

Abstract
Primary ovarian insufficiency is one of the main causes of female infertility owing to an abnormal ovarian reserve. Its relevance has increased in more recent years due to the fact that age of motherhood is being delayed in developed countries, with the risk of having either primary ovarian insufficiency or less chances of pregnancy when women consider the option of having their first baby. Several exogenous factors can lead to this event, such us viral infections, metabolomic dysfunction, autoimmune diseases, and environmental or iatrogenic factors, although in most cases the mechanism that leads to the disorder is unknown. Genetic factors represent the most commonly identified cause and the impact of sex chromosome abnormalities (e.g., Turner syndrome or X structural abnormalities), autosomal and X-linked mutations on the genesis of primary ovarian insufficiency has also been well described. Yet in most cases, the genetic origin remains unknown and there are multiple candidate genes. This review aims to collect all the genetic abnormalities and genes associated with syndromic and non syndromic primary ovarian insufficiency that have been published in the literature to date using the candidate-gene approach and a genome-wide analysis.
Keywords: Primary ovarian insufficiency, Ovarian reserve, Genetic factors, Sex chromosome abnormalities, Candidate genes
Introduction
Primary ovarian insufficiency (POI) is a condition caused by the absence, non functionality or early depletion of the ovarian reserve that leads to infertility, instead of the gradual process of follicular atresia, typical of fertile women, until the menopause is reached. It is characterized by primary or secondary amenorrhea for at least 4 months in women under the age of 40, menopausal serum FSH levels (above 40 IU/L) obtained on two occasions at least 1 month apart and low estradiol levels (below 50 pg/mL) [1]. Another indicator is the anti-Müllerian hormone (AMH), whose serum levels can help assess the state of follicular senescence, which is a possible predictor of risk for POI [2]. However, there are presently no single screening tests that can predict a woman’s reproductive lifespan [3]. Given the existing continuum of impaired ovarian function observed in this event, the term POI is more accurate than the term premature ovarian failure (POF), which has been classically used in the literature, because more often than not, there is no specific endpoint [4]. In fact, the new terminology allows us to include the following events in the definition of POI: hypergonadotropic hypogonadism, premature ovarian failure and ovarian dysgenesis.
This condition affects one in every 10,000 women before the age of 20, one in every 1,000 women before 30, and one in 100 women before 40 [5], which makes it one of the leading causes of female infertility.
Due to lowering levels of esteroid hormones, secondary to loss of ovarian function, symptoms may appear, such as hot flashes, sleep disturbances, decreased mental concentration, and loss of sexual desire or vaginal dryness. The long-term consequences of hypoestrogenism can include being at higher risk for osteoporosis or cardiovascular disease. This is the reason why hormone replacement therapy, with estrogen and progestin, is usually given. Starting it as soon as possible is necessary to avoid problems associated with estrogen deficiency, in addition to leading a healthy lifestyle that optimizes bone and cardiovascular health. However, this therapy should be strictly controlled and individualized since it is associated with development of breast cancer and numerous side effects [6]. Besides medical treatment, professional and family support is essential as POI can also adversely affect emotional health, provoke stress, loss of self-esteem, social isolation, increased shyness and anxiety.
Potential causes for POI are iatrogenic (ovarian surgery, radiotherapy or chemotherapy), environmental factors, viral infections, metabolic and autoimmune diseases, and genetic alterations, although its origin is idiopathic, and probably genetic, in most cases. In the latter cases, POI usually appears sporadically, but there can also be a family history in 10–15 % of cases [7]. Since POI occurs without warning symptoms in many cases, any woman with an affected relative should seek family planning counseling as pedigree studies suggest autosomal dominant sex-limited transmission or X-linked inheritance with incomplete penetrance [8]. In some patients, POI appears to be associated with another syndrome, syndromic POI, whose responsible genetic mutation is usually identified, but in other patients it appears to be isolated (non syndromic POI) and its cause is not usually identified. Multiple candidate genes have been described to explain the onset of POI since its function is related to reproduction or given its effect on animal models, and its mutation has also been detected in patients.
Given the importance of genetic factors and the large number of genes described in the literature at the present-date, the purpose of this review is to collect, as completely as possible, all the genetic mutations associated with syndromic and non syndromic POI, including sex chromosome abnormalities and mutations in the genes located on the X chromosome and autosomes.
Sex chromosome abnormalities
Aneuploidies
X chromosome abnormalities, namely aneuploidies and rearrangements, represent about 13 % of POI cases [9], which makes them one of the commonest genetic causes. Amongst them we highlight Turner syndrome or monosomy X (45,X), in which most women present gonadal dysgenesis with primary amenorrhea, loss of ovarian reserve before puberty, as oocytes need two active X chromosomes. Approximately 10 % of them reach menarche, a lower percentage than in the cases of 45,X/46,XX mosaicism, 40 % of whom can menstruate for several years before developing complete POI [4]. One candidate gene for gonadal dysgenesis in these patients is USP9X (Ubiquitin-Specific Protease 9), which escapes the X inactivation process and is located on chromosome Xp11.4, a critical region for ovarian development [10]; see Table 1. Other candidate genes include ZFX (Zinc Finger Protein, X-linked) and BMP15 (Bone Morphogenetic Protein 15), which will be described later. Spontaneous pregnancies can occur in 1.8 to 7.6 % of cases, with live birth rates of between 3 and 40 %. Pregnancy rates are higher following oocyte donation (between 45 and 60 %), but around half of them end in miscarriage [11], due mainly to uterine abnormalities, such as hypoplasic uterus [12]. The gestational risks in these patients lie in cardiovascular complications, mainly pre-eclampsia, aorta dissection and ventricular deficiency [13]. Maternal death from aortic dissection in pregnancies of women with Turner syndrome is estimated to be 2 %. Since possibility of pregnancy has increased significantly through the use of oocyte donation, physicians should perform very thorough previous preconception screening to estimate the patient’s cardiovascular risk during pregnancy in order to avoid the dreaded risk of sudden death [14].
Table 1.
Genes in sex chromosomes associated with gonadal dysgenesis in Turner and Swyer syndromes. See Table 2 for ZFX and BMP15 genes. Superscripts correspond to the literature reference relating each gene with POI
| Gene | Locus | Genetic disorder | Other diseases associated |
|---|---|---|---|
| USP9X [10] | Xp11.4 | Turner syndrome | Dermatomycosis |
| SRY [23] | Yp11.31 | Swyer syndrome | Gonadal dysgenesis, hermaphroditism |
| NR5A1 [23] | 9q33 | Swyer syndrome | Acute adrenal insufficiency, 46, XX gonadal dysgenesis |
| NR0B1 [24] | Xp21.2 | Swyer syndrome | X-linked adrenal hipoplasia congenita, hypogonadotropism |
| DHH [24] | 12q13.12 | Swyer syndrome | 46,XY partial gonadal dysgenesis, with minifascicularneuropathy |
As for other aneuploidies, POI may be associated with triple X syndrome or trisomy X (47,XXX). Although it does not appear in all cases, its prevalence is higher than in controls, and 21 cases of these patients with POI have been reported until 2010 [15]. In a study with 52 Indian women with POI, it was observed that in 3.7 % of cases, these women also had triple X syndrome, and that the pregnancies occurring spontaneously before loss of ovarian reserve showed more complications in this syndrome, with a high probability of premature birth and fetal malformations [16]. Continuing with other aneuploidies, POI is not an essential characteristic of X chromosome tetrasomy (48,XXXX), although some cases have been linked, and this abnormality has been detected even in a 16-year-old girl without ovaries [17], Furthermore, POI and osteoporosis have been diagnosed in another 16-year-old girl with mosaic pentasomy X/tetrasomy X syndrome (48,XXXX/49,XXXXX) [18].
X structural abnormalities
Regarding structural abnormalities, the commonest is Xq isochromosome, whose patients usually have streak gonads and Turner stigmata [19]. Deletions and translocations are more frequent on the X chromosome than in autosomes in women with POI, especially those occurring on the long arm of the X chromosome. Specifically, they occur in what is known as the critical region on the long arm of the X chromosome (Xq13,2-q27), with two main regions with the greatest number of breakpoints: one is known as POF1 (Xq26-Xqter), where deletions are more frequent, and the other is POF2 (Xq13.3-Xq21,1), where translocations are more frequent, but the commonest mutations in women with POI are balanced X-autosome translocations, particularly in the region between Xq13 and Xq27 [20]. Candidate genes for non syndromic forms of POI affected by rearrangements on the long arm of the X chromosome are: CHM (Choroideremia (Rab Escort Protein 1)), DIAPH2 (Diaphanous- Related Formin 2), DACH2 (Dachshung homolog 2 (Drosophila)), POF1B (Premature Ovarian Failure, 1B) and XPNPEP2 (X-Prolyl Aminopeptidase (Aminopeptidase P) 2, Membrane-Bound)), although their role in ovarian function remains unclear [21]. Another gene affected by translocation t(X;15)(Xq22;p11), found in a 19-year-old girl with primary amenorrhea, is NXF5 (Nuclear RNA Export Factor 5), which is responsible for the nuclear export of mRNA and shows functional homology with the FMR1 gene, a candidate gene for POI [22]. See Table 2 for further information on the locus of these genes and the associated diseases according to Genecards, in addition to POI.
