

Abstract
Understanding disease susceptibility factors and gene-environment interactions may offer valuable insights into the biological mechanisms for the etiology of rheumatic diseases. Defining the contributions of genetic and environmental factors to the pathogenesis of rheumatic diseases such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) and ankylosing spondylitis (AS), may have important implications for understanding risk prediction, pathogenic mechanisms, cellular pathways, drug discovery, and prevention strategies. However, rheumatic diseases offer distinct challenges to researchers due to heterogeneity in disease phenotypes, low disease incidence, and geographic variation in both genetic and environmental factors. Emerging research areas, including epigenetics, metabolomics, and the microbiome, may provide additional links between genetic and environmental risk factors in rheumatic disease pathogenesis. This article reviews the methods used to establish genetic and environmental risk factors and to study gene-environment interactions in rheumatic diseases and provides specific examples of successes and challenges for identifying gene-environment interactions in RA, SLE, and AS. Finally, we describe how emerging research strategies may build upon previous discoveries as well as future challenges.
Keywords: rheumatoid arthritis, systemic lupus erythematosus, ankylosing spondylitis, RA, SLE, AS, environment, genetics, interaction, smoking
INTRODUCTION
The current paradigm for autoimmune rheumatic disease etiology is that several pre-clinical stages precede the onset of clinically apparent disease. When individuals at increased genetic risk are exposed to environmental or lifestyle factors, early alterations in the immune system and the breakdown of self-tolerance ensue, eventually leading to the presentation of overt disease (Figure 1).1, 2 Indeed, several genetic and environmental risk factors have been strongly associated with the risk of incident rheumatic diseases, and many more are weakly associated or hypothesized to be related.3, 4 However, the pathogenesis and biological mechanisms for the development of autoimmune rheumatic diseases remain poorly understood.
Figure 1.
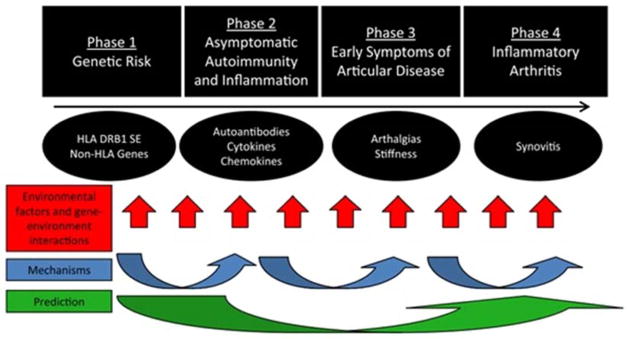
Schematic showing the pre-clinical phases of RA. Other rheumatic diseases likely follow similar phases of progression from genetic susceptibility, immune dysregulation, and sub-clinical disease to classifiable disease. Environmental factors and gene-environment interactions likely occur throughout this process of disease pathogenesis.
From Karlson EW, Deane K. Environmental and gene-environment interactions and risk of rheumatoid arthritis. Rheum Dis Clin North Am 2012;38(2):405–26; with permission.
Interactions between genetic and environmental factors may elucidate biological mechanisms for rheumatic disease susceptibility and bridge findings in several fields of research. Greater understanding of rheumatic disease etiology may provide important insights into prevention, screening, and treatment options. Therefore, rheumatic disease researchers are motivated to explore the intersection of genetic and environmental risk factors. However, rheumatic diseases present distinct challenges for the identification of gene-environment interactions (GEIs). These challenges include heterogeneous phenotypes, low disease incidence and prevalence, geographic variation in epidemiology, and the difficulty in identifying individuals at elevated disease risk prior to clinical diagnosis.
This article will serve as an overview to contextualize genetic and environmental risk factors in the development of rheumatic diseases and to highlight future research directions according to study designs and molecular approaches. We will address specific successes and challenges concerning rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and ankylosing spondylitis (AS).
Current research strategies for environmental and genetic risk factors
As in other areas of research, bias, confounding, chance, and generalizability are major threats to validity that must be considered in study designs and analyses investigating genetic and environmental risk factors (Table 1). Investigators studying environmental and lifestyle risk factors for rheumatic disease risk have utilized primarily traditional epidemiologic techniques: the case-control and prospective cohort study designs.
Table 1.
