

Abstract
Retinoblastoma is the most common intraocular malignancy in children, with a reported incidence ranging from 1 in 15,000 to 1 in 18,000 live births. It is second only to uveal melanoma in the frequency of occurrence of malignant intraocular tumors. Pawius described retinoblastoma as early as in 1597 referred to the tumor as fungus hematodes and suggested enucleation as the primary mode of management. The discovery of ophthalmoloscope in 1851 facilitated recognition of specific clinical features of retinoblastoma. Initially thought to be derived from the glial cells, it was called a glioma of the retina by Virchow (1864). Flexner (1891) and Wintersteiner (1897) believed it to be a neuroepithelioma because of the presence of rosettes. Later, there was a consensus that the tumor originated from the retinoblasts and the American Ophthalmological Society officially accepted the term retinoblastoma in 1926. Retinoblastoma was associated with near certain death just over a century ago. There has been a dramatic change in the overall management of retinoblastoma in the last decade. Specific genetic protocols have been able to make pre natal diagnosis of retinoblastoma. Early diagnosis and advancements in focal therapy have resulted in improved eye and vision salvage. This article explains the complexity of retinoblastoma, genetic association, clinical features, management and prognosis.
Keywords: Leukocoria, Endophytic, Exophytic, Chemotherapy, Brachytherapy
Introduction
Retinoblastoma is the most common intraocular malignancy in children, with a reported incidence ranging from 1 in 15,000 to 1 in 18,000 live births.1 It is second only to uveal melanoma in the frequency of occurrence of malignant intraocular tumors. There is no racial or gender predisposition for the incidence of retinoblastoma. Retinoblastoma is bilateral in about 25–35% of cases.2 The average age at diagnosis is 18 months, unilateral cases being diagnosed at around 24 months and bilateral cases before 12 months.2
Pawius described retinoblastoma as early as 1597.3 In 1809, Wardrop referred to the tumor as fungus hematodes and suggested enucleation as the primary mode of management.3 The introduction of the ophthalmoloscope in 1851 facilitated the recognition of specific clinical features of retinoblastoma. Initially thought to be derived from the glial cells, it was called a glioma of the retina by Virchow (1864).3 Flexner (1891) and Wintersteiner (1897) believed it was a neuroepithelioma because of the presence of rosettes.3 Later, the consensus was that the tumor originated from the retinoblasts and the American Ophthalmological Society officially accepted the term retinoblastoma in 1926.4
Retinoblastoma was associated with near certain death just over a century ago. Early tumor recognition aided by indirect ophthalmoscopy and refined enucleation techniques contributed to improved survival from 5% in 1896 to 81% in 1967.2 Advances in external beam radiotherapy in the 1960s and 1970s and further progress in planning and delivery provided an excellent alternative to enucleation, that enabled salvaging the eye.2 Focal therapeutic measures such as cryotherapy, photocoagulation and plaque brachytherapy allowed targeted treatment of smaller tumors that saved vision. Parallel advancements in ophthalmic diagnostics and introduction of ultrasonography, computed tomography, and magnetic resonance imaging contributed to improved diagnostic accuracy and early detection of extraocular retinoblastoma.
The recent advances such as identification of genetic mutations,5,6 replacement of external beam radiotherapy by chemoreduction as the primary modality of management, use of chemoreduction to minimize the size of the regression scar with consequent optimization of visual potential,7–11 identification of histopathologic high-risk factors following enucleation12 and provision of adjuvant therapy to reduce the incidence of systemic metastasis,13 protocol-based management of retinoblastoma with accidental perforation or intraocular surgery14,15 and aggressive multimodal therapy in the management of orbital retinoblastoma16,17 have contributed to improved outcome in terms of better survival, improved eye salvage and potential for optimal visual recovery.