Table 2.
Genes on the X chromosome associated with POI. Superscripts correspond to the literature reference relating each gene with POI
| Gene | Locus | Other diseases associated |
|---|---|---|
| CHM [21] | Xq21.1-q21.3 | Choroideremia |
| DIAPH2 [21] | Xq21.33 | − |
| DACH2 [21] | Xq21.3 | Allan-Herndon-Dudley syndrome |
| POF1B [21] | Xq21.2 | − |
| XPNPEP2 [21] | Xq25 | Angioedema |
| NXF5 [22] | Xq22.1 | Pelizaeus Merzbache disease, intelectual disability |
| FMR1 [26] | Xq27.3 | X fragile syndrome |
| FMR2 [35] | Xq28 | XE fragile syndrome, intellectual disability |
| XIST [10] | Xq13.2 | X inactivation, familial skewed, testicular cancer |
| CENP1 [20] | Xq22.1 | Scleroderma, liver disease |
| PGRMC1 [36] | Xq22-q24 | Placental choriocarcinoma, sphenoid sinusitis |
| AR [37] | Xq12 | Androgenin sensivity syndrome |
| FOXO4 [38] | Xq13.1 | Malignant spindle cell melanoma, X-linked dystonia-parkinsonism |
| AGTR2 [19] | Xq22-23 | Cerebrovascular accident, Conn’s syndrome |
| BHLHB9 [3] | Xq22.1 | Colon cancer, Alzheimer’s disease |
| BMP15 [39] | Xp11.2 | Turner syndrome |
| ZFX [10] | Xp22.1-21.3 | Retinoschisis, Turner syndrome |
| SHOX [40] | Xp22.33 | Leri weill dyschondrosteosis, Turner syndrome |
Rearrangements on the short arm of the X chromosome are also relevant, especially in the critical region located between Xp11.1 and Xp21, because approximately half of women with 46,X,del(Xp)(p11) display primary amenorrhea and gonadal dysgenesis, and some women also present deletions between Xp21.1 and Xp22.1.22 and POI [10].
See Fig. 1 for a diagram of the X-chromosome showing its relative size and banding pattern to help locate the genes and chromosomal regions associated with POI.
Fig. 1.
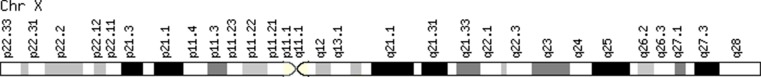
Diagram of the X-chromosome. Data extracted from Genecards
Other sex chromosome abnormalities
Another case of gonadal dysgenesis, which should be included in this review, is Swyer syndrome (46,XY) or 46,XY pure gonadal dysgenesis. Affected individuals are born with non functioning ovaries (bilateral streak gonads), and consequently they have primary amenorrhea. Ovarectomies are usually practiced to avoid ovarian cancer as these patients are more predisposed. The main gene candidates for gonadal dysgenesis in this syndrome are: SRY (Sex-Determining Region Y), located in the distal region of the short arm of the Y chromosome (Yp11.3), responsible for initiating male sex determination; NR5A1 (Nuclear receptor Subfamily 5, Group A, Member 1), located on chromosome 9, another candidate for gonadal dysgenesis cases in patients with karyotype 46,XX [23]; NR0B1 (Nuclear Receptor Subfamily 0, Group B, Member 1), located on the X chromosome, which may also influence embryo development; and DHH (Desert Hedgehog), located on chromosome 12, and essential for testes development; they are all involved in sex development disorders [24]. Using donor eggs, 15 successful pregnancies have been reported until 2013 [25]. Births in these patients often require caesarean section for various reasons, most of which appear to be related to an androgen shape of the pelvis.
See Table 1 for further information on the locus of the aforementioned gene candidates for gonadal dysgenesis in these syndromes and associated diseases according to Genecards, in addition to POI.
Mutations in genes on the X chromosome
Genes on the long arm of X chromosome
Besides the genes affected by the aforementioned rearrangements, other candidate genes located on the long arm of the X chromosome are FMR1, FMR2, XIST, CENPI, PGMRC1, AR, FOXO4, AGTR2 and BHLHB9.
The FMR1 (Fragile X Mental Retardation 1) gene, which encodes the fragile X mental retardation protein, is known to have an effect on POI. Whenever there is a mutation in which the CGG triplet (located at the FRAXXA site) is repeated more than 200 times, it produces an autosomal dominant genetic disorder called fragile X syndrome, characterized by mental retardation and other symptoms such as hyperactivity or attention and emotional problems. This is the most commonly inherited form of intellectual disability in boys. The premutation of this gene, which occurs when a number of triplet repeats falls between 55 and 200, represents the cause of POI in 3–15 % of the patients studied [26], which makes it another of the most well-known genetic causes of POI, especially in familiar cases. Moreover, the premutation range can also be manifested as the recently identified Fragile X-associated tremor ataxia syndrome (FXTAS), a late onset neurodegenerative disorder associated with problems with movement, memory and the autonomic nervous systems in male permutation carriers aged over 50 [27]. However, it has now been reported in female premutation carriers, so it is another reason to test women for this condition [28], [29]. Diminished cognitive flexibility among FXTAS women has been shown, which makes access to less typical category exemplar words more difficult [30].
There seems to be a synergistic effect of premutation and aging on cognitive impairment among older female fragile X premutation carriers, and even in those not presenting FXTAS symptoms. In any case, these deficits are relatively mild if compared to those in FXTAS males [31]. In 2013, a case of a 31-year-old patient with FMR1 premutation and a low ovarian reserve was published, in whom, as a result of the very low response of these patients to stimulation with exogenous gonadotropins, a decision was made to carry out a serial in vitro oocyte maturation (IVM) protocol. This case became the first successful pregnancy and birth in a patient with FMR1 premutation associated with a low ovarian reserve using this protocol [32]. Evidence shows that pregnancies in carriers of fragile X syndrome have no additional complications, save possible bleeding in later stages, so no special fetal monitoring is necessary [33].
In these patients, the preimplantation genetic diagnosis (PGD) technique is of special interest because having FMR1 premutation carries the risk of bearing a child with fragile X syndrome. In addition, PGD for fragile X syndrome is more difficult than for other genetic diseases because the couple needs to be informed, and women are more likely to have a low ovarian reserve, thus a poor response. However once the embryo transfer is possible, the results are comparable to other PGD cases for monogenic diseases [34].
Near the FMR1 gene, at the FRAXE locus, the FMR2 (Fragile Mental Retardation 2) gene is situated whose mutation in the form of deletions also causes mental retardation. It has been observed that the microdeletions in this gene are associated with 1.5 % of cases of POI, a higher percentage than for the remaining population [35].
The XIST (X-Inactive Specific Transcript) gene, responsible for the X- inactivation process, is located in the critical region on the X chromosome. Mutations in this gene are possible as a cause of POI because it is considered that anomalies in the X- inactivation process may affect the ovarian function [10].
The CENPI (Centromere Protein I) gene is also called FSHPRH1 (FSH Primary Response Protein 1). Given its role in the gonadal response to FSH during development, its mutation could affect ovary development and gametogenesis Hence it is another candidate gene [20].
The PGRMC1 (Progesterone Receptor Membrane Protein 1) gene encodes a progesterone-binding protein and is also considered a candidate for POI [36], since its expression alters in women with different forms of infertility. Therefore, it appears to play a role in regulating follicular development.
The AR (Androgen Receptor) gene has become a candidate after a longer length of CAG repeats was detected in women with POI than in controls [37].
The FOXO4 (Forkhead box O4) gene encodes a transcription factor that is involved in the regulation of the insulin signaling pathway, and it has also been considered a candidate, although one study conducted with Tunisian patients failed to find significant mutations [38].
The AGTR2 (Angiotensin II Receptor, Type 2) gene encodes an integral membrane protein that is highly expressed in the fetus and granulosa cells, and is associated with POI [19].
Finally, another gene which, according to the literature, is associated with POI is BHLHB9 (Basic Helix-Loop-Helix Domain Containing, Class B, 9) [3] which may play a role in controlling cellular aging and survival.
Genes on the short arm of X chromosome
Other candidate genes for non syndromic POI, which attract attention given their location in the critical region on the short arm of the X chromosome, are BMP15, ZFX and SHOX. The most important one is BMP15 (Bone Morphogenetic Protein 15) because different variants are associated with POI and with primary and secondary amenorrhea in 1.5–12 % of the cases studied, depending on the population [21]. It encodes bone morphogenetic protein 15, which stimulates folliculogenesis and is expressed in the oocyte. It belongs to the transforming growth factor-β (TGF-β) superfamily, a large family of proteins that plays a key role in regulating human ovarian functions [39].
The ZFX (Zinc Finger X) gene, homologous to ZFY, is a possible transcriptional activator, and the number of germ cells has been reported to lower in knockout mice [10].