Threats to validity in study designs.
| Case-control study | Cohort study | Genome-wide association study | |
|---|---|---|---|
|
|
|||
| Bias | Selection bias Recall bias Inappropriate matching Reverse causation |
Selection bias Misclassification Length-time bias Reverse causation Loss to follow-up |
Phenotype misclassification Inappropriate controls Genomic inflation |
| Confounding | Unmeasured confounders | Unmeasured confounders | Population stratification |
| Chance | Multiple testing Power |
Multiple testing Power |
Multiple testing False discovery rate Rare variants Power |
| Generalizability | Study-specific | Cohort-specific | Race/region-specific Phenotype-specific |
Case-control studies
identify incident disease cases and match them to controls. Advantages of case-control studies are relative efficiency, especially when cases are rare and difficult to identify prospectively. Disadvantages are inherent biases based on the retrospective nature of these studies—selection and recall bias. In particular, the selection of inappropriate controls may introduce biases that lead to erroneous conclusions. Pre-clinical disease changes might also truly influence factors, such as dietary intake, physical activity, or weight changes, introducing the potential for reverse causation. Important environmental exposures may occur early in life and may be difficult to assess. In addition, when genes are associated with established disease, it is unclear whether the effect of these genes is in disease etiology or in disease propagation. Given these nuances, multiple case-control studies in diverse populations are usually warranted to establish an association between an environmental factor and disease susceptibility. Population-based case-control studies, where inclusion is compulsory as in national registries, may overcome some of these challenges.
Cohort studies
offer solutions to some of the biases inherent in case-control studies. This study design follows populations prospectively based on a common exposure or demographic factor before individuals might develop incident disease. The major advantage of cohort studies is that data are collected without bias for all individuals prior to disease onset. Design issues such as selection or recall bias are therefore less problematic, though subtle subclinical manifestations might still introduce potential for reverse causation.5 Inclusion criteria, however, might affect generalizability. Nested case-control studies on stored samples performed within cohort studies, can evaluate biomarkers in the pre-clinical period. Confirmation of incident cases is often a limiting factor in cohort studies, since most rheumatic diseases have low incidence and require either extremely large cohorts, long follow-up, or specific populations at increased risk, such as first-degree relatives or those with pre-clinical symptoms (e.g., arthralgias). Meta-analyses of multiple large cohorts may overcome some of these limitations, however heterogeneity in study designs and populations may also be limiting. These different cohort designs (general population, unaffected relative, and symptomatic but non-classifiable individuals) may correspond to particular phases in the development of autoimmune diseases (Figure 1).1, 6
Early studies seeking to identify genetic risk factors often focused on heritability, usually in the context of familial disease. Heritability was estimated evaluating rates of disease within homozygote and heterozygote twins. Linkage analyses utilized entire families, with affected and unaffected family members. These methods were able to establish the familial heritability of a disease, but shared environmental factors might still contribute to this heritability. Linkage analyses were most useful in identifying highly penetrant genes with large effect sizes. In addition, candidate gene studies genotyped a few loci at a time, a slow process focusing on known cellular pathway mechanisms.7
Genome Wide Association Studies (GWAS)
The advent of high-throughput genotyping brought about the era of GWAS, which evaluates initially thousands (and more recently hundreds of thousands) of different genetic loci at once for association with disease.8 This hypothesis-free method is capable of evaluating the entire genome for potential disease susceptibility genes.9 However, GWAS are dependent upon the performance and composition of specific platforms of single nucleotide polymorphisms (SNPs) across the genome, which might vary widely based on race, ethnicity, and geography. Potentially associated loci may not necessarily be included on each platform and therefore would not be tested. The ability of GWAS to detect true associations is dependent upon a homogeneous disease phenotype with similar disease pathogenesis. Appropriate control selection, with similar race and genetic structure is therefore important. For example, most early GWAS in RA were performed for seropositive RA cases among whites with European heritage.10–12 Findings from these studies may not be generalizable to seronegative RA or non-white European populations. This concept presents major challenges for diseases such as SLE with heterogeneous subtypes and variation by race, ethnicity, and geography. The genetic components of rheumatic diseases might also be more pronounced in early-onset compared to older-onset disease, in which environmental or age-related factors may be more important.13, 14
GWAS studies usually offer only statistical associations with probabilities of disease development. Disease susceptibility loci detected through this method are not necessarily causal for rheumatic diseases, but may be proxies for causal loci due to linkage disequilibrium. GWAS is most successful at discovering common variants, as opposed to rare variants. The latter, however, often have high effect sizes, and therefore may provide important pathogenic information compared to common variants with lower effect sizes. For example, single mutations of genes involved in the complement cascade (C1q, C2, and C4) occur rarely but are very strongly associated with increased risk of SLE.15 Methodological issues such as genomic inflation, population stratification, false discovery rates, linkage disequilibria, and multiple comparisons are major design and computational obstacles for GWAS, requiring advanced statistical expertise.16 Despite these caveats, GWAS have rapidly and efficiently detected numerous rheumatic disease susceptibility loci.17 To date, however, findings from GWAS for rheumatic diseases have had little clinical impact as significant SNPs are may not be causal and have low effect size estimates, and thus offer little ability to classify individuals according to risk for disease development.
