH guidelines for the proper treatment of animals. Embryonic day 8 (E8) embryos were exposed to saline or ethanol via intraperitoneal injection to the mother, twice at a four hour interval with 2.8 g/kg (each dose) ethanol in saline for the ethanol group and saline only for the control group as previously described (Parnell et al., 2009). Postnatal day 7 (P7) pups were directly injected subcutaneously with saline or ethanol as described (Olney et al., 2002a, 2002b, Saito et al., 2007). Each mouse in a litter was assigned to the saline or ethanol group at an equivalent proportion of the total number of mice with a distributed gender ratio. Ethanol treatment (2.5 g/kg) was delivered twice in the same day at a two hour interval as originally described for C57BL/6 mice (Olney et al., 2002a, 2002b). After injections, pups were returned to their home cage. Pups were weaned at P28 into gender-specific group cages of littermates. Same-sex mice were housed together in cages in numbers between two and four per cage. Gender differences were not observed in P7-exposed mice when studied previously (Wilson et al., 2011, Sadrian et al., 2012), nor were any significant differences observed between genders here, so both genders were combined in the present E8/P7 developmental experiments. The total number of mice from within the seven litters examined for each condition is as follows: E8 Saline = 15 mice from seven litters (single condition of embryonic treatment for entire litter), gender ratio Male/Female (m/f) of 9m/6f; E8 Ethanol = 15 mice from seven litters (single condition of embryonic treatment for entire litter), 8m/7f; P7 Saline = 18 mice from seven litters (of which received mixed treatment, saline or ethanol as littermates), 8m/10f; P7 Ethanol = 15 mice from seven litters (the same litters from which P7 Saline mice were attained), 7m/8f. No more than two males and two females from any one litter were included. P7 ethanol treatment induced a peak blood alcohol level (BAL) of ~0.5g/dL when truncal blood was collected at 0.5, 1, 3, and 6 hours following the second ethanol injection and analyzed with an Alcohol Reagent Set (Pointe Scientific, Canton, MI) (Saito et al., 2007). Under the same P7 ethanol treatment conditions, it has been reported that initial BAL peaks are attained approximately one hour after each injection, with BAL falling below half of this level eight hours after first ethanol exposure (Wozniak et al., 2004, Young and Olney, 2006). In the E8 model, it has previously been reported that the maternal BAL reaches 0.5–0.6g/dL four hours after initial injection (Webster et al., 1983). Since BAL is considered comparable to brain alcohol level, and maternal BAL is comparable to fetal BAL (Burd et al., 2012), brain alcohol levels in E8 embryos were presumed similar to that of P7 pups, though fetal alcohol elimination rates may differ from those of neonates (Burd et al., 2012). A previous study using a similar ethanol dosage for three days (E6–8), found no significant differences in body mass throughout the embryonic trajectory (Oyedele and Kramer, 2008). Also, P7 binge exposure has been shown to cause no significant differences in body weight following ethanol administration, measured at P8 and P22 (Coleman et al., 2012). Additionally, the adult total body weights we measured did not significantly differ between groups on the date of behavioral, physiological and anatomical measures ([Condition = Males ± SEM / Females ± SEM]: E8 Saline = 25.6g±0.56 / 21.7g±0.40; E8 Ethanol = 25.0g±0.54 / 20.7g±0.53; P7 Saline = 26.1g±0.45 / 21.7g±0.25; P7 Ethanol = 25.9g±0.72 / 21.4g±0.53), as determined by ANOVA for main effects of treatment and age, with no significant interaction between the two variables.
Cresyl Violet Histology
Following in vivo electrophysiological depth recordings, mice were perfused intracardially with 0.1M phosphate buffered saline followed by 4% formalin fixative solution. The recorded (left) hemisphere of the brain was stored in the same fixative and subsequently sectioned coronally at a 40μm thickness, and stained with cresyl violet to provide the anatomical orientation of each recording.
BrdU Treatment and Tissue Preparation
Bromodeoxyuridine (BrdU) diluted in saline was injected intraperitoneally into 12 week old E8 and P7-treated mice once a day at a dose of 50μg/g for four consecutive days. Three weeks later mice were recorded and perfused in the same day with 0.1M PBS and then 4% formalin in 0.1M PBS. Dissected brain tissue from the unrecorded (right) hemisphere was post-fixed in perfusion solution and then dehydrated with 30% sucrose for sagittal sectioning at a 40μm thickness and stored at 4°C in 0.1M PBS plus 0.1% sodium azide.
Parvalbumin (PV) and bromodeoxyuridine (BrdU) immunohistochemistry and cell counts
For the immuno-histochemistry, sections were washed two times (10 minutes each) with 0.3% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA) in PBS. For DNA denaturation, sections were incubated in 2N HCl for 45 minutes at 37 °C and then washed with 0.1M sodium borate for 10 minutes. Next, sections were washed three times (10 minutes each) with 0.3% Triton X-100 in PBS and then incubated in blocking solution containing 5% normal donkey serum (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and 0.3% Triton X-100 in PBS for 1 hour at room temperature. Sections were then washed in 0.3% Triton X-100 in PBS for 10 minutes and subsequently incubated with primary antibody Mouse-anti-BrdU (BD Biosciences # 563445) and Rabbit-anti-PV (1:1000, Swant, Marly, Switzerland), in blocking solution overnight at 4°C. Then sections were washed in 0.1% Triton X-100 in PBS three times and incubated with a mixture of Alexa Fluoro 594 Goat-anti-Mouse IgG (1:1000) and Alexa Fluoro488 Goat anti-Rabbit IgG (1:1000) (both Life Technologies, Grand Island, NY) in 0.1% Triton X-100 in PBS containing 1% BSA for 1 hour at room temperature. Sections were finally washed in PBS three times, mounted, and cover-slipped using ProLong Gold Antifade Reagent (Life Technologies). All photomicrographs were taken through a 4X objective with a Nikon Eclipse TE2000 inverted microscope attached to a digital camera DXM1200F. The PV− or BrdU-positive cell number of each area of interest (AOI) and total dimensions of each AOI were measured using Image-Pro software, version 4.5 (Media Cybernetics, Silver Spring, MD). AOIs for PV-positive (PV+) cell counting were the piriform cortex, dorsal CA hippocampus, and dorsal subiculum, and AOIs for BrdU+ cell counting were specifically the dorsal dentate gyrus and the granule cell layer of the olfactory bulb. These AOIs were defined according to the Atlas of the Developing Mouse brain (Paxinos and Franklin, 2008). Then, the PV+ or BrdU+ cell densities of each AOI were calculated as the mean cell number per square millimeter, using four to five sections between 1.8–2.8mm (from mid-sagittal plane) for piriform cortex, four to six sections between 0.6–2.4mm for dorsal CA hippocampus and subiculum, four to six sections around 1.8–2.8mm for dorsal dentate gyrus, and two to three sections around 0.8–1.4mm for olfactory bulb. A total of eight to fifteen animals derived from six litters per group (Figure 1) were used for the calculations.
Behavior Experiments
An object placement spatial memory task was adapted for mice from (Luine et al., 2003, Macbeth et al., 2008) as performed in (Sadrian et al., 2012). Mice treated at either E8 or P7 aged to 14 weeks old first acclimated to an open Plexiglas arena (20cm × 40cm × 20cm) lined with fresh corn-cob bedding and marked well to provide orientation. The task occurred over the course of three days, with acclimation occurring on days 1 and 2 and training and testing occurring on day 3. First day acclimation (two, 5 min sessions with an inter-trial interval [ITI] of 10 min) was filmed to measure openfield exploration distances using Noldus Ethovision center point tracking software, and results compared by a Two-way ANOVA (treatment x age). The second day mice were again acclimated in two 5min sessions, this time with an ITI of four hours. On the third day mice were placed in the same arena for a training trial (Trial 1). The arena now contained two identical objects (amber glass vials, 2cm diameter × 5cm tall) placed equidistant to adjacent corners of the arena with openings facing inward. Mice were recorded during the 5min training trial during which they investigated the objects by sniffing. Mice were returned to their home cage immediately after training. Four hours after training trial, mice were tested (Trial 2) for 5min with one object moved diagonally to a new position adjacent to the other object, which retained its original position of Trial 1 (see Figure 6). The objects were cleaned with 70% ethanol and dried between testing of each subject during training and testing trials. The arena was rinsed with ethanol and dried and bedding refreshed between testing of each subject during acclimation, training and testing phases. All alcohol odor was evaporated before each trial. For double-blind offline analysis, total object investigation times were quantified from video recordings of the training and test trials. Object investigation times were scored as the time the nose was pointed toward the object within 2cm. Investigation times in each trial were summarized as the time the moved object was investigated as a proportion of total investigation time of both objects. Mice that did not investigate both objects during training or testing were removed from analysis (n-values: E8 Saline = 0; E8 Ethanol = 0; P7 Saline = 1; P7 Ethanol = 1). Mice that preferentially investigated one object more than the other object over three-quarters of total investigation time during the training trial were not tested in order to eliminate possible arena preference confounds (n-values: E8 Saline = 1; E8 Ethanol = 1; P7 Saline = 1; P7 Ethanol = 1). Investigation time proportions were compared within group between the training and test trials using a student’s t-test. Raw total object investigation times were compared by a Two-way ANOVA (treatment x age).