Genetics
Of newly diagnosed cases of retinoblastoma, only 6% are familial and 94% are sporadic.2,18 Bilateral retinoblastomas involve germinal mutations in all cases. Approximately 15% of unilateral sporadic retinoblastomas are caused by germinal mutations affecting only one eye while 85% are sporadic.2
In 1971, Knudson proposed the two hit hypothesis. He stated that for retinoblastoma to develop, two chromosomal mutations are needed.19 In hereditary retinoblastoma, the initial hit is a germinal mutation, which is inherited and is found in all the cells. The second hit develops in the somatic retinal cells leading to the development of retinoblastoma. Therefore, hereditary cases are predisposed to the development of monocular tumors such as osteosarcoma.
In unilateral sporadic retinoblastoma, both the hits occur during the development of the retina and are somatic mutations. Therefore there is no risk of second monocular tumors.
Genetic counseling is an important aspect in the management of retinoblastoma. In patients with a positive family history, 40% of the siblings would be at risk of developing retinoblastoma and 40% of the offspring of the affected patient may develop retinoblastoma. In patients with no family history of retinoblastoma, if the affected child has unilateral retinoblastoma, 1% of the siblings are at risk and 8% of the offspring may develop retinoblastoma. In cases of bilateral retinoblastoma with no positive family history, 6% of the siblings and 40% of the offspring have a chance of developing retinoblastoma.2
Clinical features
Table 1 lists the common presenting signs and symptoms of retinoblastoma.20 Leukocoria is the most common presenting feature of retinoblastoma, followed by strabismus, painful blind eye and loss of vision. The clinical presentation of retinoblastoma depends on the stage of the disease.10 Early lesions are likely to be missed, unless an indirect ophthalmoscopy is performed. The tumor appears as a translucent or a white fluffy retinal mass. The child may present with strabismus if the tumor involves the macula or with reduced visual acuity.10 Moderately advanced lesions usually present with leukocoria due to the reflection of light by the white mass in the fundus (Fig. 1).
Table 1.
Common presenting features of retinoblastoma.
| Leukocoria | 56% |
| Strabismus | 20% |
| Red painful eye | 7% |
| Poor vision | 5% |
| Asymptomatic | 3% |
| Orbital cellulitis | 3% |
| Unilateral Mydriasis | 2% |
| Heterochromia iridis | 1% |
| Hyphema | 1% |
Figure 1.
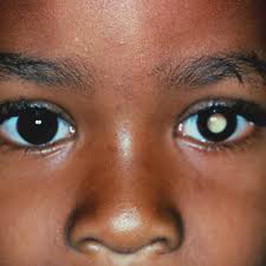
Child presenting with Leucocoria.
Three patterns are usually present:
-
•
Endophytic, in which the tumor grows into the vitreous cavity (Fig. 2). A yellow white mass progressively fills the entire vitreous cavity and vitreous seeds occur. The retinal vessels are not seen on the tumor surface.
-
•
Exophytic, in which the tumor grows toward the subretinal space (Fig. 3). Retinal detachment usually occurs and retinal vessels are seen over the tumor.
-
•
Diffuse infiltrating tumor, in which the tumor diffusely involves the retina causing just a placoid thickness of the retina and not a mass. This is generally seen in older children and usually there is a delay in the diagnosis.
Figure 2.
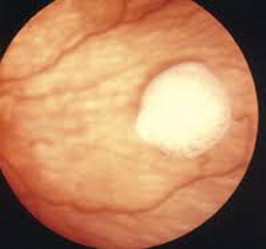
Endophytic growth.
Figure 3.
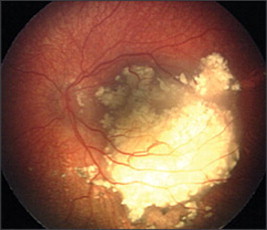
Exophytic growth.
Advanced tumors manifest with proptosis secondary to optic nerve extension or orbital extension and systemic metastasis.10 Retinoblastoma can spread through the optic nerve with relative ease especially once the lamina cribrosa is breached. Orbital extension may present with proptosis and is most likely to occur at the site of the scleral emissary veins. Systemic metastasis occurs to the brain, skull, distant bones and the lymph nodes.
Some of the atypical manifestations of retinoblastoma include pseudohypopyon, spontaneous hyphema, vitreous hemorrhage, phthisis bulbi and preseptal or orbital cellulitis.