Genetic risk scores (GRS)
have been developed to combine many validated genetic loci into a single summary variable, improving statistical power to detect potential associations.18 GRS can be calculated simply by counts of risk alleles. However, if effect sizes of individual genetic components vary appreciably, weighted genetic risk scores (wGRS) should be utilized.19 Typically, wGRS are weighted using the natural logarithm of odds ratios found in large GWAS or meta-analyses.19 Since HLA loci are often more strongly associated with rheumatic disease risk compared to non-HLA SNPs, wGRS have been used in rheumatic disease research.14, 19–22
Gene-environment interactions
The rapid emergence of genetic susceptibility loci and the identification and replication of specific environmental risk factors provide the opportunity to evaluate specific GEIs that may offer new biological mechanisms in disease pathogenesis as well as potentially provide personalized medicine approaches where an individuals risk for disease can be calculated using a combination of genetic and environmental factors.23 However, study design, statistical power and expertise are major challenges in the identification of potential GEIs.24 Since GWAS require very large samples sizes to detect associations of genetic factors with disease, often with modest effect sizes, studies with statistical power to detect GEIs may not feasible using current techniques. While GEIs may suggest biological mechanisms for disease development, it is not clear that they will be able to provide robust predictive abilities for rheumatic diseases, at least in the foreseeable future.25, 26
GEIs can be identified statistically through additive or multiplicative interactions (Table 2). An additive interaction is an effect beyond the sum of the risks associated with individual factors and can be measured by the relative excess risk due to interaction (RERI), attributable proportion due to interaction (AP), or synergy index (S).27 A multiplicative interaction is an effect greater than the multiplied effects of the individual factors and is measured by the ratio of odds ratios (ROR).27 The interpretation and suitability of interaction terms depend on the scale of the statistical model utilized. Logistic regression models are on a multiplicative scale, while linear regression models are on an additive scale. Therefore, in a logistic regression model (e.g., with an outcome of development of disease or not), a statistically significant interaction term implies a multiplicative interaction between the two factors. Additive interactions have been thought to represent biologic interaction of two factors within the same pathway.24 However, a statistical interaction does not necessarily imply a biological interaction. In studies of disease pathogenesis, a statistically significant additive GEI should be ideally replicated in independent studies and animal models in order to validate biological plausibility. GEIs usually have statistically significant main effects for both genetic and environmental factors in predicting disease onset, though this may not be a requirement if factors work exclusively in synergy.
Table 2.
Statistical measures of interaction.
| Type of interaction | Statistical measure | Interpretation | Null hypothesis | |
|---|---|---|---|---|
| Additive | RERI | Relative excess risk due to interaction | Additional risk compared to the expected risk from adding the risks for each exposure | RERI=0 |
| AP | Attributable proportion due to interaction | Proportion due to interaction of overall risk among those with both exposures | AP=0 | |
| S | Synergy index | Excess risk from combined exposures relative to the risks from each exposure | S=1 | |
| Multiplicative | ROR | Ratio of odds ratios | Additional risk compared to the expected risk from multiplying risks of each exposure | ROR=1 |
Recent advancements in fine mapping of genetic regions of interest and high throughput next-generation sequencing will result in yet more genetic loci associated with the risk of rheumatic diseases, so these issues are timely. We now provide specific examples of genetic factors, environment factors, and GEIs in RA, SLE, and AS susceptibility.