Lastly, the SHOX (Short Stature Homeobox) gene, located in the pseudoautosomal region, is related to the short stature phenotype of Turner syndrome patients, and a cryptic duplication including this gene has been found in a patient with POI [40].
See Table 2 for further information on the locus of these genes and associated diseases according to Genecards, in addition to POI.
Mutations in autosomal genes
Genes associated with syndromic POI
Autosomal genes whose mutations cause some of the syndromes associated with the development of POI are: FSHR, GNAS, FOXL2, GALT, AIRE, STAR, CYP17A1, CYP19A1, eIF2B, NOG, ATM, POLG, PMM1, BMPR1B and GJA4.
The FSHR (Follicle Stimulating Hormone Receptor) gene encodes the FSH receptor, which is essential for follicular recruitment. Women who are homozygous for an antisense mutation in this gene are diagnosed with POI given the gonadotropin- resistant ovary syndrome, in which the follicular reserve remains, but is not functional. The possibility of gene therapy using adenovirus expressing the correct copy to restore fertility is under study, which has proven successful in mice [41]. In Finland, the mutation is a common cause of POI in 46,XX females, but this does not seem to apply to other countries like USA, Germany, Brazil or Mexico [10]. In 2013, the first live birth using IVM in a woman resistant to FSH and primary amenorrhea was reported [42]. Similarly, mutations in the LHR (Luteinizing Hormone Receptor) gene encoding the LH receptor, which is essential for ovulation, result in resistance to LH, be it rare, causing primary and secondary amenorrhea, a high serum LH and LH/FSH ratio and infertility, as seen in patients with male relatives affected by Leydig cell hypoplasia [43]. The mutations in the genes encoding the β subunit of FSH and LH are related to cases of primary and secondary amenorrhea, and with low and normal FSH levels, respectively, so this is not strictly considered to be POI, but secondary ovarian insufficiency [4].
The GNAS (Guanine Nucleotide Binding Protein (G Protein) Alpha Stimulating Activity Polypeptide 1) gene encodes a G protein involved in the hormonal regulation of adenylate cyclase, whose loss of sense mutation causes a resistance hormonal condition called pseudohypoparathyroidism type 1a. Since the FSH receptor is coupled to a G protein, POI is also produced by the same mechanism, which occurs in resistance to FSH [21].
The FOXL2 (Forkhead Box L2) gene encodes a transcription regulatory protein involved in ovary development and function. The mutations in this gene are associated with blepharophimosis-ptosis-epicanthus inversus syndrome (BPES), a genetic disease characterized by a dominantly inherited malformation of the eyelids, with four main ocular characteristics: blepharophimosis, ptosis, epicanthus inversus and telecanthus. This syndrome has two types: the first is also associated with POI, while the second does not affect fertility. Moreover, it has been found that the mutations in this gene can cause POI with no malformations of the eyelids [44]. Thus, it is another candidate gene for non syndromic POI. Recently, a pregnancy in a woman affected by ovarian insufficiency associated with BPES type 1 has been reported using ovarian stimulation with gonadotropins, which could help other patients [45].
The mutations in the GALT (Galactose-1-Phosphate Transferaseuridylyl) gene produce galactosemia, a rare genetic metabolic disorder characterized by the inability to metabolize galactose into glucose. Most women also are affected by POI because, as a result of this disease, biochemical damage to the ovary occurs. Surprisingly, 50 cases of spontaneous pregnancy in affected women have been reported until 2008, and some of them even had undetectable AMH levels [46]. The ovarian tissue cryopreservation option to preserve fertility in these patients has its limitations because their ovaries are often damaged early.
The AIRE (Autoimmune Regulator) gene encodes an autoimmune regulator protein expressed in thymic medulla. Its mutation causes a disease called autoimmune syndrome type 1 or autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), whose affected individuals present three conditions: Addison’s disease, hypoparathyroidism and mucocutaneous candidiasis. Of all the affected individuals, more than half have POI due to an autoimmune response to steroidogenic enzymes and steroid ovarian cells, which end up mediating ovary destruction [47]. Type 2 is also related to POI, but its genetic cause is unknown. In those cases in which follicles still remain, the syndrome can be treated and pregnancy can be achieved, not necessarily using assisted reproduction techniques, but immunotherapy treatment with corticosteroids.
The StAR (Steroidogenic Acute Regulatory) gene encodes a protein that regulates the synthesis of steroid hormones by increasing the conversion of cholesterol into pregnenolone. This process occurs in theca cells. The mutations in this gene give rise to lipoid congenital adrenal hyperplasia. Consequently, cholesterol accumulates and damages ovarian cells, resulting in POI [48].
The CYP17A1 (Cytochrome P450, family 17, subfamily A, Polypeptide 1) gene encodes an enzyme of the cytochrome P450 superfamily that converts pregnolona and progesterone into dehydroepiandrosterone and androstenedione. Its deficiency, which causes congenital adrenal hyperplasia due to 17-alpha hydroxylase deficiency, affects ovarian steroidogenesis and provokes primary amenorrhea because follicles are unable to grow sufficiently. However, oocyte maturation and fertilization can take place using assisted reproductive (IVF and in vitro steroidogenesis) techniques [49].
The CYP19A1 (Cytochrome P450, family 19, subfamily A, Polypeptide 1) gene encodes the enzyme aromatase, which is responsible for converting androgens into estrogens. Its mutation causes the aromatase deficiency syndrome, characterized by virilization and primary amenorrhea due to a maturation arrest of follicles. In addition, polymorphisms have also been detected in the epistasis between CYP19A1 and ESR1, whose interaction may be involved in circulating estrogen levels, and between FSHR and CYP19A1, whose interaction regulates estrogen production in Korean women with POI [50].
The mutations in eIF2B (Eukaryoticinitation factors 2B) genes, particularly in EIF2B2, EIF2B4 and EIF2B5, which catalyze the exchange of GDP bound to eIF2 for GTP, are associated with ovarioleukodistrophy (degeneration of brain white matter and POI), but they have not been detected in patients only with POI [51].
The NOG (Noggin) gene is expressed in the ovary, and its product, the noggin protein, is an antagonist to bone morphogenetic proteins (BMPs), which are important for the ovarian function. Its mutation in POI patients has been found on two occasions and has always been associated with proximal symphalangism, a disease characterized by the fusion of proximal interphalangeal joints caused by the haploinsufficiency of this gene. It has been proposed that the mutation increases the odds of POI in patients with genetic predisposition given the impaired function of BMPs [52].
The ATM (Ataxia Telangiectasia Mutated) gene encodes a protein kinases member that participates in cell cycle control and metabolism, and detects DNA damage. The mutations in this gene are the underlying cause of ataxia telangiectasia, a recessive disorder that causes degeneration of the brain which controls movement and speech, and in which infertility is a common feature. An ovarian hypoplasia with no mature oocytes has been observed in some patients. Besides, knockout mice have shown lack of follicles. Yet no confirmatory studies have been conducted on the relationship between POI and the ATM gene in other patients [21].
The POLG (Polymerase (DNA Directed) Gamma) gene encodes a mitochondrial DNA γ polymerase that is responsible for mitochondrial DNA replication. The mutations in this enzyme cause progressive external ophthalmoplegia, characterized by the development of ptosis and limited eye movements. Furthermore, it has been shown that POLG mitochondrial dysfunction also causes POI and its mutation cosegregates with parkinsonism [53].
The PMM1 (Phosphomannomutase 1) gene has been related to a metabolic abnormality called carbohydrate-deficient glycoprotein deficiency, which results from the accumulation of mannose-6-phospate and is associated with POI [54].
At the end of this list, a homozygous mutation that results in a new variant of the BMPR1B (Bone Morphogenetic Protein Receptor 1B) gene, causes a new type of acromesomelic chondrodysplasia, an inherited disease with skeletal abnormalities caused by mutations in GDF5 (Growth Differentiation Factor 5), further characterized by genital abnormalities and amenorrhea (Demirhan syndrome), while patients with only the mutations in GDF5 do not present these abnormalities. Since the GDF5 protein binds with a high affinity to BMPR1B receptor, it is believed that this gene also contributes to the development of the disease by playing a role in reproduction [55].
Perrault syndrome should be mentioned, defined by the association of gonadal dysgenesis in 46,XX patients to sensorineural hearing impairment. Its genetic cause is unknown, but the connexin family represents a group of candidate genes because they explain cases of congenital deafness. Furthermore, the GJA4 (connexin 3) gene is another candidate gene for POI since follicles stopped growing in knockout mice when they reached the antral stage [56].
Other syndromes that have been occasionally associated with POI in the literature, but are beyond the power of discussion of this review are Bloom syndrome, Werner syndrome, Rothmund-Thomson syndrome, Fanconi anemia, Nijmegen syndrome, Martsolf syndrome and Cockayne syndrome [10].