In vivo olfacto-hippocampal physiology
15 week old mice were anesthetized with urethane (1.5 g / kg) and placed in a stereotaxic apparatus. Monopolar tungsten electrodes were directed by stereotaxic coordinates according to a mouse brain atlas (Paxinos et al., 2007) at the olfactory bulb (OB), anterior piriform cortex (aPCX) and dorsal hippocampus (dHipp), and recordings were made relative to a scalp reference electrode. Respiration was monitored with a piezoelectric plethysmograph placed on the animal’s back. Signals were amplified and filtered (0.3 to 3000Hz), digitized at 10kHz, and stored and analyzed with Spike2 (Cambridge Electronic Design Inc; Cambridge, England). Paired-pulse analysis of lateral olfactory tract (LOT)-evoked monosynaptic potentials in the piriform cortex consisted of two, equal intensity constant current pulses at 0.1mA with varying inter-stimulus intervals (20, 50, 100, 300 ms) with a 10s delayed between consecutive pairs. The slope of the initial positive-evoked potential was used as a measure of response amplitude and the slope of the response to the second (test pulse) was expressed as a percentage of the response to the first (conditioning) pulse. Comparisons were made using a Two-way ANOVA (treatment x age) and Bonferonni post-hoc tests. Local field potential responses to a simple, monomolecular odor stimulus (nonane; Sigma, diluted 1:100 in mineral oil, 2 second duration, 3 repeats) were recorded simultaneously in all three brain regions. The mean baseline (2 seconds prior to odor onset) and odor-evoked activity (2 seconds from odor onset) were used in the analyses (see Figure 5a). Odor-evoked local field potentials (LFP) were analyzed with Fast-Fourier Transforms (FFT) with 2.49 Hz bin resolution. OB-aPCX and OB-dHipp spontaneous LFP coherence were determined across at least 50 sec of stable activity using a “cohere” script function within Spike2 at a 5-Hz resolution. Coherence measuring pairwise functional connectivity between regions of the olfacto-hippocampal circuit were made.
Data Summaries and Statistics
All individual animal data points within each litter were quantified into litter averages. These litter representations yielded condition averages for graphic representation of each of the four groups, which were compared by treatment (Saline or Ethanol) and age of exposure (E8 or P7), using a Two-way ANOVA with Bonferroni post-hoc tests of individual condition comparisons. For behavioral experiments in spatial memory, paired t-tests were used for repeated measures comparisons of the relative proportions of investigation time spent at each of two identical objects by each animal (see Figure 6).
III. Results
Long-term parvalbumin-positive interneuron counts are differentially affected from developmentally distinct windows of acute ethanol exposure
The maturation of sensory circuit inhibition largely occurs during critical early periods of development (Hensch, 2005, Sarma et al., 2011) and varies across brain regions. To test for differential impacts of initial developmental alcohol exposure timing, we first studied the long-term effects of ethanol on the quantity of PV+ interneurons in the piriform cortex and hippocampal formation (Figure 2).
Figure 2.

Parvalbumin (PV)-positive inhibitory interneuron counts in various brain regions of adult mice treated with ethanol or saline at E8 or P7. Long-term differences were found in the piriform cortex (PCX), dorsal hippocampus CA1 (dCA), and the subiculum (Sub) as seen in (A) representative images of parvalbumin immunohistochemistry. B) Timing-specific effect of developmental alcohol exposure on PV+ cell counts, represented here as percentages relative to saline-treated animals of the same age exposure group (E8ethanol vs. E8saline and P7ethanol vs. P7saline) for each of the different brain regions measured. This illuminates regional differences of the timing-specific effects on PV+ cell counts. C) Interaction plots of statistical comparisons for treatment (saline or ethanol) and age (E8 or P7) effects on adult PV+ cell counts. Two-way ANOVA results show piriform cortex (left) had a significant treatment main effect (* p<0.01), while dCA (middle) showed a significant interaction (Treatment x Age, triple open-star = p<0.0001). The subiculum (right) also showed a significant interaction (Treatment x Age, double open-star, p<0.01). Treatment condition post-hoc comparisons in the dorsal CA and subiculum within each age condition are differentially significant based on timing (** p<0.01, ***p<0.001). Bar in A = 100μm.
In the adult piriform cortex, PV+ neuron counts were significantly decreased by either E8 or P7 ethanol exposure. We observed a significant main effect of ethanol treatment with a decrease in PV+ cells in the piriform cortex (PCX) of ethanol-treated animals in both age groups (Two-way ANOVA [treatment]PCX F[1,24]=26.09, p<0.0001; Bonferroni post-test [Saline vs. Ethanol]: Age E8, p<0.01; Age P7, p<0.01). There was also a main effect of age, ([age]PCXF[1,24]=4.75, p=0.0394) with no significant interaction between treatment and age. Neurodegeneration in the PCX has been detected before immediately following acute ethanol exposure in early postnatal rats (Balaszczuk et al., 2011) and in adult rats (Leasure and Nixon, 2010). The identity of the affected cell populations and long-term effects however, had not previously been identified.
In contrast, the effects of developmental ethanol exposure on PV+ neurons in the adult hippocampal formation were heavily dependent on the age of exposure, with a significant loss of PV+ neurons only after exposure later in development (P7). In both the dorsal CA hippocampus (CA) and the subiculum (SUB), PV+ cells were differentially affected depending on the developmental stage of exposure with a significant main effect of age ([Age]CA: F[1,24]=8.84, p=0.0066; [Age]SUB: F[1,24]=5.77, p=0.0245; Bonferroni post-test [E8 vs. P7]: p<0.01), and each region showing significantly variable interactions (Two-way ANOVA [Treatment x Age]CA: F[1,24]=32.53, p<0.0001; [Treatment x Age]SUB: F[1,24]=6.47, p=0.0178). There were no significant treatment effects for either the dorsal CA, nor the subiculum areas; however, post-hoc t-tests between treatment conditions at each age are significant in an age-dependent manner in both the dorsal CA ([E8 Saline vs. E8 Ethanol]CA: t =3.38, p<0.01 and [P7 Saline vs. P7 Ethanol]CA: t=4.68, p<0.001) and the subiculum ([E8 Saline vs. E8 Ethanol]SUB: t=0.493, not significant, and [P7 Saline vs. P7 Ethanol]SUB: t=3.11, p<0.01). Interestingly, the PV+ cell counts were significantly higher for E8 ethanol exposed adults than saline treated controls. These data collectively suggests that changes in PV+ cell number caused by early ethanol exposure vary based on the developmental period of initial exposure and brain region measured, with the earlier developing piriform cortex more sensitive to E8 exposure than the later developing hippocampal formation.
Differential long-term deficits in olfactory physiology based on the initial timing of ethanol exposure
We next examined physiological effects that may be associated with the long-term anatomical differences in the PCX presented above. We performed acute in vivo local field potential (LFP) recordings (please see Figure 3a) in the same adult mice from which brain tissue was prepared for the immunohistochemical procedures described above. We selected for physiological metrics of olfactory processing known to be supported by the same brain regions (Neville and Haberly, 2004, Franks and Isaacson, 2006) that we found to be anatomically impacted by early ethanol. As we described previously (Wilson et al., 2011), ethanol exposure at P7 resulted in odor-evoked LFP hyperactivation in the adult PCX (Figure 3b). Here we find that both P7 and E8 exposure induce PCX hyper-excitability. Comparing long-term effects of initial exposure at E8 or P7 yielded a non-significant main effect for age, but a significant main effect for treatment with elevated odor-evoked power in the beta range in the adult PCX (Two-way ANOVA: [treatment]Beta F[1,24]=96.02, p=0.0222); post-hoc analysis indicates P7 (and not E8) ethanol-treated animals were significantly elevated. Evoked gamma-band activity was also significantly increased over saline controls for P7 exposed adult mice (Two-way ANOVA: [treatment]Gamma F[1,24]=5.78, p=0.0247. These results imply long-term effects on PCX odor-evoked beta hyperactivity that is more dramatic after ethanol exposure at P7 than at E8. Olfactory bulb and dorsal hippocampus LFP analyses showed no significant differences in odor-evoked amplitude or spontaneous activity (data not shown).
Figure 3.