Rheumatoid arthritis
The study of genetic and environmental risk factors for RA is the most developed among the rheumatic diseases, owing to a relatively homogeneous disease phenotype and high disease prevalence.28 Despite these advantages, differences in the risk factor assessment for RA are appreciated based on RA serologic status.29 Early studies of RA risk factors were stratified by the presence of absence of rheumatoid factor (RF). However, RF positivity may be seen in other diseases or as a consequence of aging.30 Recent studies have utilized anti-citrullinated peptide antibody (ACPA) to classify RA, which is more specific for RA so has less potential for misclassification.31 Despite validated classification criteria for RA, atypical presentations of other systemic rheumatic diseases have the potential to be misclassified as seronegative RA.32, 33
RA genetic risk factors: The shared epitope and beyond
Genetic variants in the major histocompatibility complex (MHC) region on chromosome 6 were identified to be potently associated with RA susceptibility and were deemed the “shared epitope.”34 Polymorphisms in three human leukocyte antigen (HLA) genes (HLA-DRB1, HLA-DPB1, HLA-B) in the MHC region are highly associated with RA.35 Polymorphisms in HLA-DRB1, in particular the classical shared epitope genotypes of *0401 and *0404, are strongly associated with RA (odds ratios of approximately 4 for risk variants).36 Specific amino acid positions (11, 71 and 74) that correspond to the HLA-DRB1 shared epitope classical genotypes are located in the peptide-binding groove of the protein HLA-DRβ1, offering a potential biological mechanism for this potent RA genetic risk factor.35
Large genetic consortia have identified many non-HLA SNPs associated with RA using GWAS.4, 11, 37, 38 Most recently, a large trans-ethnic GWAS associated 101 genetic loci with RA across European and Asian populations.4 However, most of the non-HLA SNPs are only modestly associated with RA.37 An exception is a SNP near the gene PTPN22 (encoding a tyrosine-phosphatase protein expressed in lymphoid tissue), which is strongly associated with RA (odds ratio 1.78).37 SNPs near the genes TNFAIP3 and TYK2 also have relatively large effect sizes for RA risk.17
Most early genetic studies focused on seropositive RA among Caucasian or Japanese populations.4 In a recent large genetic consortium study, Han and colleagues made special efforts to correctly classify ACPA-positive and APCA-negative RA cases by using a highly specific ACPA assay.39 This study identified new genetic risk factors associated with ACPA-negative RA: serine or leucine at amino acid position 11 in HLA-DRB1 and aspartate in position 9 in HLA-B.39 These findings provide further evidence that ACPA-positive and ACPA-negative RA are genetically distinct and thus may have separate pathogeneses.4, 17, 39, 40
Despite the significance of the HLA shared epitope to RA risk and the growing number of non-HLA SNPs associated with RA, most genetic heritability remains unexplained. The HLA shared epitope explains only 12% of the genetic variance for RA, while only about 4% is explained by all other non-HLA SNPs.37 Gene-gene interactions for RA may also be present, in particular an association between the HLA shared epitope and PTPN22, specifically based on presence of the R620W allele, for the development of seropositive RA.41, 42 Studies among familial RA and twins suggest that environmental factors have an important role in the development of RA and RA-related antibodies, so GEIs may be important to link these discoveries.20, 43
RA environmental risk factors: Cigarette smoking and more
Strong epidemiologic evidence supports cigarette smoking as an environmental risk factor for RA development. Multiple studies have shown an increased RA risk with smoking among men and women, in varied populations.44–53 Furthermore, there is a dose-dependent effect between smoking pack-years and RA development.46, 54 After twenty years of smoking cessation, RA risk of former smokers returns to the RA risk of the general population.46 Murine models evaluating cigarette smoke exposure have induced inflammatory arthritis, providing further biological evidence.55 This association is particularly strong for the development of seropositive RA.44 Smoking may contribute up to 25% of RA risk, and up to 35% of risk for ACPA-positive RA.56 However, studies of smoking and seronegative RA have revealed less powerful associations.44, 47
Other environmental factors have been associated with the development of RA. It is beyond the scope of this article to review these factors extensively, but they include occupational exposures such as silica, reproductive factors in women, and excess body mass.57–59 Dietary factors that might be protective for RA include intake of alcohol and fish.60–65
Given the knowledge of a period of preclinical RA autoimmunity where autoantibodies are present in the absence of clinically apparent joint inflammation, there is growing interest that the induction of RA may occur at extra-articular sites. The induction of autoimmunity in RA may specifically occur at mucosal surfaces in the respiratory tract, mouth, or gut.66 Mucosal involvement in the lung has been posited as a possible site for RA initiation due to the increased risk seen in both smoking and particulate silica exposure.44, 62 Citrullination of peptides occurs in the lung due to inflammation, and this is most pronounced in smokers.66, 67 Interstitial lung disease is a well known non-articular manifestation of RA and may occur even without articular involvement. Periodontitis has also been associated with both prevalent and incident RA, although smoking and secondary Sjögren’s syndrome may confound these associations.68, 69 Porphyromonas gingivalis, a causative bacterium for periodontitis, expresses the enzyme peptidylarginine deaminase, which can citrullinate both enolase and fibrinogen, self-antigens thought to be involved in the joint-specificity of RA.70 A study composed of RA first-degree relatives reported higher concentrations of P. gingivalis antibodies in RA relatives who had developed RA-related antibodies, arguing that pre-clinical infection, or its immune response, may be important in the development of altered immunity in RA.71
RA gene-environment interaction: HLA-DRB1-cigarette smoking interaction
The strong association of smoking with RA risk and evidence for induction of autoimmunity and citrullination in mucosa led to a hypothesized biological mechanism for the role of smoking in the development of RA. Studies initially performed by Klareskog and colleagues demonstrated a strongly significant multiplicative GEI between cigarette smoking and HLA-DRB1 in determining RA risk.42, 72–76 This association was strongest for seropositive RA and has been replicated in several populations, including US women, and Swedish, and Malaysian populations.72–74 The presence of heavy smoking and two HLA-DRB1 genes increased the odds for RA by 23-fold compared to those with neither risk factor.42, 73
Transgenic mouse models have demonstrated a strong immune response between HLA-DRβ1 and citrullinated self-antigens implicated in smoking, offering more evidence of biologic plausibility for this GEI.77 Smoking in particular, but other inflammatory exposures as well, are now thought to cause citrullination of a large array of peptides. The specific responses to these citrullinated peptides are likely genetically determined, generating a diversity of ACPAs to different citrullinated antigens, which arise prior to clinical disease onset and have varying pathogenicity. The HLA-smoking interaction may contribute to citrullination processes and autoimmunity and might explain the seroconversion noted in pre-clinical RA (see Figure 2).78, 79 This GEI seems to apply only to seropositive RA, and possibly in particular to those with antibodies to citrullinated α-enolase and vimentin.80 Other genes (GSTT1 and HMOX1) in which polymorphisms might interfere with the metabolism of cigarette smoke are hypothesized to also enhance RA susceptibility.81
Figure 2.