Genes associated with non syndromic POI
For cases in which POI occurs isolated and the cause is a mistery, there have been proposed many candidate genes for different reasons, including: INHA, GDF9, TGFBR3, NOBOX, NANOS3, FIGLα, FOXO1, FOXO3, ESR1, WT1, PTEN, CDKN1B, CITED2, SF1, WNT4, DMC1 and MSH5.
The INHA (Inhibin, alpha) gene encodes the inhibin α subunit, which is important for down-regulating the secretion of gonadotropins and belongs to the TGF-β superfamily, like the aforementioned BMP15. Based on its function, it is considered a strong candidate and its mutation was present in 7 % of 38 New Zealand patients, 10.5 % of 133 Indian patients and 4.5 % of 157 Italian patients [57]. Yet it is more likely to be a susceptibility factor to POI rather than the single genetic cause.
The GDF9 (Growth Differentiation Factor 9) gene also encodes a protein member of the TGF-β superfamily, and may act by forming a heterodimer with BMP15. GDF9 is expressed in oocytes, appears essential for folliculogenesis and its mutation has been detected significantly in Indian, Chinese and Caucasian women with POI. So it is another strong candidate [10].
The TGFBR3 (Transforming Growth Factor, Beta Receptor III) gene encodes transforming growth factor betatype III or β-glucan receptor, which binds to TGF-beta, and may be involved in its presentation to signaling receptors. Its mutation in the germline may cause loss of inhibin-mediated FSH regulation, and it has been detected in a significant number of Indian women with POI [58].
The NOBOX (NOBOX oogenesis homeobox) gene encodes a transcription factor that regulates oocyte-specific genes and is expressed until the metaphase II stage. Knockout mice do not perform oogenesis. In addition to Chinese and Caucasian, but not Japanese, women, the mutation has been detected in 6.2 % of 178 women with POI in a study from France [59].
The NANOS3 (Nanos homolog 3 (Drosophila)) gene encodes an RNA-binding protein, and knockout mice lack germ cells. Its mutation, which was already described, has been detected in 80 Chinese women and 88 Caucasian women with POI [10].
The FIGLα (Factor In the Germline alpha) gene is also important for oogenesis. It encodes an oocyte-specific transcription factor, which regulates the genes related to folliculogenesis and those responsible for zona pellucida formation. It has been associated with POI since no primordial follicles form in knockout mice, and two different mutations have been detected in two women with POI who had not been included in the 340 controls [60].
The FOXO1 (Forkhead box O1) and FOXO3 (Forkhead box O3) genes belong to the forkhead gene family, as does FOXL2. In knockout mice for FOXO3, follicles are activated prematurely, causing sterility. The mutations in FOXO1 and FOXO3 have been identified in one and two women with POI, respectively, of 90. However, it would be necessary to analyze more patients or to perform functional studies [61].
The ESR1 (Estrogen Receptor 1) gene, together with the HK3 (Hexokinase 3) and BRSK1 (BR Serine/threonine Kinase 1) genes, have been associated with POI and natural menopause after the detection of variants in Chinese, Korean and Dutch patients, but not in Serbs [62]. In addition, polymorphisms alone in the ESR1 gene appear to be a risk factor for POI in Chinese women, but further studies in other populations are required [63].
The WT1 (Wilms tumor 1) gene encodes a transcription factor expressed in granulosa cells. For this gene, knockout mice have shown a similar phenotype to POI, and its mutation has been seen to inhibit FSHR, aromatase and the 3β-HSD (3β- hydroxysteroid dehydrogenase) expression, leading to defects in granulosa cell polarity, which could explain the origin of POI [64].
The PTEN (Phosphatase And Tensin Homolog) gene encodes a tumor suppressor protein. Studies in mammals have suggested that it may be related to POI, and a point variation in exon 7 has been reported in some patients [65].
The CDKN1B (Cyclin-Dependent Kinase inhibitor 1B (P27, Kip1)) gene plays a key role during mouse ovarian development and a novel mutation has been potentially related to the phenotype since it has been found in patients from Tunisia [66].
The CITED2 (Cbp/p300-interacting transactivator, with Glu/Asp-rich carboxy- terminal domain, 2) gene causes anomalies in gonadal development in knockout mice and its mutations have been found in patients of Tunisian and Australian origin. Thus it could be involved in POI pathogenesis [67].
The SF1 (Splicing Factor 1) gene plays an important role in ovarian development and it seems to cause POI in Tunisian women by reducing estradiol levels [68].
The WNT4 (Wingless-type MMTV Integration Site Family, Member 4) gene plays an important role in female sex determination, differentiation, ovary maintenance and follicle survival, and variation has been detected in Han Chinese patients, although it did not change the amino acid sequence [69].
Finally, two other potential candidate genes involved in meiosis are DMC1 (DNA Meiotic Recombinase 1), whose mutation has been detected in an African woman with POI, and MSH5 (MutS Homolog 5), whose mutation was detected in two Caucasian women with POI. Hence these mutations might be an uncommon cause [70].
See Table 3 for further information on the locus of these genes and associated diseases according to Genecards, in addition to POI. Table 3 also shows other genes associated with POI (DAZL, EIF5B, YBX2) and the pathways of apoptosis in ovary or ovarian follicular dysfunction (BAX, SOHLH1) [3], which do not abound in the literature.
Table 3.
Autosomal genes associated with syndromic and non syndromic POI. Superscripts correspond to the literature reference relating each gene with POI
| Gene | Locus | Other diseases associated |
|---|---|---|
| FSHR [41, 10] | 2p21-p16 | Ovarian hyperstimulations yndrome, mucinous cystadenocarcinoma |
| LHR [43] | 2p21 | Precocious puberty, Leydig cells hypoplasia |
| FSHβ [4] | 11p13 | Follicle-stimulating hormone deficiency, isolated, pandas |
| LHβ [4] | 19q13.32 | Frozen shoulder, hypogonadism |
| GNAS [21] | 20q13.3 | Pseudohypoparathyroidism, Mccune-albrightsyndrome |
| FOXL2 [44] | 3q22-q23 | Blepharophimosis, blepharophimosis, ptosis and epicanthus inversus syndrome |
| GALT [46] | 9p13 | Galactosemia, galactokinasedeficiency |
| AIRE [47] | 21q22.3 | Candidiasis, autoinmunepolyendocrinesyndrome |
| StAR [48] | 8p11.2 | Lipoid adrenal hyperplasia |
| CYP17A1 [49] | 10q24.3 | 17-alpha-hydroxilase-deficient congenitaladrenalhyperplasia |
| CYP19A1 [71], [50] | 15q21.1 | Aromatasedeficiency, Leydig cell tumor |
|
EIF2B2 [51] EIF2B4 [51] EIF2B5 [51] |
14q24.3 2p23.3 3q27 |
Childhood ataxia with central nervous system hypomyelination/vanishing white matter, ovarioleukodystrophy |
| NOG [52] | 17q22 | Tarsal carpal coalition syndrome, stapes ankylosis with broad thumbs and toes |
| ATM [21] | 10q22.3 | Ataxia telangiectasia, chromosome 11q deletion |
| POLG [53] | 15q25 | Alperssyndrome, status epilepticus |
| PMM1 [54] | 22q13.2 | congenital disorder of glycosylation type 1A, anoxia |
| BMPR1B [55] | 4q22-q24 | Chrondrodisplasya, acromesomelic, with genital anomalies |
| GJA1 [56] | 1p34.3 | Erythrokeratodermia variabilis, myocardial infarction |
| INHA [57] | 2q33-q36 | Sertolicell tumor, multi-drugresistant tuberculosis |
| GDF9 [10] | 5q23.2 | Polycysticovarysyndrome, blepharophimosis |
| TGFBR3 [58] | 1p33-p32,1 | Eisenmengersyndrome, priapism |
| NOBOX [59] | 7q35 | Oophoritis |
| NANOS3 [10] | 19p13.13 | Seminoma |
| FIGLα [60] | 2p13.3 | − |
| FOXO1 [61] | 13q14.1 | Alveolarrhabdomyosarcoma |
| FOXO3 [61] | 6p21 | Rhabdomyosarcoma, acuteleukemia |
| ESR1 [62, 63] | 6p35 | Estrogen resistance syndrome, uterine disease |
| WT1 [64] | 11p13 | Wilms tumor, Denys-Drash syndrome |
| PTEN [65] | 10q23.31 | Endometrial carcinoma, Cowden disease |
| CDKN1B [66] | 12p13.1 | Mature T-cell and Nk-cell neoplasm, blasticplasmacytoid dendritic cell |
| CITED2 [67] | 6q24.1 | Single ventricular septaldefect, atrial septal defect sinusvenosus |
| SF1 [68] | 11q13.1 | Familial isolated pituitary adenoma, Wermer syndrome |
| WNT4 [69] | 1p36.12 | Eaf, Serkal syndrome |
| DMC1 [70] | 22q13.1 | Keratoderma, pterygium |
| MSH5 [70] | 6p21.3 | Common variable immunodeficiency, immnoglobulin alpha deficiency |
| DAZL [3] | 3p24.3 | Azoospermia, infertility |
| EIF5B [3] | 2q11.2 | Von Hippel-Lindaudisease, glucoseinterolerance |
| YBX2 [3] | 17p13.1 | Azoospermia, germ cell tumors |
| BAX [3] | 19q13.33 | Malignant teratoma, ovary adenocarcinoma |
| SOHLH1 [3] | 9q34.3 | Azoospermia |
Autosomal rearrangements
Rearrangements in women with POI are very rare in autosomes and only three cases have been detected: two translocations between chromosomes 2 and 15 in two women with karyotype 46,XX,t(2, 15)(q32.3,q13.3) [72]; one translocation between chromosomes 13 and 14 in a woman with karyotype 45,XX,t(13, 14) [73].