Diagnosis
A thorough clinical evaluation with careful attention to details, supplemented with B-scan ultrasonography helps in the diagnosis (Fig. 4).[10] Computed tomography (Fig. 5) and magnetic resonance imaging are generally reserved for cases with atypical manifestations and diagnostic dilemmas and where extraocular or intracranial tumor extension is suspected.10
Figure 4.
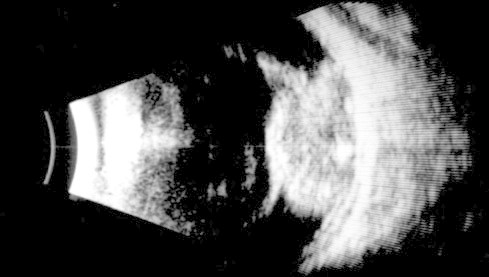
B scan showing echogenic mass fiiling the vitreous cavity- confirming diagnosis of retinoblastoma.
Figure 5.
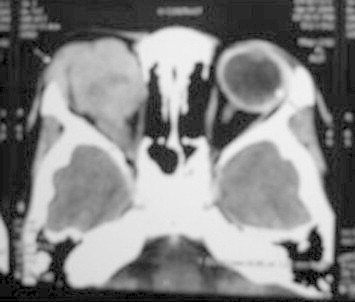
CT orbit showing intracranial tumor extension.
A child with suspected retinoblastoma warrants a complete ophthalmic evaluation including a dilated fundus examination under anesthesia.10 The intraocular pressure is measured and the anterior segment is examined for neovascularization, pseudohypopyon, hyphema, and signs of inflammation.10
Bilateral fundus examination with 360° scleral depression is mandatory. Direct visualization of the tumor by an indirect ophthalmoscope is diagnostic for retinoblastoma in over 90% of cases.20 RetCam is a wide-angle fundus camera, useful in accurately documenting retinoblastoma and monitoring response to therapy. Ultrasonography B-scan shows a rounded or irregular intraocular mass with high internal reflectivity representing typical intralesional calcification.10 Computed tomography delineates extraocular extension and can detect an associated pinealoblastoma.10 Magnetic resonance imaging is specifically indicated if optic nerve invasion or intracranial extension is suspected.10 On fluorescein angiography, smaller retinoblastoma shows minimally dilated feeder vessels in the arterial phase, blotchy hyperfluorescence in the venous phase and late staining.10
Management
The primary goal of management of retinoblastoma is to save the patient’s life. Salvage of the organ (eye) and function (vision) are the secondary and tertiary goals, respectively. The management of retinoblastoma needs a multidisciplinary team approach including an ocular oncologist, pediatric oncologist, radiation oncologist, radiation physicist, geneticist and an ophthalmic oncopathologist. The management strategy depends on the stage of the disease such as, intraocular retinoblastoma, retinoblastoma with high-risk characteristics, orbital retinoblastoma and metastatic retinoblastoma.
The management of retinoblastoma is highly individualized and is based on several considerations including, age at presentation, laterality, tumor location, tumor staging, visual prognosis, systemic condition, family and societal perception, and, to a certain extent, the overall prognosis and cost-effectiveness of treatment in a given economic situation.
The majority of children with retinoblastoma manifest at the stage when the tumor is confined to the eye. About 90–95% of children in developed countries are present with intraocular retinoblastoma while 60–70% present at this stage in the developing world.10 Diagnosis of retinoblastoma at this stage and appropriate management are crucial to reduce mortality, save the eye and possibly save the remaining vision.
There are several methods to manage intraocular retinoblastoma – focal (cryotherapy, laser photocoagulation, transpupillary thermotherapy, transcleral thermotherapy, plaque brachytherapy), local (external beam radiotherapy, enucleation), and systemic (chemotherapy). While primary focal measures are mainly reserved for small tumors, local and systemic modalities are used to treat advanced retinoblastoma.