In vivo local field potential (LFP) recording of olfacto-hippocampal circuit physiology in the olfactory bulb, piriform cortex and hippocampus. A) Example trace of an evoked response to odor delivery in anaesthetized mice. Sampled data was digitally filtered into designated frequency bands for analysis. Example electrode tract recording locations in the olfactory bulb (OB), dorsal hippocampus (dHipp) and anterior piriform cortex (aPCX) are shown to the right (Bars = 500μm). FFT analysis of odor-evoked power measured in the piriform cortex (PCX) were acquired for the frequency spectrum 0–80Hz as shown in (B), and broken down into individual frequency bands defined as Delta (0–5Hz), Theta (5–15Hz), Beta (15–35Hz) and Gamma (35Hz–80Hz) for Two-way ANOVA (Age x Treatment) translated to cognate interaction line plots. A significant main effect for ethanol treatment in the piriform cortex (PCX) was found in both beta and gamma bands, with an average relative power increase in the beta and gamma bands over those of saline controls in both ages, of which only P7 ethanol treated adults was significant (* p<0.05).
GABAergic interneurons support and help coordinate circuit function by enhancing temporal precision of both odor-evoked and spontaneous local activity in the PCX (Selby et al., 2007, Suzuki and Bekkers, 2012). The significant differences in LFP were in the hyper-activated direction compared to controls in both age exposure groups, which coincides with the decreases in PV+ cell number we found in the PCX. We therefore examined paired-pulse effects in the PCX, which are known to be partially dependent on synaptic inhibition (Patneau and Stripling, 1992). In our paired-pulse analysis we gave an electrical stimulation conditioning pulse to the lateral olfactory tract (LOT) immediately followed by a second test pulse, while recording evoked responses to each in the PCX (see Figure 4a for schematic). Second pulse response slopes (test) were measured and quantified as a percentage of the initial conditioning pulse response (baseline) slope (Figure 4b). Relatively short (20ms) inter-pulse intervals (IPI) used at the start of the experiment were progressively increased to longer IPIs. The amplitude of the test response reflects both changes in pre-synaptic excitatory transmitter release and feedback and feed-forward synaptic inhibition (Ketchum and Haberly, 1993, Best and Wilson, 2004). Saline-treated controls in both E8 and P7 age groups exhibited typical paired-pulse inhibition (Figure 4c and 4d). Ethanol-treated animals from both E8 or P7 exposures exhibited paired-pulse facilitation, which was significantly different than their respective age saline-treated controls (One-way repeated measures ANOVA [20–100ms IPI]: F[3,15]=8.43, p=0.0056, Bonferroni post-test [E8: Saline vs. Ethanol]q=4.52, p<0.05; [P7: Saline vs. Ethanol] q=5.49, p<0.05), yet with no significant differences between age groups. The enhanced paired-pulse facilitation may indicate a long-term reduction in local feedback inhibition in the ethanol-treated mouse PCX, independent of the initial age of exposure. These results provide further physiologic evidence for synaptic inhibition dysfunction in ethanol-treated mice, regardless of whether the developmental age of exposure was at E8 or P7. The shared deficit in local feedback inhibition is conceptually aligned with the decrease of PV+ inhibitory interneurons in both age ethanol exposure groups described above.
Figure 4.
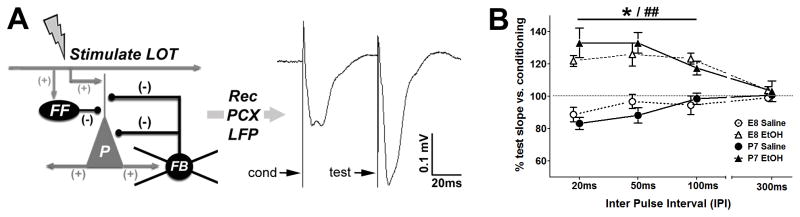
Paired-pulse analysis performed to examine local circuit synaptic inhibition in adult mice. A) Diagram illustrates the experimental paradigm used. Pairs of lateral olfactory tract (LOT) electrical stimulation (0.5mA each) originating in the olfactory bulb creates a change in local field potential (LFP) measurable at the next synaptic contact in the piriform cortex (PCX), which contains associative excitatory (+ sign) pyramidal cells (P, grey) and local inhibitory (− sign) interneurons (black) that provide both feedfoward (FF) and feedback (FB) inhibition. Initial conditioning pulse stimulation of PCX pyramidal cells conventionally generates feedback inhibition from local interneurons that dampen successive responsiveness lasting a limited period (~20–100ms), after which synaptic inhibition is not detectable. Arrows indicate artifacts produced by both conditioning and test electric stimulation events while the remainder of the trace demonstrates the resulting average LFP waveforms in the PCX. (B) Both E8-treated and P7-treated mice show a significant shift from paired-pulse depression to paired-pulse facilitation when comparing response curve slopes at inter-pulse intervals (IPI) between 20ms and 100ms (** E8 EtOH p<0.01, #P7 EtOH p <0.05, ##P7 EtOH p<0.01).
Negative impacts on adult cell proliferation caused by varied ethanol exposure timing
Deficits in brain function are not only associated with neurodegenerative cell loss (Seeley et al., 2009), but also with decreases in neurogenesis that would otherwise support circuit refinement over time (Lledo et al., 2006). Newly born neurons are continually provided to the olfactory bulb (from the subventricular zone [SVZ]) and hippocampus (from the dentate gyrus [DG]) to integrate and support persisting refinement toward adaptive memory (Gould et al., 1999, Sahay et al., 2011, Alonso et al., 2012). A decrease in DG neurogenesis found in rodents exposed to ethanol during early postnatal development has been shown to persist into adulthood (Klintsova et al., 2007, Burd et al., 2012). We sought to determine if earlier episodes of ethanol exposure also produce a similar change in long-term cell proliferation within a behaviorally measureable circuit, and also to test whether the effects were expressed in the other major site of adult neurogenesis- the olfactory bulb (Lledo and Saghatelyan, 2005). We performed BrdU immunohistochemistry to quantify adult cell proliferation in the olfactory bulb (OB) and hippocampus dentate gyrus (DG) of cells that also concomitantly survived the three week period between initial labeling and tissue fixation (Figure 5a). There was no decrease in the number of adult-born olfactory bulb cells in the granule cell layer of mice exposed to ethanol at either E8 or P7 (Two-way ANOVA [Treatment]OB F[1,25]=0.049, p=0.83, Figure 5b and 5c). In contrast, adult-born DG cells were significantly reduced by early ethanol exposure. BrdU-positive cell counts showed a significant main effect for treatment with a decrease in the ethanol treated mice compared to saline controls (Two-way ANOVA [Treatment]DG F[1,37]=27.36, p<0.0001, Figure 5b and 5d) with significant post-hoc test results at both ages (Bonferroni post-tests [E8: Saline vs. Ethanol] t=2.76, p<0.05, [P7: Saline vs. Ethanol] t=3.49, p<0.01, Figure 5b).
Figure 5.

Long-term changes in cell proliferation as a result of early ethanol exposure. A) Representative images of BrdU immunohistochemistry in the hippocampal dentate gyrus. B) Relative comparisons of ethanol treatment mice with same-aged saline treatment controls (E8ethanol/E8saline and P7ethanol/P7saline) in each brain region examined (OB=olfactory bulb, DG=dentate gyrus) show decreases in newborn cells stained with BrdU in the dentate gyrus that have also survived to the point of measurement, three weeks after the final injection. Cell counts were made at adulthood (3 months after the latest P7 saline or ethanol exposure, please see Figure 1 for diagram). C) Counts of BrdU-positive cells in the OB showed no relative difference between saline and ethanol conditions at either E8 or P7 exposure age. D) BrdU-positive cell counts in the dentate gyrus were significantly lower in the ethanol treated groups for both age exposures than that of saline controls at either age (***p<0.001).
Hippocampal-dependent spatial memory and object investigation are deficient in adult mice exposed to ethanol at P7, but not in adults exposed at E8
The above results support previous findings (Klintsova et al., 2007) of sustained decreases in neurogenesis rates in the hippocampus of early ethanol-treated animals. Activity in subregions of the hippocampus have long been known to predict spatial memory retention (Segal et al., 1988), while neurogenesis is considered integral to specific types of memory formation (Deng et al., 2010). In addition, we and others have previously shown that acute postnatal ethanol exposure results in immediate apoptotic neurodegeneration in the hippocampus (Olney et al., 2002, Wozniak et al., 2004), and P7 ethanol-induced neurodegeneration in this region is correlated with deficits in spatial memory task performance (Wozniak et al., 2004, Wilson et al., 2011, Sadrian et al., 2012). We accordingly tested spatial memory performance in the same adult mice used for immunohistochemical analysis of PV+ and BrdU-positive cell counts described above. Spatial memory was tested using an object placement task, where performance was quantified as the percentage of investigation time of a moved object during a five minute test period (please see Methods and Figure 6a).
Figure 6.

Spatial memory performance in adult mice using an object placement task. A) Diagram illustrating the spatial memory behavior task used. Functional spatial memory was determined by moved object investigation time (sniffing object <2cm away) in the test/recall trial (Trial 2) versus the training trial (Trial 1). B) Trial 1 training investigation of the two novel identical objects averaged around 50% (random chance – horizontal dotted line) for each object in all treatment/age groups. Trial 2 saline-treated control animals in both age categories showed a significant increase in moved object investigation time proportion versus that same object in its original location during Trial 1, indicating spatial memory recall (see methods section). Mice treated with ethanol at E8 also showed a significantly higher proportion of moved object investigation (* p<0.05, ** p<0.01). Adult mice treated with ethanol at P7 however, showed no difference between trials, indicating a spatial memory deficit. C) Total raw investigation times of both objects during Trial 1 were not significantly higher in this same spatial memory-deficient P7 ethanol-treated group as compared to all other treatment/age groups during either trial. D) Openfield exploration times during first exposure to spatial task arena acclimation showed no significant differences between groups.