Novel candidate POI genes identified by genome-wide analysis
Apart from the candidate gene approach to date, another strategy that has identified new genes that might be associated with POI is the genome-wide analysis, which has two variants: genome-wide association studies (GWAS) and linkage analyses. GWAS analyses polymorphisms (SNPs or CNVs) in affected individuals, if compared with a control group, and has proposed new candidate genes through different studies (Table 4). Linkage analyses, based on following the inheritance of a genetic marker (SNPs or microsatellites) linked to a region of interest in related individuals, are difficult to carry out for POI because it is not easy to find a sufficient number of patients. In 2008, the first linkage analysis in POI patients was conducted in a Dutch family with seven affected women, which enabled the detection of a region of interest on chromosome 5q14.1-q15, where some genes (see Table 4) could play a role in ovarian failure [74]. Recently, other analyses using exome sequencing, in a family with nine affected women [75], and in a consanguineous Middle-Eastern family with five affected women [76, 77], have suggested new candidate genes (Table 4).
Table 4.
Novel candidate POI genes identified by the genome-wide approach. Superscripts correspond to the literature reference relating each gene with POI
| Gene | Initials | Locus |
|---|---|---|
| PTHB1 [81] | Parathiroid-Hormone Responsive B1 | 7p14 |
| BCKDHB [82] | Branched Chain Keto Acid Dehydrogenase E1, Beta Polypeptide | 6q14.1 |
| ACSL6 [83] | Acyl-CoA Synthetase Long-Chain Family Member 6 | 5q31 |
| HK3 [84] | Hexokinase 3 | 5q35.2 |
| BRSK1 [84] | BR Serine/Threonine Kinase 1 | 19q13.4 |
| ADAMTS19 [85] | ADAM Metallopeptidase With Thrombospondin Type 1 Motif, 19 | 5q23.3 |
| LAMC1 [86] | Laminin, Gamma 1 (Formerly LAMB2) | 1q31 |
| PCMT1 [87] | Protein-L-Isoaspartate (D-Aspartate) O-Methyltransferase | 6q24-q25 |
| DNAH5 [88] | Dynein, Axonemal, Heavy Chain 5 | 5p15.2 |
| NAIP [88] | NLR Family, Apoptosis Inhibitory Protein | 5q13.2 |
| DUSP22 [88] | Dual SpecificityPhosphatase 22 | 6p25.3 |
| NUPR1 [88] | Nuclear Protein, TranscriptionalRegulator, 1 | 16p11.2 |
| AKT1 [88] | V-Akt Murine Thymoma Viral Oncogene Homolog 1 | 14q32.32 |
| UTP14A [89] | U3 Small NucleolarRibonucleoprotein, Homolog A | Xq26.1 |
| PCDH19 [89] | Protocadherin 19 | Xq13.3 |
| VCX [89] | Variable Charge, X-Linked | Xp22 |
| STS [89] | Steroid Sulfatase (Microsomal), Isozyme S | Xp22.32 |
| BCORL1 [89] | BCL6 Corepressor-Like 1 | Xq26.1 |
| TSPAN7 [89] | Tetraspanin 7 | Xp11.4 |
| AIFM1 [89] | Apoptosis-Inducing Factor, Mitochondrion-Associated, 1 | Xq26.1 |
| SYCE1 [90, 91] | Synaptonemal Complex Central Element Protein 1 | 10q26.3 |
| CPEB1 [91] | Cytoplasmic Polyadenylation Element Binding Protein 1 | 15q25.2 |
| CCBE1 [91] | Collagen And Calcium Binding EGF Domains 1 | 18q21.32 |
| PMAIP1 [91] | Phorbol-12-Myristate-13-Acetate-Induced Protein 1 | 18q21.32 |
| CTNNA3 [91] | Catenin (Cadherin-Associated Protein), Alpha 3 | 10q21.3 |
| ANKRD22 [91] | AnkyrinRepeatDomain 22 | 10q23.31 |
| STAMBPL1 [91] | STAM BindingProtein-Like 1 | 10q23.31 |
| HSD3B2 [91] | Hydroxy-Delta-5-Steroid Dehydrogenase, 3-Betaand Steroid Delta-Isomerase2 | 1p12 |
| HAO2 [91] | Hydroxyacid Oxidase 2 (Long Chain) | 1p12 |
| CYP2E1 [90] | Cytochrome P450, Family 2, Subfamily E, Polypeptide 1 | 10q26.3 |
| NRXN1 [90] | Neurexin 1 | 2p16.3 |
| PARK2 [90] | Parkin RBR E3 UbiquitinProtein Ligase | 6q26 |
| CARD11 [90] | Caspase Recruitment Domain Family, Member 11 | 7p22.2 |
| DHFR [74] | DihydrofolateReductase | 5q14.1 |
| NR2F1 [74] | Nuclear Receptor Subfamily 2, Group F, Member 1 | 5q14 |
| MEF2C [74] | MADS Box Transcription Enhancer Factor 2, Polypeptide C | 5q14.3 |
| CCNH [74] | Cyclin H | 5q13.3-q14 |
| SSBP2 [74] | Single-Stranded DNA Binding Protein 2 | 5q14.1 |
| CSPG2 [74] | Chondroitin Sulfate Proteoglycan 2 | 5q14.2 |
| EIF4ENIF1 [75] | Eukaryotic Translation Initiation Factor 4E Nuclear Import Factor 1 | 22q12.2 |
| DLX5 [76] | Distal-LessHomeobox 5 | 7q21.3 |
| DLX6 [76] | Distal-Less Homeobox 6 | 7q21.3 |
| DSS1 [76] | Split Hand/Foot Malformation (Ectrodactyly) Type 1 | 7q21.3 |
| STAG3 [77] | Stromal Antigen 3 | 7q22.1 |
Moreover, recent studies relate the regulatory function of microRNAs (miRNAs) in oocyte maturation and folliculogenesis, and different miRNA expression profiles in the plasma of POI patients and fertile women have been detected when a miRNA microarray analysis was performed [78]. Similar results have been observed in an animal model in which a different profile for differentially expressed miRNAs has been observed between normal rats and 4-vinylcyclohexene diepoxide (VCD)-induced rat POF when miRNA microarrays were used [79]. A recent study performed in Korean women observed that the XPO5 rs2257082 T variant allele occurs more frequently in POI patients than in controls, suggesting that this allele may be associated with increased POI risk [80].
Conclusions
This review shows the weight and variability of genetic factors in the origin of POI, as ovarian function depends on the expression of multiple genes, and it includes nearly all the genetic abnormalities and genes associated with POI that have been detected by different techniques.
In short, the most commonly known genetic causes of POI are X chromosome abnormalities (13 % of cases) [9], especially Turner syndrome, and FMR1 premutations (3–15 % of cases) [26]. Then, when a woman is diagnosed with POI, in the absence of other more obvious causes, it should be carried out a cytogenetic analysis, although there is also an important immune factor that could be rule out with the detection of autoantibodies. Other possible genetic causes of POI isolated are mutations in FMR2 (1.5 % of cases) [35], PGRMC1 and BMP15 genes on the X chromosome (1.5–12 % of cases) [21], and NR5A1, FOXL2, INHA (4.5–10.5 % of New Zealand, Indian and Italian patients) [57], GDF9, NOBOX (5.2 % of French patients) [59], NANOS3 and FIGLα genes in autosomes, although their degree of association depends on the study population. Mutations in all the other genes mentioned in this review are population-specific, not common or rare causes. It should also be contemplated that the origin of POI development may not be due to a single mutation in a candidate gene, but an interaction of low-frequency polymorphisms or mutations in different genes in the same woman, together with the effect of the environment on the phenotype.
Among the genes associated with POI, only a few (FMR1, BMP15, GDF9) have been incorporated as diagnosis biomarkers [92], therefore more research is needed before using other genes as a routine tool. In order to automate the identification of specific genetic causes, sensitive arrays with all the candidate genes represented, or at least the most important ones, could be designed (a POI genetic diagnostic tool). Identifying the genetic causes and increasing the sensitivity of genetic screening would allow professionals to design tests to predict menopausal age in women at increased risk for POI, which is the aim of many groups [21].
In case of POI prediction, women should consider the option of preserving their fertility by means of assisted reproduction techniques with considerable results, such as oocyte cryopreservation [93]. For that purpose, healthcare physicians must be aware of this possibility of oocyte vitrification and advise patients so they have the chance to be mothers in the future by mainly taking into account that, nowadays, most women delay maternity, and when they decide to have a baby, POI is almost established.