Cryotherapy
Cryotherapy is performed for small equatorial and peripheral retinal tumors measuring up to 4 mm in basal diameter and 2 mm in thickness.2,10 Triple freeze thaw cryotherapy is applied at 4–6 week intervals until complete tumor regression. Cryotherapy produces a scar much larger than the tumor. Complications of cryotherapy include transient serous retinal detachment, retinal tear and rhegmatogenous retinal detachment. Cryotherapy administered 2–3 h prior to chemotherapy can increase the delivery of chemotherapeutic agents across the blood retinal barrier and thus has a synergistic effect.10
Laser photocoagulation
Laser photocoagulation is used for small posterior tumors 4 mm in basal diameter and 2 mm in thickness.2,10 The treatment is directed to delimit the tumor and coagulate the blood supply to the tumor by surrounding it with two rows of overlapping laser burns. Complications include transient serous retinal detachment, retinal vascular occlusion, retinal hole, retinal traction, and preretinal fibrosis. It is less often employed now with the advent of thermotherapy. In fact, laser photocoagulation is contraindicated while the patient is on an active chemoreduction protocol.10
Thermotherapy
In thermotherapy, focused heat generated by infrared radiation is applied to tissues at subphotocoagulation levels to induce tumor necrosis.21 The goal is to achieve a slow and sustained temperature range of 40 to 60 °C within the tumor, thus sparing damage to the retinal vessels. The standard treatment for transpupillary thermotherapy involves using infrared radiation from a semiconductor diode laser delivered with a 1300-micron large spot indirect ophthalmoscope delivery system. Alternatively transpupillary delivery can be performed through an operating microscope or via a transscleral route with a diopexy probe. Thermotherapy provides satisfactory control for small tumors – 4 mm in basal diameter and 2 mm in thickness. Complete tumor regression can be achieved in over 85% of tumors using 3–4 sessions of thermotherapy.21 The common complications are focal iris atrophy, focal paraxial lens opacity, retinal traction and serous retinal detachment.
Plaque brachytherapy
Plaque brachytherapy involves the placement of a radioactive implant on the sclera corresponding to the base of the tumor to trans-scleral irradiation of the tumor.22 Commonly used radioactive materials include Ruthenium 106 and Iodine 125. The advantages of plaque brachytherapy are focal delivery of radiation with minimal damage to the surrounding normal structures, minimal periorbital tissue damage, absence of cosmetic abnormality because of retarded bone growth in the field of irradiation as it occurs with external beam radiotherapy, reduced risk of second malignant neoplasm and shorter duration of treatment. Plaque brachytherapy requires precise tumor localization and measurement of its basal dimensions. The tumor thickness is measured by ultrasonography. The data are used for dosimetry on a three-dimensional computerized tumor modeling system. The plaque design is chosen depending on the basal tumor dimensions, its location, and configuration. The dose to the tumor apex ranges from 4000 to 5000 cGy. The plaque is sutured to the sclera after confirming tumor centration and is left in situ for the duration of exposure, generally ranging from 36 to 72 h. The common complications are radiation papillopathy and radiation retinopathy.22
External beam radiotherapy
External beam radiotherapy was the preferred form of management of moderately advanced retinoblastoma in the late 1900s.23,24 Presently it is indicated in eyes where primary chemotherapy and local therapy had failed, or rarely when chemotherapy is contraindicated.10
Enucleation
Primary enucleation continues to be the treatment of choice for advanced intraocular retinoblastoma with neovascularization of iris, secondary glaucoma, anterior chamber tumor invasion, tumors occupying >75% of the vitreous volume, necrotic tumors with secondary orbital inflammation, and tumors associated with hyphema or vitreous hemorrhage where the tumor characteristics cannot be visualized, especially when only one eye is involved.10
Chemotherapy
Chemoreduction, defined as the process of reduction in the tumor volume with chemotherapy, has become an integral part of the current management of retinoblastoma.