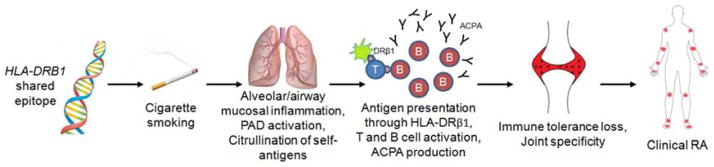
Schematic of HLA-DRB1-cigarette smoking interaction in the development of seropositive RA. Individuals with one or more HLA-DRB1 shared epitope alleles smoke cigarettes, which induces inflammation in the airways and alveolar mucosa. Peptidylarginine deiminase (PAD) is activated, which citrullinates self-antigens. Antigen-presenting cells stimulate T cells through aberrant HLA-DRβ1 interactions and stimulate B cells to produce anti-citrullinated peptide antibodies (ACPA). Immune tolerance is lost specifically in joints, which manifests clinically as synovitis with signs and symptoms compatible with RA.
From Klareskog L, Ronnelid J, Lundberg K, Padyukov L, Alfredsson L. Immunity to citrullinated proteins in rheumatoid arthritis. Annu Rev Immunol 2008;26:651–75; with permissions.
Although HLA-smoking GEIs have led to robust findings about the pathogenesis of ACPA-positive RA among those with both risk factors, they confer little ability to discriminate between RA cases and controls in risk modeling, emphasizing the need for continued research of other biological pathways involved in RA pathogenesis.20, 21
Systemic lupus erythematosus
Due to its elevated morbidity and mortality, there is interest in identifying risk factors for the development of SLE.82 SLE has a heterogeneous clinical phenotype, ranging from mild mucocutaneous or musculoskeletal involvement to severe, life-threatening neurologic or renal manifestations.83 The American College of Rheumatology (ACR) classification criteria for SLE considers these varied manifestations as a single disease entity.84, 85 This presents methodological challenges as different SLE subtypes may have separate pathogeneses and therefore different genetic and environmental risk factors. This further partitions an already rare disease, decreasing the statistical power of even large studies with long follow-up periods. Despite these challenges, specific autoantibody and biomarker profiles offer insight into defining distinct SLE subtypes.
SLE genetic risk factors
Like RA and other autoimmune diseases, HLA genes are thought to have a central role in SLE susceptibility. The haplotypes HLA-DRB1*03:01 and *15:01 are strong genetic risk factors for SLE in European populations.36 GWAS have associated over 40 genetic loci with SLE susceptibility.86 Several genes, such as PTPN22, are implicated in SLE and other autoimmune diseases, RA in particular.87 However, many other genes are specifically associated with SLE susceptibility.88
SLE-associated genetic loci have been categorized according to putative functionality.86 These categories include DNA degradation and cellular debris, immune complex clearance, toll-like receptors, interferon regulation, NF-κB, and regulation of B-cells, T-cells, monocytes and neutrophils.86 The role of DNA repair genes, in particular ATG5, TREX1 and DNASE1, may be of particular interest given the nearly ubiquitous presence of antinuclear antibodies in SLE.86 A wGRS for SLE, composed of 22 SNPs including HLA-DRB1, has been developed and used to calculate GRS of particular SLE subtypes based on validated SNPs for SLE and ACR criteria (such as renal involvement and presence of anti-dsDNA antibodies).22 This wGRS is associated with earlier-onset SLE, consistent with the notion that increased genetic burden may correspond to earlier disease onset.22 Gene-gene interactions may also be present in SLE, specifically between CTLA4, IRF5, and ITGAM with HLA-DRB1 as well as between PDCD1 and IL21, among others.89
SLE environmental risk factors
Smoking and exposure to ultraviolet light have been implicated as possible environmental factors for SLE susceptibility.90–93 Ultraviolet-B light is well known to cause SLE exacerbations and its potential pathogenic mechanism in aberrant apoptosis and the removal of cellular debris make this a key candidate in the etiological role for SLE, particularly for cutaneous manifestations.94 Epstein-Barr (EBV) virus has been suggested to be involved in the pathogenesis of SLE based on high EBV viral loads in pediatric SLE patients compared to controls, with posited molecular mimicry of EBV antigens and autoantigens targeted by lupus autoantibodies.95, 96 However, the association of EBV and risk of SLE remains controversial. Finally, the female predominance in SLE argues strongly for hormonal and reproductive influences. Oral contraceptives and postmenopausal hormones have both been related to increased risk of SLE, in particular among women taking higher doses of ethinyl estradiol.97–100