Both objects and their orientation in the arena were novel during the training trial (Trial 1) and therefore the investigation proportions of each object averaged near random chance (50:50). Saline controls of both age exposure groups demonstrated successful spatial memory behavior, defined as a statistically significant increase in investigation of the moved object during the testing trial (Trial 2) versus that same object in its original location during the preceding training trial (paired t-test within subjects [trial 2 vs. trial 1] E8-Saline: t=4.54, df=14, p=0.0005; P7-Saline: t=2.36, df=17, p=0.031). P7 ethanol-exposed adults showed a spatial memory deficit with non-significant difference for investigation of the moved object during the testing trial (P7 Ethanol: t=1.32, df=14, p=0.207). Interestingly, E8 ethanol-exposed adults had no apparent spatial memory deficit (t=7.20, df=14, p<0.0001, Figure 6b). There were no significant differences in total raw object investigation time between experimental groups during the training (Trial 1) nor the test trial (Trial 2), in which spatial memory was assessed based on relative investigation of the moved object. Investigation proportions were not confounded by ambulatory variance between groups during training or testing trials, as openfield exploration during initial behavior arena acclimation was similar across all groups (Figure 6d). Together these results indicate a temporally specific window of development in which spatial memory deficit develops as a result of acute ethanol exposure, implications of which are discussed below.
Long-term effects on spontaneous LFP coherence within the olfacto-hippocampal circuit vary depending on the age of developmental ethanol exposure
We have thus far identified distinct profiles of ethanol-induced change in piriform cortex anatomy and physiology as well as in hippocampal anatomy and hippocampus-dependent behavior that are each exposure time-dependent. Furthermore, we have identified changes in a specific class of inhibitory interneurons within these structures. Local inhibitory activity generated by interneurons within a local microcircuit establishes the oscillatory activity that facilitates communication between distant brain regions (Buzsaki and Draguhn, 2004). We therefore examined how these functional/anatomical effects of developmental ethanol exposure might associate with functional connectivity (coherence) between regions within the olfacto-hippocampal pathway. The olfactory bulb projects to the piriform cortex, and both the olfactory bulb and piriform cortex project to the entorhinal cortex, which is the primary cortical input to the hippocampal formation via the lateral perforant path (Turner et al., 1998). Additionally, the olfactory bulb and piriform cortex receive indirect feedback from the CA1 hippocampus via the entorhinal cortex (van Groen and Wyss, 1990, Martin et al., 2007). Using the adult mice of each age exposure group, we measured spontaneous LFP coherence between the olfactory bulb and dorsal CA1 hippocampus (OB-dHipp) as well as between the olfactory bulb and PCX (OB-PCX). Coherence was determined across a frequency spectrum from 0–80Hz over a 50 second period of spontaneous baseline activity. Frequency bins were grouped as Delta (0–5Hz), Theta (5–15Hz), Beta (15–35Hz), and Gamma (35–80Hz) band averages for statistical analysis. Each age group of ethanol exposure showed specialized changes in both spontaneous OB-dHipp and OB-PCX coherence compared to saline-injected controls (Figure 7).
Figure 7.

Inter-regional spontaneous coherence comparisons in adult mice treated with either saline or ethanol at either age E8 or P7. A) Spontaneous coherence measures between the OB and piriform cortex (PCX). Upper panels left and right show normalized coherence across frequencies 0–80Hz, with the grouped bar graph panel summarizing average coherences within each frequency band, defined as Delta (0–5Hz), Theta (5–15Hz), Beta (15–35Hz) and Gamma (35Hz–80Hz). There was a significant main effect for treatment in spontaneous theta band OB-PCX coherence (** p<0.01). When general gamma coherence was subdivided (inset plots) into slow (39–49Hz) and fast (75–80Hz) ranges, P7 ethanol (*p<0.05), but not E8 ethanol-treated adult mice show a significant main effect for treatment with elevated coherence over saline controls. B) Coherence between the olfactory bulb (OB) and Hippocampus with data presentation formatted as in (A). Lower panels of interaction plots display age-related differences between saline and ethanol treated groups, with additional insets for slow and fast gamma subdivisions. Spontaneous delta coherence between the OB and hippocampus was variable as a result of ethanol treatment based on timing of delivery with a a significant interaction between variables (Treatment x Age: ☆ p<0.05). Gamma coherence was elevated in adult mice treated with ethanol at age E8, most dramatically in the fast gamma frequency range (*p<0.05).
Spontaneous OB-PCX gamma coherence was modified by developmental ethanol exposure (Figure 7a). OB-PCX effects were present in P7-exposed adults (Two-way ANOVA [OB-PCX, Treatment x Age]θ: F[1,24]=5.61, p=0.0263; Bonferroni post-test [E8: Saline vs. Ethanol] t=3.334, p<0.01), but not E8-treated adults. Spontaneous gamma coherence was not significant for any conditional comparison when examining the entire band (35–80Hz); however, when subdivided into slow gamma (39–49Hz) and fast gamma (75–80Hz) ranges, spontaneous fast gamma showed a statistical significant main effect for treatment (F[1,24]=5.54, p=0.0271), specifically for P7-treated adults.
Spontaneous coherence between the OB and dorsal CA hippocampus was also significantly altered in a developmentally dependent manner, with a significant interaction (Treatment x Age) in the delta frequency range (Two-way ANOVA [OB-dHipp, Treatment]Δ: F[1,24]=8.197, p=0.0086; Figure 7b). Additionally, adult mice treated at E8 specifically showed a significant increase in gamma band and fast gamma band coherence (Two-way ANOVA [Treatment]γ: F[1,24]=7.87, p=0.0098). Post-hoc analysis revealed only animals treated with ethanol at E8 are statistically significant different from controls (Bonferroni post-test [E8: Saline vs. Ethanol] t=2.43, p<0.05; [P7: Saline vs. Ethanol] t=1.54, p>0.05 ns).
IV. Discussion and Conclusions
The present study took advantage of the distinct developmental trajectories of two components within the olfacto-hippocampal pathway in order to explore age-dependence of developmental ethanol exposure effects. This study also begins an exploration of a potential role for a specific class of inhibitory interneuron in long-term neural circuit functional disruption induced by early ethanol. Developmental ethanol exposure reduced parvalbumin-positive inhibitory interneuron number and enhanced excitability of both the piriform cortex and the hippocampal formation in adults. The earlier developing piriform cortex was more strongly affected by E8 ethanol exposure, while the later developing hippocampal formation was more strongly affected by P7 ethanol exposure. Although both the olfactory bulb and the hippocampal dentate gyrus display cell growth through adulthood, only adult cell growth in the dentate gyrus was affected by developmental ethanol (please see Table 1 and Figure 8 for data summaries). Together, the results suggest there are commonalities across systems in terms of the cell types affected and the resulting functional disturbance. Importantly, timing matters (Tran and Kelly, 2003, Nunes et al., 2011, Komitova et al., 2013), and treatments for FASD may have to be individualized to match the age of maximal developmental exposure.
Table 1.
Table summarizing long-term impacts in adult mice measured after single day binge ethanol exposure at the specified time-point.
| Long-term outcomes versus saline-injected controls | |||
|---|---|---|---|
| E8 EtOH exposure | → Common ← | P7 EtOH exposure | |
| PV-positive cell count | |||
| Piriform | ↓↓ | ||
| Dorsal CA | ↑ | ↓↓ | |
| Subiculum | = | ↓ | |
| Odor-Evoked LFP (PCX) | |||
| Delta | = | ||
| Theta | = | ||
| Beta | ↑ | ||
| Gamma | = | ↑ | |
| Paried-Pulse Response | |||
| 20ms | ↑↑↑ | ||
| 50ms | ↑↑↑ | ||
| 100ms | ↑↑↑ | ||
| 300ms | = | ||
| BrdU-positive cells | |||
| Olfactory Bulb | = | ||
| Dentate Gyrus | ↓ | ↓↓ | |
| Spatial Memroy Task | |||
| % moved object inv. time | = | ↓ | |
| OB-Hipp Coherence | |||
| Delta | = | ↑ | |
| Theta | = | ||
| Beta | = | ||
| Gamma | ↑ | = | |
| Slow Gamma | = | ||
| Fast Gamma | ↑ | = | |
| OB-Piriform Coherence | |||
| Delta | = | ||
| Theta | = | ↑ | |
| Beta | = | ||
| Gamma | = | ||
| Slow Gamma | = | ||
| Fast Gamma | ↑ | = | |
Figure 8.