| Chemoreduction regimen and doses for intraocular retinoblastoma |
| Day 1: Vincristine + Etoposide + Carboplatin |
| Day 2: Etoposide |
| Standard dose (3 weekly, 6 cycles): Vincristine 1.5 mg/m22 (0.05 mg/kg for children <36 months of age and maximum dose <2 mg), Etoposide 150 mg/m (5 mg/kg for children <36 months of age), Carboplatin 560 mg/m2 (18.6 mg/kg for children <36 months of age) |
| High-dose (3 weekly, 6–12 cycles): Vincristine 0.025 mg/kg, Etoposide 12 mg/kg, Carboplatin 28 mg/kg |
Adjuvant therapy
Studies on the efficacy of adjuvant therapy to minimize the risk of metastasis initiated in the 1970s were marked by variable results and provided no firm recommendation.17 A recent study with a long-term follow-up provides useful information.13,28 It included a subset of patients with unilateral sporadic retinoblastoma who underwent primary enucleation. The study used specific predetermined histopathologic characteristics for patient selection. A minimum follow-up of 1 year was allowed to include metastatic events that generally occur at a mean of 9 months following enucleation.13 The incidence of metastasis was 4% in those who received adjuvant therapy compared to 24% in those who did not. The study found that administration of adjuvant therapy significantly reduced the risk of metastasis in patients with high-risk histopathology characteristics. Adjuvant therapy may include systemic chemotherapy and orbital external beam radiotherapy.
With improved understanding of the risk factors predictive of metastasis,29,30and the availability of effective chemotherapy regimens for intraocular retinoblastoma,30,31 it would seem logical to consider adjuvant chemotherapy following enucleation to prevent metastasis in high-risk cases. However, the utility of adjuvant chemotherapy in such cases remains debatable, and its role has yet to be clearly defined.29,30 Adjuvant orbital external beam radiotherapy following enucleation is recommended in patients with tumor invasion of optic nerve transection, scleral and extrascleral extension, spontaneous or accidental ocular perforation, and intraocular surgery for unrecognized retinoblastoma. The role of such therapy, however, is not well established. The controversy regarding adjuvant therapy is compounded by disagreement over the histopathologic prognostic factors that define “high-risk” for developing metastasis.31 The overall rarity of retinoblastoma, including the unusual finding of extraretinal involvement, has limited the experience with adjuvant therapy.
Follow-up schedule
The usual protocol is to schedule the first examination 3–6 weeks after the initial therapy. In cases where chemoreduction therapy has been administered, the examination should be done every 3 weeks with each cycle of chemotherapy. Patients under focal therapy are evaluated and treated every 4–8 weeks until complete tumor regression. Following tumor regression, subsequent examination should be every 3 months for the first year, every 6 months for three years or until the child reaches 6 years of age, and yearly thereafter.
High risk retinoblastoma
Systemic metastasis is the main cause for mortality in patients with retinoblastoma. Although the survival of patients with retinoblastoma has dramatically improved over the last three decades, with a reported survival of more than 90% in developed countries,25 mortality is still as high as 50% in the developing nations.26,27 Reduction in the rate of systemic metastasis by identification of high-risk factors and appropriate adjuvant therapy may help improve survival.
Conclusion
There has been a dramatic change in the overall management of retinoblastoma in the last decade. Specific genetic protocols have been able to make prenatal diagnosis of retinoblastoma. Early diagnosis and advancements in focal therapy have resulted in improved eye and vision salvage. Chemoreduction has become the standard of care for the management of moderately advanced intraocular retinoblastoma. Periocular chemotherapy is now an additional useful tool in saving eyes with vitreous seeds. Enucleation continues to be the preferred primary treatment approach in unilateral advanced retinoblastoma. Post-enucelation protocol, including identification of histopathologic high risk characteristics and provision of adjuvant therapy has resulted in substantial reduction in the incidence of systemic metastasis. The vexing orbital retinoblastoma now seems to be finally cureable with an aggressive multimodal approach. The future holds promise for further advancement in focal therapy and targeted drug delivery.
Conflict of interest
The author declared that there is no conflict of interest.
Footnotes
Peer review under responsibility of Saudi Ophthalmological Society, King Saud University.