SLE gene-environment interactions
Unlike RA, there is not yet strong evidence associating particular genetic and environmental factors to SLE susceptibility, which makes GEIs for SLE challenging to identify. However, GEIs for both smoking and ultraviolet-B exposure have been reported.101, 102 In the Carolina Lupus Study, women with null homozygous genotypes for GSTM1 and more than two years of occupational sun exposure had a three-fold increased odds for SLE, with a trend towards statistical significance.102 Other genes involved in DNA-repair (such as ATG5, TREX1, and DNASE1) have not been specifically studied for a GEI with ultraviolet-B light for SLE development. A Japanese case- control study found that women who were smokers and had the slow acetylator N-acetyl transferase-2 (NAT2) genotype had six-fold increased odds of SLE, compared to never smokers with the rapid acetylator form of NAT2. This interaction had a significant additive interaction, an attributable proportion due to interaction of 50%, suggesting that metabolism of oxidants from cigarette smoke may play a role in SLE pathogenesis.101 In addition, genes for toll-like receptors (such as IRF5 and TLR7) may engage microbes inappropriately resulting in SLE autoimmunity. Infections with a number of potential organisms may trigger immune responses that go awry in genetically predisposed individuals leading to SLE autoimmunity, although particular pathogens and interactions have not yet been identified.
Ankylosing spondylitis
The incidence and prevalence of AS varies markedly by geography, perhaps due to the prevalence of HLA-B27 in these populations.103 The identification of at-risk individuals therefore may be comparably easier for AS than other rheumatic diseases based on geography, HLA-B27-positivity, or AS relatives. However, AS classification criteria are still evolving, which makes consistent phenotyping challenging.104, 105 There is considerable clinical overlap between AS and other HLA-B27 associated diseases, such as reactive arthritis, psoriatic arthritis, and inflammatory bowel disease. Inconsistent phenotyping could hinder the identification genetic and environmental associations in AS. Finally, since AS patients often present in adolescence or go undiagnosed for many years, identifying exposure windows prior to disease onset is challenging. Cohort studies used in rheumatic diseases are typically not large enough to detect AS in exposed compared to unexposed and most do not follow children or adolescents, who might go on to develop AS in early adulthood.
AS genetic risk factors: HLA-B27 dominates
Unlike other rheumatic diseases, AS has long been associated with a gene with very large effect size.106 HLA-B27 positivity confers an odds ratio of approximately 90 for developing AS compared to HLA-B27 negative individuals.107 HLA-B27 is present in about 90% of patients with AS, but only about 5% of HLA-B27-positive individuals develop AS or another form of spondyloarthritis.107 This illustrates the difficulty in the clinical implementation of genetic testing for rheumatic disease susceptibility. A recent large GWAS associated 31 genetic loci with AS.108 However, these non-HLA loci are overwhelmed by the influence of HLA-B27. The overall contribution of HLA-B27 to AS heritability is estimated to be 20% and about 4% is due to other loci.108 Other genes implicated in AS risk include IL23R and IL1R2 as well as the intergenic region at 2p15.107 These associations offer insight into the role of cytokines IL-17/IL-23 and IL-1 in AS pathogenesis.
Several possible explanations might explain the striking association of HLA-B27 with AS development. The arthritogenic peptide hypothesis states that similarity between microbial peptides and HLA-B27-specific CD8-positive lymphocytes could induce autoimmunity.109 The heavy chain homodimer hypothesis states that HLA-B27 dimers are resistant to normal degradation and engage natural killer receptors inappropriately and result in autoimmunity.110 Finally, the protein misfolding hypothesis states that unfolded HLA-B27 accumulates in the endoplasmic reticulum and stimulates the release of proinflammatory cytokines is most widely accepted.111, 112
AS environmental risk factors: Microbial and gut influences?