Diagram summarizing the measured long-term differences of single day ethanol exposure that were both similar and divergent among the various parameters, depending on the developmental age of exposure.
Synaptic balance is maintained in part by inhibitory interneuron populations that provide feed-forward and feedback inhibition toward temporal control of excitatory pyramidal cell firing (Buzsaki et al., 2007, Bekkers and Suzuki, 2013). We have previously observed long-term deficits of circuit function in olfactory cortical processing that are indicative of synaptic excitation/inhibition imbalance (reviewed in Sadrian et al., 2013). This hypothesized imbalance is a mechanistic foundation for many of the neurobehavioral deficits found in FASD and related cognitive disorders (Sadrian et al., 2013). How long-term synaptic imbalance is initiated by early ethanol exposure is unclear, but is likely connected to the many well-documented effects of ethanol on brain function, such as: apoptotic neurodegeneration (Ikonomidou et al., 2000, Chakraborty et al., 2008), altered synaptic transmission in specific cell populations (Sanderson et al., 2009, Wang et al., 2013), skewed neurotransmitter profiles (Sari et al., 2010), disruption of glutamatergic receptor subunit regulation (Nixon et al., 2002), dendritic tree reduction of PV+ interneurons (De Giorgio et al., 2012), and decreased neurogenesis (Burd et al., 2012); all found immediately following insult and some shown lasting into adulthood. These effects have each been demonstrated after acute or short-term ethanol exposure and are therefore often attributed to disruption of developmental processes occurring within that period. In accord with this, we have found that outcomes of developmental ethanol exposure can vary simply due to the timing of single-day binge ethanol. However, in parallel to these findings we have also found several common long-lasting deficits in both E8 and P7-exposed animals alike, therefore indicating that some foundations of anatomical integrity and neurobehavioral circuit function are persistently vulnerable to ethanol toxicity across a wider span of development.
Animals exposed to ethanol at either E8 or P7 each showed a long-term significant decrease in PV+ inhibitory interneuron count in the PCX, with a corresponding facilitation response to paired electrical stimulation of the LOT. This result is at least partially indicative of local feedback inhibition dysfunction (Ketchum and Haberly, 1993). P7-exposed adults also showed hyperactive beta and gamma LFP power in response to odor stimulation. These are physiologic indicators of an insufficiently regulated neuronal circuit, identified in the same mice that also have decreased cortical PV+ interneurons. Interneurons are not universal in their susceptibility to damage and cell death. A recent study of hypoxic-reared mice found decreased expression of PV and somatostatin (SST), an additional marker of GABAergic interneurons, yet showed an increase in reelin (RLN)-expressing interneurons and no change in total interneuron number (Komitova et al., 2013).
A decline in interneuron numbers does not necessarily imply lower inhibitory capacity. Our finding of PV+ cell reduction is rather an anatomical correlate for the physiological changes we observed. The present data does not determine whether the decrease in PV+ cells is a source of the excitation/inhibition LFP imbalance measured, a result of it, or simply coincidental. Persistent decreases in tonic GABA inhibition into adulthood have in any case been measured after acute ethanol exposure during adolescent equivalent stages (reviewed in Spear and Swartzwelder, 2014). Additionally, early alcohol exposure has been postulated to increase ontological risk for aberrant GABAergic inhibitory capacity in the prefrontal cortex, which is a commonly characterization in schizophrenia (Volk and Lewis, 2013).
The significance of PV+ interneurons and ethanol-induced behavioral dysfunction was illustrated in a recent study showing that a reduction of PV+ cells induced by embryonic ethanol exposure were replenished through successful transplantation of neural stem cell progenitors in the rat cingulate cortex and amygdala, which coincidentally rescued the social interaction deficits otherwise found in mice with an ethanol-induced PV+ cell deficit (Shirasaka et al., 2012). The development of behaviorally relevant circuits is associated with early windows of elevated plasticity, outside of which circuit refinement is less likely (Moriceau and Sullivan, 2005, Bavelier et al., 2010). We have demonstrated here that the timing of ethanol exposure is very influential to specific spatial memory task performance. This dichotomy of behavioral deficit correlates with exposure outcomes of PV+ cell counts in the adult dorsal CA hippocampus, which varied based on alcohol exposure timing.
We observed an increase of PV+ cells in E8-exposed adults in the dorsal CA. A short term increase in PV+ neurons in the parietal cortex has been reported in response to acute ethanol exposure during embryonic (E11–14) development (Isayama et al., 2009). However, this study also found no long-term changes in PV+ cell counts when adult tissue was examined, in contrast to what we found here in the hippocampus. Additionally, P4-10 ethanol exposure study yielded no long-term differences in PV+ cell number in the medial septum nor anterior cingulate cortex (Mitchell et al., 2000), also suggesting a brief increase in PV+ interneurons that ultimately brought net changes to null. The discrepancy with our data may simply be due to the different regions analyzed in these studies and/or the different exposure schedules used. P7 ethanol exposure has been shown to generate long-term decreases in PV+ cell number in the M2 prefrontal motor cortex (Coleman et al., 2012), so long-term changes in subpopulation cell counts have previously been shown to persist into adulthood.
A major function of interneuron local inhibitory activity is to contribute toward field oscillations that facilitate communication (functional connectivity) between distinct brain regions (Buzsaki and Draguhn, 2004). Functional connectivity is a measure of network cohesion and information flow. Pathologies such as schizophrenia (Tang et al., 2013), ADHD (Hoekzema et al., 2013), depression (Greicius, 2008, Whitfield-Gabrieli and Ford, 2012) and Alzheimer’s disease (Wesson et al., 2011) are associated with hypo- and hyper-connectivity. PV-interneurons have been found integral to the establishment of gamma oscillations in the hippocampus (Tukker et al., 2007, Cardin et al., 2009). We examined whether the age-profiled effects discussed above were associated with changes in interregional communication within the olfacto-hippocampal circuit. We found spontaneous coherence between the OB and hippocampus to be elevated specifically in delta band (0–5Hz) frequencies in P7-exposed adults, and not E8-exposed adults. Between the OB and PCX, P7-exposed animals showed increased coherence in the theta band (5–15Hz). In contrast, E8-exposed adults and not P7-exposed adults showed elevated coherence between the OB and PCX in the gamma band (35–80Hz), specifically in the fast gamma frequency range (75Hz–80Hz). Select synchronicity at slow gamma and fast gamma frequencies has been shown between afferent inputs of the hippocampal formation based on the brain region of origin (Colgin et al., 2009). Again this timing-based variation indicates specific developmental processes that are specifically vulnerable to ethanol insult during these corresponding periods, yielding dichotomous trajectories of circuit dysfunction. While PV+ interneurons play an integral role in generating local and interregional oscillatory activity, it may be that the increases in coherence we found at specified frequency bands were due to other ethanol-induced perturbations, such as synaptic efficiency, that are not a result of PV-interneuron deficit, but rather coincide with it.
Our LFP results of odor-evoked power raise questions about a functional meaning. The long-lasting ethanol-induced abnormalities measured in the olfacto-hippocampal circuit that varied depending on age of exposure, did so with associated differences in the frequency bands affected. For instance, odor-evoked LFP’s in P7 (but not E8) exposed adults was elevated in the beta and gamma bands. Oscillatory activity in these same frequency ranges are coincidentally known to be highly dynamic during olfactory task training in awake behaving animals (Lowry and Kay, 2007, Martin et al., 2007). Specifically, behaviorally relevant odor sampling under training produces an increase in beta oscillatory activity with a concomitant reduction in spontaneous gamma oscillations (Martin and Ravel, 2014). Of course, it is problematic to relate these findings toward a functional significance in our data, as all of our physiological recordings were done in anaesthetized animals. What is discernable from our findings, is a clear divergence in physiological consequence that can be attributed to the initial age of binge alcohol exposure. How the ethanol-induced changes in olfacto-hippocampal circuit LFP might affect fine odor discrimination tasks, odor memory and hedonics evaluation are important questions for future study.
Finally, our results further support the previously reported differences between adult cell proliferation in the subventricular zone and the hippocampal formation. Adult neurogenesis in both systems has been shown to be sensitive to experience (Rochefort et al., 2002, Brown et al., 2003, Mandairon et al., 2003) and adult generated neurons contribute to memory (Rochefort et al., 2002, Leuner et al., 2006, Lledo et al., 2006) and pattern separation (Sahay et al., 2011). However, while adult exercise increases, and early life stress reduces, adult neurogenesis in the dentate gyrus, neither impact neurogenesis in the subventricular zone which supplies the olfactory bulb (Brown et al., 2003, Belnoue et al., 2013). Similarly, we find here that adult cell growth in the dentate gyrus is reduced by ethanol exposure at either E8 or P7, while cell growth in the olfactory system is unaffected by developmental ethanol exposure. Differences in developmental mechanisms between these two stem cell populations are unknown. Given the reported decrease of neurogenesis and survival of hippocampal-born neurons as a result of ethanol exposure (Ehlers et al., 2013), and the known component of adult hippocampal neurogenesis deficit in mood disorders such as depression (Samuels and Hen, 2011), further investigation of the link between impaired cell proliferation and memory and mood disorders in FASD is warranted.