References
- 1.Bishop J.O., Madsen E.C. Retinoblastoma: review of current status. Surv Ophthalmol. 1975;19:342–366. [PubMed] [Google Scholar]
- 2.Shields J.A., Shields C.L. WB Saunders Company; Philadelphia, PA, USA: 1992. Intraocular tumors – a text and Atlas. [Google Scholar]
- 3.Albert D.M. Historic review of retinoblastoma. Ophthalmology. 1987;94:654–662. doi: 10.1016/s0161-6420(87)33407-4. [DOI] [PubMed] [Google Scholar]
- 4.Jackson E. Report of the committee to investigate and revise the classification of certain retinal conditions. Trans Am Ophthalmol Soc. 1926;24:38–39. [Google Scholar]
- 5.Ata-ur-Rasheed M., Vemuganti G., Honavar S., Ahmed N., Hasnain S., Kannabiran C. Mutational analysis of the RB1gene in Indian patients with retinoblastoma. Ophthalmic Genet. 2002;23:1218. doi: 10.1076/opge.23.2.121.2211. [DOI] [PubMed] [Google Scholar]
- 6.Kiran V.S., Kannabiran C., Chakravarthi K., Vemuganti G.K., Honavar S.G. Mutational screening of the RB1 gene in Indian patients with retinoblastoma reveals eight novel and several recurrent mutations. Hum Mutat. 2003;22:339. doi: 10.1002/humu.9181. [DOI] [PubMed] [Google Scholar]
- 7.Shields C.L., Honavar S.G., Shields J.A., Demirci H., Meadows A.T., Naduvilath T.J. Factors predictive of recurrence of retinal tumors, vitreous seeds, and sub retinal seeds following chemoreduction for retinoblastoma. Arch Ophthalmol. 2002;120:460–464. [PubMed] [Google Scholar]
- 8.Shields C.L., Honavar S.G., Meadows A.T., Shields J.A., Demirci H., Singh A. Chemoreduction plus focal therapy for retinoblastoma: factors predictive of need for treatment with external beam radio therapy or enucleation. Am J Ophthalmol. 2002;133:657–664. doi: 10.1016/s0002-9394(02)01348-x. [DOI] [PubMed] [Google Scholar]
- 9.Shields C.L., Honavar S.G., Meadows A.T., Shields J.A., Demirci H., Naduvilath T.J. Chemoreduction for unilateral retinoblastoma. Arch Ophthalmol. 2002;120:1653–1658. doi: 10.1001/archopht.120.12.1653. [DOI] [PubMed] [Google Scholar]
- 10.Murthy R., Honavar S.G., Naik M.N., Reddy V.A. Retinoblastoma. In: Dutta L.C., editor. Modern ophthalmology. Jaypee Brothers; New Delhi, India: 2004. p. 849859. [Google Scholar]
- 11.Honavar S.G., Singh A.D. Management of advanced retinoblastoma. Ophthalmol Clin North Am. 2005;18:6573. doi: 10.1016/j.ohc.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Vemuganti G., Honavar S.G., John R. Clinicopathological profile of retinoblastoma in Asian Indians. Invest Ophthalmol Visual Sci. 2000;41(S):790. [Google Scholar]
- 13.Honavar S.G., Singh A.D., Shields C.L., Meadows A., Shields J.A. Does adjuvant chemotherapy prevent metastasis in high-risk retinoblastoma? Invest Ophthalmol Visual Sci. 2000;41(S):790. [Google Scholar]
- 14.Honavar S.G., Rajeev B. Needle tract tumor cell seeding following fine needle aspiration biopsy for retinoblastoma. Invest Ophthalmol Visual Sci. 1998;39(S):658. [Google Scholar]
- 15.Shields C.L., Honavar S., Shields J.A., Demirci H., Meadows A.T. Vitrectomy in eyes with unsuspected retinoblastoma. Ophthalmology. 2000;107:2250–2255. doi: 10.1016/s0161-6420(00)00427-9. [DOI] [PubMed] [Google Scholar]