Microbes have been postulated to trigger altered immunity in AS through molecular mimicry. Some suggest this might specifically occur in the gut through the microbiome. The etiologic role of chlamydiae species and enterobacteria in reactive arthritis has been posited to also apply to AS pathogenesis.113, 114 Transgenic HLA-B27 murine models have also demonstrated that the introduction of bacteria is necessary for the development of spondyloarthropathy.115 Inflammation in the gut has been consistently observed in spondyloarthritis.116 Cigarette smoking has also been implicated in AS susceptibility, underscoring its role in multiple inflammatory and autoimmune diseases.117 Unlike most other rheumatic diseases, males are more likely than females to develop AS, so male-specific factors such as testosterone may also be involved in the pathogenesis of AS.118
AS gene-environment interactions: Engaging HLA-B27
Given the large influence of HLA-B27 on AS susceptibility, any GEI will need to have biological plausibility with HLA-B27 functionality.112 In this sense, finding significant GEIs may be more straightforward in AS than in RA and SLE (Table 3). A proteomic analysis of Chlamydia trachomatis identified peptides that interact with HLA-B27 in mouse models and also stimulate T cells from patients with spondyloarthritis.119 The role of environmental factors in AS pathogenesis and why many HLA-B27-positive individuals never develop AS or other forms of spondyloarthritis are yet unsolved.120, 121
Table 3.
Selected genetic, environment, and gene-environment interactions in RA, SLE, and AS.
| Phenotype | MHC gene (variance explained) | Number of non-HLA SNPs associated Ref | Selected non-HLA genes/loci | Key environmental factors | Proposed gene-environment interactions |
|---|---|---|---|---|---|
| RA |
HLA-DRB1 HLA-DPB1 HLA-B (12%) |
1014 |
PTPN22 TNAIF3 TYK2 |
Cigarette smoking Reproductive factors and hormonal exposures Silica Periodontitis Excess body mass Alcohol intake Fish intake |
HLA-DRB1-smoking PTPN22- smoking |
| SLE |
HLA-DRB1 HLA-DPB1 HLA-G |
>4086 |
ATG5 TREX1 DNASE1 IRF5 TLR7 FCGR2A |
Ultraviolet-B light Current cigarette smoking Silica Reproductive factors and hormonal exposures Alcohol intake Epstein-Barr virus |
DNA repair genes-UV-B light and smoking TLR genes-infections |
| AS | HLA-B27 (20%) | 30108 |
IL23R IL1R2 2p15 |
Gut microbiome Cigarette smoking Testosterone |
HLA-B27-infections |
Abbreviations: AS, ankylosing spondylitis; HLA, human leukocyte antigen; MHC, major histocompatibility complex; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; SNPs, single nucleotide polymorphisms; TLR, toll-like receptor; UV-B, ultraviolet-B.
Emerging research strategies
Epigenetics
Epigenetic regulation is thought to be largely responsible for the cell-specific expression of genes. Epigenetic changes are heritable from one cell cycle to the next, potentially reversible, and do not alter nucleotide sequences.122 Biochemical modifications involved in epigenetics include acetylation, histone modifications, methylation, phosphorylation, sumoylation, and microRNA (miRNA).123
Epigenetic modifications provide an important potential link between genes and the environment, since environmental factors influence epigenetic processes, such as methylation and acetylation, while other epigenetic factors are inherited.124 It also adds many layers of complexity to our notions of inheritance of complex disease phenotypes. Epigenetic responses to environmental exposures are likely in part genetically determined themselves and can be inherited as stable traits and act as bona fide epigenetic quantitative trait loci, perhaps responsible for a great deal of unexplained heritability. This leads to added complexity in understanding the interplay between genetic factors and environmental exposures, and epigenetic control and modification and subsequent genetic expression, in determining disease susceptibility.125
Studies performed in rheumatic diseases have mostly focused on inflammatory pathways in distinct immune cell types.126 Demethylation has been linked to drug-induced lupus in humans and murine models.127, 128 Sophisticated techniques are required to systematically identify and isolate cells of the same immunophenotype. The differential expression of epigenetic markers between cell types and innumerable immunophenotypes make population-based epigenetic studies difficult to implement. At present, specific hypotheses, often based on known genetic findings and immune function, are often needed for epigenetic investigations. As the technological aspects of high-throughput epigenetic screening evolve, population-based studies utilizing epigenetics are likely to become increasingly feasible. For example, a study examined epigenetic patterns in over 200 neonates to investigate the roles of genetic and maternal factors.129 Another study performed a genome-wide methylation analysis of peripheral blood mononuclear cells in a cohort of African American women to find and identified over 900 statistically significant loci potentially related to cigarette smoking.130 Methylation patterns have also been implicated as an intermediary of genetic risk seen in ACPA-positive RA.131 Methods for epigenome-wide association studies are currently being refined and may be ready for large-scale utilization in the near future.132
Microbiome
The microbiome refers to the symbiosis and interaction of individuals and their bacterial flora. It has long been recognized that bacteria occupy the mucosal surfaces in the gut, mouth, respiratory system, and skin. Recent findings implicating autoimmunity at these sites have heightened the search for imbalances of the microbiome and rheumatic disease susceptibility. The relationship of microbiota to diseases such as acute rheumatic fever and reactive arthritis provide a precedent for the potential importance of microbes to other rheumatic diseases.