Footnotes
Capsule
Genetic factors are the most commonly identified cause of Primary Ovarian Insufficiency. This review aims to collect all the genetic abnormalities and genes related with this condition.
References
- 1.Nelson LM. Clinical practice. Primary ovarian insufficiency. N Engl J Med. 2009;360:606–14. doi: 10.1056/NEJMcp0808697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Visser JA, Schipper I, Laven JS, Themmen AP. Anti-Mullerian hormone: an ovarian reserve marker in primary ovarian insufficiency. Nat Rev Endocrinol. 2012;8:331–41. doi: 10.1038/nrendo.2011.224. [DOI] [PubMed] [Google Scholar]
- 3.De Vos M, Devroey P, Fauser BC. Primary ovarian insufficiency. Lancet. 2010;376:911–21. doi: 10.1016/S0140-6736(10)60355-8. [DOI] [PubMed] [Google Scholar]
- 4.Welt CK. Primary ovarian insufficiency: a more accurate term for premature ovarian failure. Clin Endocrinol (Oxf) 2008;68:499–509. doi: 10.1111/j.1365-2265.2007.03073.x. [DOI] [PubMed] [Google Scholar]
- 5.Coulam CB, Adamson SC, Annegers JF. Incidence of premature ovarian failure. Obstet Gynecol. 1986;67:604–6. [PubMed] [Google Scholar]
- 6.Aubuchon M, Santoro N. Lessons learned from the WHI: HRT requires a cautious and individualized approach. Geriatrics. 2004;59:22–6. [PubMed] [Google Scholar]
- 7.van Kasteren YM, Hundscheid RD, Smits AP, Cremers FP, van Zonneveld P, Braat DD. Familial idiopathic premature ovarian failure: an overrated and underestimated genetic disease? Hum Reprod. 1999;14:2455–9. doi: 10.1093/humrep/14.10.2455. [DOI] [PubMed] [Google Scholar]
- 8.Beck-Peccoz P, Persani L. Premature ovarian failure. Orphanet J Rare Dis. 2006;1:9. doi: 10.1186/1750-1172-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cordts EB, Christofolini DM, Dos Santos AA, Bianco B, Barbosa CP. Genetic aspects of premature ovarian failure: a literature review. Arch Gynecol Obstet. 2011;283:635–43. doi: 10.1007/s00404-010-1815-4. [DOI] [PubMed] [Google Scholar]
- 10.Simpson JL. Genetic and phenotypic heterogeneity in ovarian failure: overview of selected candidate genes. Ann N Y Acad Sci. 2008;1135:146–54. doi: 10.1196/annals.1429.019. [DOI] [PubMed] [Google Scholar]
- 11.Chakhtoura Z, Touraine P. Fertility on women with Turner syndrome. Presse Med. 2013;42:1508–12. doi: 10.1016/j.lpm.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 12.Fenichel P, Brucker-Davis F. Environmental endocrine disruptors and breast cancer: new risk factors? Gynecol Obstet Fertil. 2008;36:969–77. doi: 10.1016/j.gyobfe.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Hagman A, Kallen K, Bryman I, Landin-Wilhelmsen K, Barrenas ML, Wennerholm UB. Morbidity and mortality after childbirth in women with Turner karyotype. Hum Reprod. 2013;28:1961–73. doi: 10.1093/humrep/det113. [DOI] [PubMed] [Google Scholar]
- 14.Fenichel P, Letur H. Procreation in Turner’s syndrome: which recommendations before, during and after pregnancy? Gynecol Obstet Fertil. 2008;36:891–7. doi: 10.1016/j.gyobfe.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 15.Otter M, Schrander-Stumpel CT, Curfs LM. Triple X syndrome: a review of the literature. Eur J Hum Genet. 2010;18:265–71. doi: 10.1038/ejhg.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goswami R, Goswami D, Kabra M, Gupta N, Dubey S, Dadhwal V. Prevalence of the triple X syndrome in phenotypically normal women with premature ovarian failure and its association with autoimmune thyroid disorders. Fertil Steril. 2003;80:1052–4. doi: 10.1016/S0015-0282(03)01121-X. [DOI] [PubMed] [Google Scholar]
- 17.Collen RJ, Falk RE, Lippe BM, Kaplan SA. A 48, XXXX female with absence of ovaries. Am J Med Genet. 1980;6:275–8. doi: 10.1002/ajmg.1320060404. [DOI] [PubMed] [Google Scholar]
- 18.Wood A, Kleis L, Toriello H, Cemeroglu AP. Mosaic pentasomy X/tetrasomy X syndrome and premature ovarian failure. Indian Pediatr. 2011;48:402–4. [PubMed] [Google Scholar]
- 19.Simpson JL. Gonadal dysgenesis and abnormalities of the human sex chromosomes: current status of phenotypic-karyotypic correlations. Birth Defects Orig Artic Ser. 1975;11:23–59. [PubMed] [Google Scholar]
- 20.Goswami D, Conway GS. Premature ovarian failure. Hum Reprod Update. 2005;11:391–410. doi: 10.1093/humupd/dmi012. [DOI] [PubMed] [Google Scholar]
- 21.Persani L, Rossetti R, Cacciatore C. Genes involved in human premature ovarian failure. J Mol Endocrinol. 2010;45:257–79. doi: 10.1677/JME-10-0070. [DOI] [PubMed] [Google Scholar]
- 22.Bertini V, Ghirri P, Bicocchi MP, Simi P, Valetto A. Molecular cytogenetic definition of a translocation t(X;15) associated with premature ovarian failure. Fertil Steril. 2010;94:1097.e5–e8. doi: 10.1016/j.fertnstert.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 23.Bashamboo A, Ravel C, Brauner R, McElreavey K. NR5A1 and ovarian failure. Med Sci (Paris) 2009;25:809–13. doi: 10.1051/medsci/20092510809. [DOI] [PubMed] [Google Scholar]
- 24.Paliwal P, Sharma A, Birla S, Kriplani A, Khadgawat R, Sharma A. Identification of novel SRY mutations and SF1 (NR5A1) changes in patients with pure gonadal dysgenesis and 46, XY karyotype. Mol Hum Reprod. 2011;17:372–8. doi: 10.1093/molehr/gar002. [DOI] [PubMed] [Google Scholar]
- 25.Polakova M, Alexander D, Sulc J, Zetova L, Vlk R, Krepelova A, et al. Pregnancy and delivery in a patient with pure 46, XY karyotype. Summary of actual knowledge about XY women. Ceska Gynekol. 2013;78:443–7. [PubMed] [Google Scholar]
- 26.Wittenberger MD, Hagerman RJ, Sherman SL, McConkie-Rosell A, Welt CK, Rebar RW, et al. The FMR1 premutation and reproduction. Fertil Steril. 2007;87:456–65. doi: 10.1016/j.fertnstert.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 27.Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–30. doi: 10.1212/WNL.57.1.127. [DOI] [PubMed] [Google Scholar]
- 28.Hagerman RJ, Leavitt BR, Farzin F, Jacquemont S, Greco CM, Brunberg JA, et al. Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am J Hum Genet. 2004;74:1051–6. doi: 10.1086/420700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Utari A, Adams E, Berry-Kravis E, Chavez A, Scaggs F, Ngotran L, et al. Aging in fragile X syndrome. J Neurodev Disord. 2010;2:70–6. doi: 10.1007/s11689-010-9047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang JC, Simon C, Schneider A, Seritan AL, Hamilton L, Hagerman PJ, et al. Abnormal semantic processing in females with fragile X-associated tremor/ataxia syndrome. Genes Brain Behav. 2014;13:152–62. doi: 10.1111/gbb.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang JC, Simon C, Niu YQ, Bogost M, Schneider A, Tassone F, et al. Phenotypes of hypofrontality in older female fragile X premutation carriers. Ann Neurol. 2013;74:275–83. doi: 10.1002/ana.23933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nayot D, Chung JT, Son WY, Ao A, Hughes M, Dahan MH. Live birth following serial vitrification of embryos and PGD for fragile X syndrome in a patient with the premutation and decreased ovarian reserve. J Assist Reprod Genet. 2013;30:1439–44. doi: 10.1007/s10815-013-0079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kallinen J, Korhonen K, Kortelainen S, Heinonen S, Ryynanen M. Pregnancy outcome in carriers of fragile X. BJOG. 2000;107:969–72. doi: 10.1111/j.1471-0528.2000.tb10398.x. [DOI] [PubMed] [Google Scholar]
- 34.Platteau P, Sermon K, Seneca S, Van Steirteghem A, Devroey P, Liebaers I. Preimplantation genetic diagnosis for fragile Xa syndrome: difficult but not impossible. Hum Reprod. 2002;17:2807–12. doi: 10.1093/humrep/17.11.2807. [DOI] [PubMed] [Google Scholar]