A major confounding obstacle to understanding FASD etiology is the variability with which neurobehavioral pathology surfaces. In order to tailor early detection and/or intervention methods in human FASD, we must better understand the kinship between major variables, such as exposure timing, and the mechanisms of toxicity through which behavioral pathology is generated. Valuable bioinformatics tools are now available and allow for stratified comparisons of early brain growth and function to be made between multiple species, including rodents and humans (Clancy et al., 2007, Workman et al., 2013). This platform not only helps determine developmental equivalency between various animal models, but also allows for prediction of toxic effects like ethanol-induced neurodegeneration that are collectively based upon previous experimental findings (Gohlke et al., 2005). Carefully modeled research can now be coordinated to provide the additional raw data needed to continually refine these computer-modeled approaches. Such coordinated efforts could in turn feedback to bench research with optimized guidelines for future investigation and FASD-targeted treatments.
Highlights.
Early alcohol exposure timing creates variable long-term outcomes in mice
Adult olfacto-hippocampal circuit function varies based on age of alcohol exposure
Early alcohol exposure reduces interneuron number in adult piriform and hippocampus
Long-term neurogenesis decline in the dentate gyrus after P7 exposure, not E8
P7, but not E8, alcohol exposure induces adult spatial memory deficits
Acknowledgments
The authors were supported by grants T32-MH067763 from the NIMH to B.A.S., R01-DC003906 from the NIDCD to D.A.W., R01 AA015355 from the NIAAA to M.S., and R01-AA023181 to M.S. and D.A.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
VI. Works Cited
- Abel EL. Fetal alcohol syndrome: the ‘American Paradox’. Alcohol Alcohol. 1998;33:195–201. doi: 10.1093/oxfordjournals.alcalc.a008382. [DOI] [PubMed] [Google Scholar]
- Alonso M, Lepousez G, Sebastien W, Bardy C, Gabellec MM, Torquet N, Lledo PM. Activation of adult-born neurons facilitates learning and memory. Nat Neurosci. 2012;15:897–904. doi: 10.1038/nn.3108. [DOI] [PubMed] [Google Scholar]
- Balaszczuk V, Bender C, Pereno GL, Beltramino CA. Alcohol-induced neuronal death in central extended amygdala and pyriform cortex during the postnatal period of the rat. Int J Dev Neurosci. 2011;29:733–742. doi: 10.1016/j.ijdevneu.2011.05.011. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Suzuki N. Neurons and circuits for odor processing in the piriform cortex. Trends Neurosci. 2013;36:429–438. doi: 10.1016/j.tins.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Belnoue L, Grosjean N, Ladeveze E, Abrous DN, Koehl M. Prenatal stress inhibits hippocampal neurogenesis but spares olfactory bulb neurogenesis. PloS one. 2013;8:e72972. doi: 10.1371/journal.pone.0072972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best AR, Wilson DA. Coordinate synaptic mechanisms contributing to olfactory cortical adaptation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:652–660. doi: 10.1523/JNEUROSCI.4220-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonthius DJ, West JR. Acute and long-term neuronal deficits in the rat olfactory bulb following alcohol exposure during the brain growth spurt. Neurotoxicology and teratology. 1991;13:611–619. doi: 10.1016/0892-0362(91)90044-w. [DOI] [PubMed] [Google Scholar]
- Brown J, Cooper-Kuhn CM, Kempermann G, Van Praag H, Winkler J, Gage FH, Kuhn HG. Enriched environment and physical activity stimulate hippocampal but not olfactory bulb neurogenesis. The European journal of neuroscience. 2003;17:2042–2046. doi: 10.1046/j.1460-9568.2003.02647.x. [DOI] [PubMed] [Google Scholar]
- Brunjes PC. Unilateral naris closure and olfactory system development. Brain research Brain research reviews. 1994;19:146–160. doi: 10.1016/0165-0173(94)90007-8. [DOI] [PubMed] [Google Scholar]
- Burd L, Blair J, Dropps K. Prenatal alcohol exposure, blood alcohol concentrations and alcohol elimination rates for the mother, fetus and newborn. J Perinatol. 2012;32:652–659. doi: 10.1038/jp.2012.57. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926–1929. doi: 10.1126/science.1099745. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Kaila K, Raichle M. Inhibition and brain work. Neuron. 2007;56:771–783. doi: 10.1016/j.neuron.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin JA, Carlen M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty G, Saito M, Mao RF, Wang R, Vadasz C. Lithium blocks ethanol-induced modulation of protein kinases in the developing brain. Biochem Biophys Res Commun. 2008;367:597–602. doi: 10.1016/j.bbrc.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy B, Finlay BL, Darlington RB, Anand KJ. Extrapolating brain development from experimental species to humans. Neurotoxicology. 2007;28:931–937. doi: 10.1016/j.neuro.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman LG, Jr, Oguz I, Lee J, Styner M, Crews FT. Postnatal day 7 ethanol treatment causes persistent reductions in adult mouse brain volume and cortical neurons with sex specific effects on neurogenesis. Alcohol. 2012;46:603–612. doi: 10.1016/j.alcohol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgin LL, Denninger T, Fyhn M, Hafting T, Bonnevie T, Jensen O, Moser MB, Moser EI. Frequency of gamma oscillations routes flow of information in the hippocampus. Nature. 2009;462:353–357. doi: 10.1038/nature08573. [DOI] [PubMed] [Google Scholar]
- Cudd TA. Animal model systems for the study of alcohol teratology. Exp Biol Med (Maywood) 2005;230:389–393. doi: 10.1177/15353702-0323006-06. [DOI] [PubMed] [Google Scholar]
- De Giorgio A, Comparini SE, Intra FS, Granato A. Long-term alterations of striatal parvalbumin interneurons in a rat model of early exposure to alcohol. J Neurodev Disord. 2012;4:18. doi: 10.1186/1866-1955-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci. 2010;11:339–350. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- Dunty WC, Jr, Chen SY, Zucker RM, Dehart DB, Sulik KK. Selective vulnerability of embryonic cell populations to ethanol-induced apoptosis: implications for alcohol-related birth defects and neurodevelopmental disorder. Alcoholism, clinical and experimental research. 2001;25:1523–1535. [PubMed] [Google Scholar]
- Ehlers CL, Oguz I, Budin F, Wills DN, Crews FT. Peri-adolescent ethanol vapor exposure produces reductions in hippocampal volume that are correlated with deficits in prepulse inhibition of the startle. Alcoholism, clinical and experimental research. 2013;37:1466–1475. doi: 10.1111/acer.12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks KM, Isaacson JS. Strong single-fiber sensory inputs to olfactory cortex: implications for olfactory coding. Neuron. 2006;49:357–363. doi: 10.1016/j.neuron.2005.12.026. [DOI] [PubMed] [Google Scholar]
- Freeman JH, Jr, Barone S, Jr, Stanton ME. Cognitive and neuroanatomical effects of triethyltin in developing rats: role of age of exposure. Brain Res. 1994;634:85–95. doi: 10.1016/0006-8993(94)90261-5. [DOI] [PubMed] [Google Scholar]
- Galindo R, Zamudio PA, Valenzuela CF. Alcohol is a potent stimulant of immature neuronal networks: implications for fetal alcohol spectrum disorder. Journal of neurochemistry. 2005;94:1500–1511. doi: 10.1111/j.1471-4159.2005.03294.x. [DOI] [PubMed] [Google Scholar]
- Gohlke JM, Griffith WC, Faustman EM. A systems-based computational model for dose-response comparisons of two mode of action hypotheses for ethanol-induced neurodevelopmental toxicity. Toxicol Sci. 2005;86:470–484. doi: 10.1093/toxsci/kfi209. [DOI] [PubMed] [Google Scholar]
- Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ. Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci. 1999;2:260–265. doi: 10.1038/6365. [DOI] [PubMed] [Google Scholar]
- Greicius M. Resting-state functional connectivity in neuropsychiatric disorders. Curr Opin Neurol. 2008;21:424–430. doi: 10.1097/WCO.0b013e328306f2c5. [DOI] [PubMed] [Google Scholar]
- Harris KR, Bucens IK. Prevalence of fetal alcohol syndrome in the Top End of the Northern Territory. Journal of paediatrics and child health. 2003;39:528–533. doi: 10.1046/j.1440-1754.2003.00208.x. [DOI] [PubMed] [Google Scholar]
- Hensch TK. Critical period mechanisms in developing visual cortex. Current topics in developmental biology. 2005;69:215–237. doi: 10.1016/S0070-2153(05)69008-4. [DOI] [PubMed] [Google Scholar]