- 16.Honavar S.G., Reddy V.A.P., Murthy R., Naik M., Vemuganti G.K. XI International Congress of Ocular Oncology; Hyderabad, India: 2004. Management of orbital retinoblastoma. [p. 51] [Google Scholar]
- 17.Stallard H.B. The conservative treatment of retinoblastoma. Trans Am Ophthalmol Soc UK. 1962;82:473–534. [PubMed] [Google Scholar]
- 18.Murphree A.L., Benedict W.F. Retinoblastoma: clues to human oncogenesis. Science. 1984;223:1028–1033. doi: 10.1126/science.6320372. [DOI] [PubMed] [Google Scholar]
- 19.Knudson A.G. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abramson D.H., Frank C.M., Susman M., Whalen M.P., Dunkel I.J., Boyd N.W., 3rd. Presenting signs of retinoblastoma. J Pediatr. 1998;132:505–508. doi: 10.1016/s0022-3476(98)70028-9. [DOI] [PubMed] [Google Scholar]
- 21.Shields C.L., Santos M.C., Diniz W. Thermotherapy for retinoblastoma. Arch Ophthalmol. 1999;117:885–893. doi: 10.1001/archopht.117.7.885. [DOI] [PubMed] [Google Scholar]
- 22.Shields C.L., Shields J.A., Cater J. Plaque radiotherapy for retinoblastoma, long term tumor control and treatment complications in 208 tumors. Ophthalmology. 2001;108:2116–2121. doi: 10.1016/s0161-6420(01)00797-7. [DOI] [PubMed] [Google Scholar]
- 23.Ellsworth R.M. Retinoblastoma. Mod Prob Ophthalmol. 1977;96:1826–1830. [PubMed] [Google Scholar]
- 24.Hungerford J.L., Toma N.M.G., Plowman P.N., Kingston J.E. External beam radiotherapy for retinoblastoma: I, whole eye technique. Br J Ophthalmol. 1995;79:112–117. doi: 10.1136/bjo.79.2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abramson D.H., Niksarli K., Ellsworth R.M., Servodidio C.A. Changing trends in the management of retinoblastoma: 1951–1965 vs. 1966–1980. J Pediatr Ophthalmol Strabismus. 1994;31:32–37. doi: 10.3928/0191-3913-19940101-07. [DOI] [PubMed] [Google Scholar]
- 26.Singh A.D., Shields C.L., Shields J.A. Prognostic factors in retinoblastoma. J Pediatr Ophthalmol Strab. 2000;37:13441. doi: 10.3928/0191-3913-20000501-04. [DOI] [PubMed] [Google Scholar]
- 27.Ajaiyeoba I.A., Akang E.E., Campbell O.B., Olurin I.O., Aghadiuno P.U. Retinoblastomas in Ibadan: treatment and prognosis. West Afr J Med. 1993;12:223–227. [PubMed] [Google Scholar]
- 28.Honavar S.G., Singh A.D., Shields C.L., Demirci H., Smith A.F., Shields J.A. Postenucleation prophylactic chemotherapy in high-risk retinoblastoma. Arch Ophthalmol. 2002;120:923–931. doi: 10.1001/archopht.120.7.923. [DOI] [PubMed] [Google Scholar]
- 29.Shields C.L., DePotter P., Himelstein B.P. Chemoreduction in the initial management of intraocular retinoblastoma. Arch Ophthalmol. 1996;114:1330–1338. doi: 10.1001/archopht.1996.01100140530002. [DOI] [PubMed] [Google Scholar]
- 30.Kingston J.E., Hungerford J.L., Madreperla S.A., Plowman P.N. Results of combined chemotherapy and radiotherapy for advanced intraocular retinoblastoma. Arch Ophthalmol. 1996;114:1339–1343. doi: 10.1001/archopht.1996.01100140539004. [DOI] [PubMed] [Google Scholar]
- 31.Gallie B.L., Budning A., DeBoer G. Chemotherapy with focal therapy can cure intraocular retinoblastoma without radiation. Arch Ophthalmol. 1996;114:1321–1328. doi: 10.1001/archopht.1996.01100140521001. [DOI] [PubMed] [Google Scholar]