- 35.Murray A, Webb J, Dennis N, Conway G, Morton N. Microdeletions in FMR2 may be a significant cause of premature ovarian failure. J Med Genet. 1999;36:767–70. doi: 10.1136/jmg.36.10.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dixit H, Rao L, Padmalatha V, Raseswari T, Kapu AK, Panda B, et al. Genes governing premature ovarian failure. Reprod Biomed Online. 2010;20:724–40. doi: 10.1016/j.rbmo.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 37.Chatterjee S, Singh R, Kadam S, Maitra A, Thangaraj K, Meherji P, et al. Longer CAG repeat length in the androgen receptor gene is associated with premature ovarian failure. Hum Reprod. 2009;24:3230–5. doi: 10.1093/humrep/dep296. [DOI] [PubMed] [Google Scholar]
- 38.Fonseca DJ, Garzon E, Lakhal B, Braham R, Ojeda D, Elghezal H, et al. Screening for mutations of the FOXO4 gene in premature ovarian failure patients. Reprod Biomed Online. 2012;24:339–41. doi: 10.1016/j.rbmo.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 39.Peng C. The TGF-beta superfamily and its roles in the human ovary and placenta. J Obstet Gynaecol Can. 2003;25:834–44. doi: 10.1016/s1701-2163(16)30674-0. [DOI] [PubMed] [Google Scholar]
- 40.Tachdjian G, Aboura A, Portnoi MF, Pasquier M, Bourcigaux N, Simon T, et al. Cryptic Xp duplication including the SHOX gene in a woman with 46, X, del(X)(q21.31) and premature ovarian failure. Hum Reprod. 2008;23:222–6. doi: 10.1093/humrep/dem358. [DOI] [PubMed] [Google Scholar]
- 41.Ghadami M, El-Demerdash E, Salama SA, Binhazim AA, Archibong AE, Chen X, et al. Toward gene therapy of premature ovarian failure: intraovarian injection of adenovirus expressing human FSH receptor restores folliculogenesis in FSHR(−/−) FORKO mice. Mol Hum Reprod. 2010;16:241–50. doi: 10.1093/molehr/gaq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grynberg M, Peltoketo H, Christin-Maitre S, Poulain M, Bouchard P, Fanchin R. First birth achieved after in vitro maturation of oocytes from a woman endowed with multiple antral follicles unresponsive to follicle-stimulating hormone. J Clin Endocrinol Metab. 2013;98:4493–8. doi: 10.1210/jc.2013-1967. [DOI] [PubMed] [Google Scholar]
- 43.Arnhold IJ, Latronico AC, Batista MC, Izzo CR, Mendonca BB. Clinical features of women with resistance to luteinizing hormone. Clin Endocrinol (Oxf) 1999;51:701–7. doi: 10.1046/j.1365-2265.1999.00863.x. [DOI] [PubMed] [Google Scholar]
- 44.Harris SE, Chand AL, Winship IM, Gersak K, Aittomaki K, Shelling AN. Identification of novel mutations in FOXL2 associated with premature ovarian failure. Mol Hum Reprod. 2002;8:729–33. doi: 10.1093/molehr/8.8.729. [DOI] [PubMed] [Google Scholar]
- 45.Roth LW, Alvero R. Pregnancy in a woman with premature ovarian insufficiency associated with blepharophimosis, ptosis, epicanthus inversus syndrome type I. A case report. J Reprod Med. 2014;59:87–9. [PubMed] [Google Scholar]
- 46.Gubbels CS, Land JA, Rubio-Gozalbo ME. Fertility and impact of pregnancies on the mother and child in classic galactosemia. Obstet Gynecol Surv. 2008;63:334–43. doi: 10.1097/OGX.0b013e31816ff6c5. [DOI] [PubMed] [Google Scholar]
- 47.Fierabracci A, Bizzarri C, Palma A, Milillo A, Bellacchio E, Cappa M. A novel heterozygous mutation of the AIRE gene in a patient with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome (APECED) Gene. 2012;511:113–7. doi: 10.1016/j.gene.2012.09.029. [DOI] [PubMed] [Google Scholar]
- 48.Bhangoo A, Buyuk E, Oktay K, Ten S. Phenotypic features of 46, XX females with StAR protein mutations. Pediatr Endocrinol Rev. 2007;5:633–41. [PubMed] [Google Scholar]
- 49.Pellicer A, Miro F, Sampaio M, Gomez E, Bonilla-Musoles FM. In vitro fertilization as a diagnostic and therapeutic tool in a patient with partial 17,20-desmolase deficiency. Fertil Steril. 1991;55:970–5. doi: 10.1016/s0015-0282(16)54308-8. [DOI] [PubMed] [Google Scholar]
- 50.Kim S, Pyun JA, Cha DH, Ko JJ, Kwack K. Epistasis between FSHR and CYP19A1 polymorphisms is associated with premature ovarian failure. Fertil Steril. 2011;95:2585–8. doi: 10.1016/j.fertnstert.2010.12.042. [DOI] [PubMed] [Google Scholar]
- 51.Fogli A, Gauthier-Barichard F, Schiffmann R, Vanderhoof VH, Bakalov VK, Nelson LM, et al. Screening for known mutations in EIF2B genes in a large panel of patients with premature ovarian failure. BMC Womens Health. 2004;4:8. doi: 10.1186/1472-6874-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kosaki K, Sato S, Hasegawa T, Matsuo N, Suzuki T, Ogata T. Premature ovarian failure in a female with proximal symphalangism and Noggin mutation. Fertil Steril. 2004;81:1137–9. doi: 10.1016/j.fertnstert.2003.08.054. [DOI] [PubMed] [Google Scholar]
- 53.Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004;364:875–82. doi: 10.1016/S0140-6736(04)16983-3. [DOI] [PubMed] [Google Scholar]
- 54.Cox L, Liu JH. Primary ovarian insufficiency: an update. Int J Womens Health. 2014;6:235–43. doi: 10.2147/IJWH.S37636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Demirhan O, Turkmen S, Schwabe GC, Soyupak S, Akgul E, Tastemir D, et al. A homozygous BMPR1B mutation causes a new subtype of acromesomelic chondrodysplasia with genital anomalies. J Med Genet. 2005;42:314–7. doi: 10.1136/jmg.2004.023564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simon AM, Goodenough DA, Li E, Paul DL. Female infertility in mice lacking connexin 37. Nature. 1997;385:525–9. doi: 10.1038/385525a0. [DOI] [PubMed] [Google Scholar]
- 57.Chand AL, Harrison CA, Shelling AN. Inhibin and premature ovarian failure. Hum Reprod Update. 2010;16:39–50. doi: 10.1093/humupd/dmp031. [DOI] [PubMed] [Google Scholar]
- 58.Dixit H, Rao KL, Padmalatha VV, Kanakavalli M, Deenadayal M, Gupta N, et al. Mutational analysis of the betaglycan gene-coding region in susceptibility for ovarian failure. Hum Reprod. 2006;21:2041–6. doi: 10.1093/humrep/del107. [DOI] [PubMed] [Google Scholar]
- 59.Bouilly J, Bachelot A, Broutin I, Touraine P, Binart N. Novel NOBOX loss-of-function mutations account for 6.2 % of cases in a large primary ovarian insufficiency cohort. Hum Mutat. 2011;32:1108–13. doi: 10.1002/humu.21543. [DOI] [PubMed] [Google Scholar]
- 60.Zhao H, Chen ZJ, Qin Y, Shi Y, Wang S, Choi Y, et al. Transcription factor FIGLA is mutated in patients with premature ovarian failure. Am J Hum Genet. 2008;82:1342–8. doi: 10.1016/j.ajhg.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Watkins WJ, Umbers AJ, Woad KJ, Harris SE, Winship IM, Gersak K, et al. Mutational screening of FOXO3A and FOXO1A in women with premature ovarian failure. Fertil Steril. 2006;86:1518–21. doi: 10.1016/j.fertnstert.2006.03.054. [DOI] [PubMed] [Google Scholar]
- 62.Qin Y, Vujovic S, Li G, Li J, Dalgleish R, Simpson JL, et al. Ethnic specificity of variants of the ESR1, HK3, BRSK1 genes and the 8q22.3 locus: No association with premature ovarian failure (POF) in Serbian women. Maturitas. 2014;77:64–7. doi: 10.1016/j.maturitas.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 63.Liu L, Tan R, Cui Y, Liu J, Wu J. Estrogen receptor alpha gene (ESR1) polymorphisms associated with idiopathic premature ovarian failure in Chinese women. Gynecol Endocrinol. 2013;29:182–5. doi: 10.3109/09513590.2012.731113. [DOI] [PubMed] [Google Scholar]
- 64.Gao F, Zhang J, Wang X, Yang J, Chen D, Huff V, et al. Wt1 functions in ovarian follicle development by regulating granulosa cell differentiation. Hum Mol Genet. 2014;23:333–41. doi: 10.1093/hmg/ddt423. [DOI] [PubMed] [Google Scholar]