- Hoekzema E, Carmona S, Ramos-Quiroga JA, Richarte Fernandez V, Bosch R, Soliva JC, Rovira M, Bulbena A, Tobena A, Casas M, Vilarroya O. An independent components and functional connectivity analysis of resting state fMRI data points to neural network dysregulation in adult ADHD. Human brain mapping. 2013 doi: 10.1002/hbm.22250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Isayama RN, Leite PE, Lima JP, Uziel D, Yamasaki EN. Impact of ethanol on the developing GABAergic system. Anat Rec (Hoboken) 2009;292:1922–1939. doi: 10.1002/ar.20966. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Kitabayashi R, Funatsu M, Izumi M, Yuede C, Hartman RE, Wozniak DF, Zorumski CF. A single day of ethanol exposure during development has persistent effects on bi-directional plasticity, N-methyl-D-aspartate receptor function and ethanol sensitivity. Neuroscience. 2005;136:269–279. doi: 10.1016/j.neuroscience.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Ketchum KL, Haberly LB. Membrane currents evoked by afferent fiber stimulation in rat piriform cortex. I. Current source-density analysis. Journal of neurophysiology. 1993;69:248–260. doi: 10.1152/jn.1993.69.1.248. [DOI] [PubMed] [Google Scholar]
- Kim CK, Kalynchuk LE, Kornecook TJ, Mumby DG, Dadgar NA, Pinel JP, Weinberg J. Object-recognition and spatial learning and memory in rats prenatally exposed to ethanol. Behavioral neuroscience. 1997;111:985–995. doi: 10.1037//0735-7044.111.5.985. [DOI] [PubMed] [Google Scholar]
- Klintsova AY, Helfer JL, Calizo LH, Dong WK, Goodlett CR, Greenough WT. Persistent impairment of hippocampal neurogenesis in young adult rats following early postnatal alcohol exposure. Alcoholism, clinical and experimental research. 2007;31:2073–2082. doi: 10.1111/j.1530-0277.2007.00528.x. [DOI] [PubMed] [Google Scholar]
- Komitova M, Xenos D, Salmaso N, May Tran K, Brand T, Schwartz ML, Ment L, Vaccarino FM. Hypoxia-induced developmental delays of inhibitory interneurons are reversed by environmental enrichment in the postnatal mouse forebrain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:13375–13387. doi: 10.1523/JNEUROSCI.5286-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leasure JL, Nixon K. Exercise neuroprotection in a rat model of binge alcohol consumption. Alcoholism, clinical and experimental research. 2010;34:404–414. doi: 10.1111/j.1530-0277.2009.01105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuner B, Gould E, Shors TJ. Is there a link between adult neurogenesis and learning? Hippocampus. 2006;16:216–224. doi: 10.1002/hipo.20153. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Alonso M, Grubb MS. Adult neurogenesis and functional plasticity in neuronal circuits. Nat Rev Neurosci. 2006;7:179–193. doi: 10.1038/nrn1867. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Saghatelyan A. Integrating new neurons into the adult olfactory bulb: joining the network, life-death decisions, and the effects of sensory experience. Trends Neurosci. 2005;28:248–254. doi: 10.1016/j.tins.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Lowry CA, Kay LM. Chemical factors determine olfactory system beta oscillations in waking rats. Journal of neurophysiology. 2007;98:394–404. doi: 10.1152/jn.00124.2007. [DOI] [PubMed] [Google Scholar]
- Luine VN, Jacome LF, Maclusky NJ. Rapid enhancement of visual and place memory by estrogens in rats. Endocrinology. 2003;144:2836–2844. doi: 10.1210/en.2003-0004. [DOI] [PubMed] [Google Scholar]
- Macbeth AH, Gautreaux C, Luine VN. Pregnant rats show enhanced spatial memory, decreased anxiety, and altered levels of monoaminergic neurotransmitters. Brain Research. 2008;1241:136–147. doi: 10.1016/j.brainres.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SE, Chen WJ, Miller JA, West JR. Fetal alcohol exposure and temporal vulnerability regional differences in alcohol-induced microencephaly as a function of the timing of binge-like alcohol exposure during rat brain development. Alcoholism, clinical and experimental research. 1997;21:1418–1428. doi: 10.1111/j.1530-0277.1997.tb04471.x. [DOI] [PubMed] [Google Scholar]
- Mandairon N, Jourdan F, Didier A. Deprivation of sensory inputs to the olfactory bulb up-regulates cell death and proliferation in the subventricular zone of adult mice. Neuroscience. 2003;119:507–516. doi: 10.1016/s0306-4522(03)00172-6. [DOI] [PubMed] [Google Scholar]
- Martin C, Beshel J, Kay LM. An olfacto-hippocampal network is dynamically involved in odor-discrimination learning. Journal of neurophysiology. 2007;98:2196–2205. doi: 10.1152/jn.00524.2007. [DOI] [PubMed] [Google Scholar]
- Martin C, Ravel N. Beta and gamma oscillatory activities associated with olfactory memory tasks: different rhythms for different functional networks? Frontiers in behavioral neuroscience. 2014;8:218. doi: 10.3389/fnbeh.2014.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PA, Fiorentino D, Coriale G, Kalberg WO, Hoyme HE, Aragon AS, Buckley D, Stellavato C, Gossage JP, Robinson LK, Jones KL, Manning M, Ceccanti M. Prevalence of children with severe fetal alcohol spectrum disorders in communities near Rome, Italy: new estimated rates are higher than previous estimates. International journal of environmental research and public health. 2011;8:2331–2351. doi: 10.3390/ijerph8062331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PA, Gossage JP. Estimating the prevalence of fetal alcohol syndrome. A summary. Alcohol research & health : the journal of the National Institute on Alcohol Abuse and Alcoholism. 2001;25:159–167. [PMC free article] [PubMed] [Google Scholar]
- May PA, Gossage JP. Maternal risk factors for fetal alcohol spectrum disorders: not as simple as it might seem. Alcohol research & health : the journal of the National Institute on Alcohol Abuse and Alcoholism. 2011;34:15–26. [PMC free article] [PubMed] [Google Scholar]
- Miller SS, Spear NE. Olfactory learning in the rat immediately after birth: Unique salience of first odors. Dev Psychobiol. 2009;51:488–504. doi: 10.1002/dev.20388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JJ, Paiva M, Heaton MB. Effect of neonatal ethanol exposure on parvalbumin-expressing GABAergic neurons of the rat medial septum and cingulate cortex. Alcohol. 2000;21:49–57. doi: 10.1016/s0741-8329(99)00101-9. [DOI] [PubMed] [Google Scholar]
- Moriceau S, Sullivan RM. Unique neural circuitry for neonatal olfactory learning. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:1182–1189. doi: 10.1523/JNEUROSCI.4578-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neville KR, Haberly LB. The synaptic organization of the brain. Oxford; New York: Oxford University Press; 2004. [Google Scholar]
- Nixon K, Hughes PD, Amsel A, Leslie SW. NMDA receptor subunit expression following early postnatal exposure to ethanol. Brain Res Dev Brain Res. 2002;139:295–299. doi: 10.1016/s0165-3806(02)00515-1. [DOI] [PubMed] [Google Scholar]
- Nunes F, Ferreira-Rosa K, dos Pereira MS, Kubrusly RC, Manhaes AC, Abreu-Villaca Y, Filgueiras CC. Acute administration of vinpocetine, a phosphodiesterase type 1 inhibitor, ameliorates hyperactivity in a mice model of fetal alcohol spectrum disorder. Drug and alcohol dependence. 2011;119:81–87. doi: 10.1016/j.drugalcdep.2011.05.024. [DOI] [PubMed] [Google Scholar]
- O’Leary CM, Nassar N, Kurinczuk JJ, de Klerk N, Geelhoed E, Elliott EJ, Bower C. Prenatal alcohol exposure and risk of birth defects. Pediatrics. 2010;126:e843–850. doi: 10.1542/peds.2010-0256. [DOI] [PubMed] [Google Scholar]
- Olney JW, Tenkova T, Dikranian K, Qin YQ, Labruyere J, Ikonomidou C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res Dev Brain Res. 2002;133:115–126. doi: 10.1016/s0165-3806(02)00279-1. [DOI] [PubMed] [Google Scholar]
- Oyedele OO, Kramer B. Acute ethanol administration causes malformations but does not affect cranial morphometry in neonatal mice. Alcohol. 2008;42:21–27. doi: 10.1016/j.alcohol.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Parnell SE, O’Leary-Moore SK, Godin EA, Dehart DB, Johnson BW, Allan Johnson G, Styner MA, Sulik KK. Magnetic resonance microscopy defines ethanol-induced brain abnormalities in prenatal mice: effects of acute insult on gestational day 8. Alcoholism, clinical and experimental research. 2009;33:1001–1011. doi: 10.1111/j.1530-0277.2009.00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patneau DK, Stripling JS. Functional correlates of selective long-term potentiation in the olfactory cortex and olfactory bulb. Brain Res. 1992;585:219–228. doi: 10.1016/0006-8993(92)91210-6. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The Mouse Brain in Stereotaxic Coordinates. New York: Academic Press; 2008. [Google Scholar]
- Paxinos G, Halliday G, Watson C, Koutcherov Y, Wang HQ. Atlas of the developing mouse brain at E17.5, P0 and P6. Burlington, MA: Academic Press; 2007. [Google Scholar]
- Raineki C, Holman PJ, Debiec J, Bugg M, Beasley A, Sullivan RM. Functional emergence of the hippocampus in context fear learning in infant rats. Hippocampus. 2010;20:1037–1046. doi: 10.1002/hipo.20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochefort C, Gheusi G, Vincent JD, Lledo PM. Enriched odor exposure increases the number of newborn neurons in the adult olfactory bulb and improves odor memory. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22:2679–2689. doi: 10.1523/JNEUROSCI.22-07-02679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy JW. Ontogeny of context-specific latent inhibition of conditioned fear: implications for configural associations theory and hippocampal formation development. Dev Psychobiol. 1994;27:367–379. doi: 10.1002/dev.420270605. [DOI] [PubMed] [Google Scholar]
- Sadrian B, Subbanna S, Wilson DA, Basavarajappa BS, Saito M. Lithium prevents long-term neural and behavioral pathology induced by early alcohol exposure. Neuroscience. 2012;206:122–135. doi: 10.1016/j.neuroscience.2011.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadrian B, Wilson D, Saito M. Long-Lasting Neural Circuit Dysfunction Following Developmental Ethanol Exposure. Brain Sciences. 2013;3:704–727. doi: 10.3390/brainsci3020704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay A, Wilson DA, Hen R. Pattern separation: a common function for new neurons in hippocampus and olfactory bulb. Neuron. 2011;70:582–588. doi: 10.1016/j.neuron.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Chakraborty G, Mao RF, Paik SM, Vadasz C. Tau phosphorylation and cleavage in ethanol-induced neurodegeneration in the developing mouse brain. Neurochemical research. 2010;35:651–659. doi: 10.1007/s11064-009-0116-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Mao RF, Wang R, Vadasz C, Saito M. Effects of gangliosides on ethanol-induced neurodegeneration in the developing mouse brain. Alcoholism, clinical and experimental research. 2007;31:665–674. doi: 10.1111/j.1530-0277.2007.00351.x. [DOI] [PubMed] [Google Scholar]
- Samuels BA, Hen R. Neurogenesis and affective disorders. The European journal of neuroscience. 2011;33:1152–1159. doi: 10.1111/j.1460-9568.2011.07614.x. [DOI] [PubMed] [Google Scholar]
- Sanderson JL, Donald Partridge L, Valenzuela CF. Modulation of GABAergic and glutamatergic transmission by ethanol in the developing neocortex: an in vitro test of the excessive inhibition hypothesis of fetal alcohol spectrum disorder. Neuropharmacology. 2009;56:541–555. doi: 10.1016/j.neuropharm.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sari Y, Hammad LA, Saleh MM, Rebec GV, Mechref Y. Alteration of selective neurotransmitters in fetal brains of prenatally alcohol-treated C57BL/6 mice: quantitative analysis using liquid chromatography/tandem mass spectrometry. Int J Dev Neurosci. 2010;28:263–269. doi: 10.1016/j.ijdevneu.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma AA, Richard MB, Greer CA. Developmental dynamics of piriform cortex. Cereb Cortex. 2011;21:1231–1245. doi: 10.1093/cercor/bhq199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62:42–52. doi: 10.1016/j.neuron.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M, Greenberger V, Israeli M, Biegon A. A correlation between regional acetylcholinesterase activity in rat brain and performance in a spatial task. Behavioural brain research. 1988;30:215–219. doi: 10.1016/0166-4328(88)90150-7. [DOI] [PubMed] [Google Scholar]
- Selby L, Zhang C, Sun QQ. Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neuroscience letters. 2007;412:227–232. doi: 10.1016/j.neulet.2006.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seress L. Comparative anatomy of the hippocampal dentate gyrus in adult and developing rodents, non-human primates and humans. Prog Brain Res. 2007;163:23–41. doi: 10.1016/S0079-6123(07)63002-7. [DOI] [PubMed] [Google Scholar]
- Shirasaka T, Hashimoto E, Ukai W, Yoshinaga T, Ishii T, Tateno M, Saito T. Stem cell therapy: social recognition recovery in a FASD model. Translational psychiatry. 2012;2:e188. doi: 10.1038/tp.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP, Swartzwelder HS. Adolescent alcohol exposure and persistence of adolescent-typical phenotypes into adulthood: A mini-review. Neuroscience and biobehavioral reviews. 2014;45C:1–8. doi: 10.1016/j.neubiorev.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Bekkers JM. Microcircuits mediating feedforward and feedback synaptic inhibition in the piriform cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:919–931. doi: 10.1523/JNEUROSCI.4112-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Liao Y, Song M, Gao JH, Zhou B, Tan C, Liu T, Tang Y, Chen J, Chen X. Aberrant default mode functional connectivity in early onset schizophrenia. PloS one. 2013;8:e71061. doi: 10.1371/journal.pone.0071061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TD, Kelly SJ. Critical periods for ethanol-induced cell loss in the hippocampal formation. Neurotoxicology and teratology. 2003;25:519–528. doi: 10.1016/s0892-0362(03)00074-6. [DOI] [PubMed] [Google Scholar]
- Tukker JJ, Fuentealba P, Hartwich K, Somogyi P, Klausberger T. Cell type-specific tuning of hippocampal interneuron firing during gamma oscillations in vivo. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:8184–8189. doi: 10.1523/JNEUROSCI.1685-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DA, Buhl EH, Hailer NP, Nitsch R. Morphological features of the entorhinal-hippocampal connection. Progress in neurobiology. 1998;55:537–562. doi: 10.1016/s0301-0082(98)00019-7. [DOI] [PubMed] [Google Scholar]
- Uecker A, Nadel L. Spatial but not object memory impairments in children with fetal alcohol syndrome. American journal of mental retardation : AJMR. 1998;103:12–18. doi: 10.1352/0895-8017(1998)103<0012:SBNOMI>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- van Groen T, Wyss JM. Extrinsic projections from area CA1 of the rat hippocampus: olfactory, cortical, subcortical, and bilateral hippocampal formation projections. J Comp Neurol. 1990;302:515–528. doi: 10.1002/cne.903020308. [DOI] [PubMed] [Google Scholar]
- Viljoen DL, Gossage JP, Brooke L, Adnams CM, Jones KL, Robinson LK, Hoyme HE, Snell C, Khaole NC, Kodituwakku P, Asante KO, Findlay R, Quinton B, Marais AS, Kalberg WO, May PA. Fetal alcohol syndrome epidemiology in a South African community: a second study of a very high prevalence area. Journal of studies on alcohol. 2005;66:593–604. doi: 10.15288/jsa.2005.66.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk DW, Lewis DA. Prenatal ontogeny as a susceptibility period for cortical GABA neuron disturbances in schizophrenia. Neuroscience. 2013;248C:154–164. doi: 10.1016/j.neuroscience.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Dubois DW, Tobery AN, Griffith WH, Brandt P, Frye GD. Long-lasting distortion of GABA signaling in MS/DB neurons after binge-like ethanol exposure during initial synaptogenesis. Brain Res. 2013;1520:36–50. doi: 10.1016/j.brainres.2013.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster WS, Walsh DA, McEwen SE, Lipson AH. Some teratogenic properties of ethanol and acetaldehyde in C57BL/6J mice: implications for the study of the fetal alcohol syndrome. Teratology. 1983;27:231–243. doi: 10.1002/tera.1420270211. [DOI] [PubMed] [Google Scholar]
- Wesson DW, Borkowski AH, Landreth GE, Nixon RA, Levy E, Wilson DA. Sensory network dysfunction, behavioral impairments, and their reversibility in an Alzheimer’s beta-amyloidosis mouse model. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:15962–15971. doi: 10.1523/JNEUROSCI.2085-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield-Gabrieli S, Ford JM. Default mode network activity and connectivity in psychopathology. Annu Rev Clin Psychol. 2012;8:49–76. doi: 10.1146/annurev-clinpsy-032511-143049. [DOI] [PubMed] [Google Scholar]
- Wilson DA, Peterson J, Basavaraj BS, Saito M. Local and regional network function in behaviorally relevant cortical circuits of adult mice following postnatal alcohol exposure. Alcoholism, clinical and experimental research. 2011;35:1974–1984. doi: 10.1111/j.1530-0277.2011.01549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman AD, Charvet CJ, Clancy B, Darlington RB, Finlay BL. Modeling transformations of neurodevelopmental sequences across mammalian species. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:7368–7383. doi: 10.1523/JNEUROSCI.5746-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, Muglia LJ. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol Dis. 2004;17:403–414. doi: 10.1016/j.nbd.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Young C, Olney JW. Neuroapoptosis in the infant mouse brain triggered by a transient small increase in blood alcohol concentration. Neurobiology of disease. 2006;22:548–554. doi: 10.1016/j.nbd.2005.12.015. [DOI] [PubMed] [Google Scholar]