- 65.Shimizu Y, Kimura F, Takebayashi K, Fujiwara M, Takakura K, Takahashi K. Mutational analysis of the PTEN gene in women with premature ovarian failure. Acta Obstet Gynecol Scand. 2009;88:824–5. doi: 10.1080/00016340902971458. [DOI] [PubMed] [Google Scholar]
- 66.Ojeda D, Lakhal B, Fonseca DJ, Braham R, Landolsi H, Mateus HE, et al. Sequence analysis of the CDKN1B gene in patients with premature ovarian failure reveals a novel mutation potentially related to the phenotype. Fertil Steril. 2011;95:2658. doi: 10.1016/j.fertnstert.2011.04.045. [DOI] [PubMed] [Google Scholar]
- 67.Fonseca DJ, Ojeda D, Lakhal B, Braham R, Eggers S, Turbitt E, et al. CITED2 mutations potentially cause idiopathic premature ovarian failure. Transl Res. 2012;160:384–8. doi: 10.1016/j.trsl.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 68.Lakhal B, Ben-Hadj-Khalifa S, Bouali N, Braham R, Hatem E, Saad A. Mutational screening of SF1 and WNT4 in Tunisian women with premature ovarian failure. Gene. 2012;509:298–301. doi: 10.1016/j.gene.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 69.Chen B, Suo P, Wang B, Wang J, Yang L, Zhou S, et al. Mutation analysis of the WNT4 gene in Han Chinese women with premature ovarian failure. Reprod Biol Endocrinol. 2011;9:75. doi: 10.1186/1477-7827-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mandon-Pepin B, Touraine P, Kuttenn F, Derbois C, Rouxel A, Matsuda F, et al. Genetic investigation of four meiotic genes in women with premature ovarian failure. Eur J Endocrinol. 2008;158:107–15. doi: 10.1530/EJE-07-0400. [DOI] [PubMed] [Google Scholar]
- 71.Kim S, Pyun JA, Kang H, Kim J, Cha DH, Kwack K. Epistasis between CYP19A1 and ESR1 polymorphisms is associated with premature ovarian failure. Fertil Steril. 2011;95:353–6. doi: 10.1016/j.fertnstert.2010.07.1067. [DOI] [PubMed] [Google Scholar]
- 72.Hens L, Devroey P, Van Waesberghe L, Bonduelle M, Van Steirteghem AC, Liebaers I. Chromosome studies and fertility treatment in women with ovarian failure. Clin Genet. 1989;36:81–91. doi: 10.1111/j.1399-0004.1989.tb03169.x. [DOI] [PubMed] [Google Scholar]
- 73.Kawano Y, Narahara H, Matsui N, Miyakawa I. Premature ovarian failure associated with a Robertsonian translocation. Acta Obstet Gynecol Scand. 1998;77:467–9. [PubMed] [Google Scholar]
- 74.Oldenburg RA, van Dooren MF, de Graaf B, Simons E, Govaerts L, Swagemakers S, et al. A genome-wide linkage scan in a Dutch family identifies a premature ovarian failure susceptibility locus. Hum Reprod. 2008;23:2835–41. doi: 10.1093/humrep/den278. [DOI] [PubMed] [Google Scholar]
- 75.Kasippillai T, MacArthur DG, Kirby A, Thomas B, Lambalk CB, Daly MJ, et al. Mutations in eIF4ENIF1 are associated with primary ovarian insufficiency. J Clin Endocrinol Metab. 2013;98:E1534–9. doi: 10.1210/jc.2013-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Caburet S, Zavadakova P, Ben-Neriah Z, Bouhali K, Dipietromaria A, Charon C, et al. Genome-wide linkage in a highly consanguineous pedigree reveals two novel loci on chromosome 7 for non-syndromic familial Premature Ovarian Failure. PLoS One. 2012;7:e33412. doi: 10.1371/journal.pone.0033412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Caburet S, Arboleda VA, Llano E, Overbeek PA, Barbero JL, Oka K, et al. Mutant cohesin in premature ovarian failure. N Engl J Med. 2014;370:943–9. doi: 10.1056/NEJMoa1309635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang X, Zhou Y, Peng S, Wu L, Lin HY, Wang S, et al. Differentially expressed plasma microRNAs in premature ovarian failure patients and the potential regulatory function of mir-23a in granulosa cell apoptosis. Reproduction. 2012;144:235–44. doi: 10.1530/REP-11-0371. [DOI] [PubMed] [Google Scholar]
- 79.Kuang H, Han D, Xie J, Yan Y, Li J, Ge P. Profiling of differentially expressed microRNAs in premature ovarian failure in an animal model. Gynecol Endocrinol. 2014;30:57–61. doi: 10.3109/09513590.2013.850659. [DOI] [PubMed] [Google Scholar]
- 80.Rah H, Jeon YJ, Lee BE, Kim JO, Shim SH, Lee WS, et al. Association of polymorphisms in microRNA machinery genes (DROSHA, DICER1, RAN, and XPO5) with risk of idiopathic primary ovarian insufficiency in Korean women. Menopause. 2013;20:1067–73. doi: 10.1097/GME.0b013e3182883907. [DOI] [PubMed] [Google Scholar]
- 81.Kang H, Lee SK, Kim MH, Song J, Bae SJ, Kim NK, et al. Parathyroid hormone-responsive B1 gene is associated with premature ovarian failure. Hum Reprod. 2008;23:1457–65. doi: 10.1093/humrep/den086. [DOI] [PubMed] [Google Scholar]
- 82.Kang H, Lee SK, Cho SW, Lee SH, Kwack K. Branched chain alpha-keto acid dehydrogenase, E1-beta subunit gene is associated with premature ovarian failure. Fertil Steril. 2008;89:728–31. doi: 10.1016/j.fertnstert.2007.03.063. [DOI] [PubMed] [Google Scholar]
- 83.Kang H, Lee SK, Kim MH, Choi H, Lee SH, Kwack K. Acyl-CoA synthetase long-chain family member 6 is associated with premature ovarian failure. Fertil Steril. 2009;91:1339–43. doi: 10.1016/j.fertnstert.2008.03.035. [DOI] [PubMed] [Google Scholar]
- 84.Qin Y, Sun M, You L, Wei D, Sun J, Liang X, et al. ESR1, HK3 and BRSK1 gene variants are associated with both age at natural menopause and premature ovarian failure. Orphanet J Rare Dis. 2012;7:5. doi: 10.1186/1750-1172-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Knauff EA, Franke L, van Es MA, van den Berg LH, van der Schouw YT, Laven JS, et al. Genome-wide association study in premature ovarian failure patients suggests ADAMTS19 as a possible candidate gene. Hum Reprod. 2009;24:2372–8. doi: 10.1093/humrep/dep197. [DOI] [PubMed] [Google Scholar]
- 86.Pyun JA, Cha DH, Kwack K. LAMC1 gene is associated with premature ovarian failure. Maturitas. 2012;71:402–6. doi: 10.1016/j.maturitas.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 87.Pyun JA, Kang H, Lee SK, Kim MH, Kwack K. Association between polymorphisms in the protein L-isoaspartate (D-aspartate) O-methyltransferase gene and premature ovarian failure. Fertil Steril. 2009;91:1362–5. doi: 10.1016/j.fertnstert.2008.03.078. [DOI] [PubMed] [Google Scholar]
- 88.Aboura A, Dupas C, Tachdjian G, Portnoi MF, Bourcigaux N, Dewailly D, et al. Array comparative genomic hybridization profiling analysis reveals deoxyribonucleic acid copy number variations associated with premature ovarian failure. J Clin Endocrinol Metab. 2009;94:4540–6. doi: 10.1210/jc.2009-0186. [DOI] [PubMed] [Google Scholar]
- 89.Quilter CR, Karcanias AC, Bagga MR, Duncan S, Murray A, Conway GS, et al. Analysis \of X chromosome genomic DNA sequence copy number variation associated with premature ovarian failure (POF). Hum Reprod. 2010;25:2139–50. [DOI] [PMC free article] [PubMed]
- 90.Zhen XM, Sun YM, Qiao J, Li R, Wang LN, Liu P. Genome-wide copy number scan in Chinese patients with premature ovarian failure. Beijing Da Xue Xue Bao. 2013;45:841–7. [PubMed] [Google Scholar]
- 91.McGuire MM, Bowden W, Engel NJ, Ahn HW, Kovanci E, Rajkovic A. Genomic analysis using high-resolution single-nucleotide polymorphism arrays reveals novel microdeletions associated with premature ovarian failure. Fertil Steril. 2011;95:1595–600. doi: 10.1016/j.fertnstert.2010.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jin M, Yu Y, Huang H. An update on primary ovarian insufficiency. Sci China Life Sci. 2012;55:677–86. doi: 10.1007/s11427-012-4355-2. [DOI] [PubMed] [Google Scholar]
- 93.Cobo A, Garcia-Velasco JA, Domingo J, Remohi J, Pellicer A. Is vitrification of oocytes useful for fertility preservation for age-related fertility decline and in cancer patients? Fertil Steril. 2013;99:1485–95. doi: 10.1016/j.fertnstert.2013.02.050. [DOI] [PubMed] [Google Scholar]