Scher and colleagues examined the stool from RA cases and healthy controls and found a potential role for the bacteria Prevotella copri in RA disease susceptibility.133 While provocative, this study requires replication as factors related to having RA (such as socioeconomic status, healthcare utilization and medications) might explain this association. Studies are currently exploring mechanisms to link specific bacteria in the microbiome and disease susceptibility. Comparisons between high-risk, asymptomatic individuals, the general population, and patients with established disease could elucidate the microbiome’s role in rheumatic disease etiology. Exploration of the microbiome of other mucosal surfaces, such as the mouth and lung, are ongoing in RA and other diseases. High-throughput methods to assess microbiota need to be developed so that large population-based studies can be performed in diverse populations and geographic areas, incorporating knowledge of other environmental factors and host genetics.
Metabolomics
Metabolomics is the study of metabolites and their relationship to disease. Importantly, metabolomics may serve as the bridge between host genetics and environmental exposures. Unlike epigenetics and the microbiome, high-throughput methods involving mass spectrometry already exist for metabolomics.134 However, metabolites levels may be depend on many factors such as sleep, fasting status, comorbidities, and medication usage making study design challenging and sensitive to many variables. Metabolomic studies require sophisticated statistical support to allow for multiple comparisons, mutual adjustments, and principal components analysis.
Despite these qualifications, studies have been performed to investigate metabolomic profiles in rheumatic diseases. Serum metabolite profiles related to lipids and lactate were related to the inflammatory burden in RA patients.135 Urine metabolite profiles in RA patients could distinguish responders from non-responders to tumor necrosis factor inhibitors.136 While provocative, it is unclear whether these methods provide clinically meaningful data beyond currently available biomarkers or whether metabolite dysregulation occurs in the pre-clinical disease period. Identification of novel metabolite biomarkers in the pre-clinical phase may prove to be helpful in identifying individuals at risk for autoimmune rheumatic disease and contribute to the understanding of pathophysiology. Thus, metabolomics may be a powerful tool in identifying biomarker patterns associated with drug response and could influence treatment decisions.
Future considerations and summary
In conclusion, genetic and environmental factors, and interactions between them, some of which may be mediated by epigenetic modification followed by post-translational effects, are associated with the development of rheumatic diseases. The discovery and validation of the HLA-DRB1-smoking interaction in RA is a model among rheumatic diseases for GEI studies. For SLE, genetics have elucidated pathways that may lead to the differentiation of disease subtypes and form hypotheses that integrate environmental risk factors. In AS, potential etiologic mechanisms must consider HLA-B27. Despite these advances, rheumatic disease etiology remains enigmatic. GEIs have and could continue to provide insights into biological mechanisms that link genetic and environmental risk factors. However, much of the risk for rheumatic disease susceptibility remains unexplained. Future research will investigate whether this unexplained risk can be explained by rare genetic variants, epigenetic controls and modifications, novel environmental factors, gene-gene interactions, or GEIs. In addition, understanding which genes may be involved in the development of autoimmunity, and those that lead to progression of disease will be important to evauate in longitudinal studies. Emerging research strategies including epigenetics, metabolomics, and the microbiome will likely further elucidate rheumatic disease susceptibility.
Key points.
Genetic and environmental risk factors have been identified for rheumatic diseases using case-control, cohort, and genome-wide association studies.
The identification of gene-environment interactions (GEIs) may elucidate biological mechanisms for rheumatic diseases, by causally linking established genetic and environmental risk factors.
The most well studied example of GEIs in rheumatic disease susceptibility is for cigarette smoking and the HLA-DRB1 for seropositive RA; the presence of both risk factors greatly increases the risk for RA development.
Due to the relative rarity of systemic lupus erythematosus (SLE), comprehensive studies of GEIs have not yet been performed for (SLE). However, there is some evidence that genes may interact with smoking and ultraviolet-B light exposure in increasing SLE risk.
HLA-B27 is the most potent genetic risk factor for ankylosing spondylitis, and there are suggestions that molecular mimicry by gut microbes might stimulate autoimmunity through GEIs with HLA-B27.
Emerging research frontiers such as epigenetics, metabolomics, and the study of the oral, respiratory, and gastrointestinal microbiome may provide new biologic mechanisms to link genetic and environmental risk factors in rheumatic disease pathogenesis.
Acknowledgments
Funding: This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (grants AR057327 and AR059073 to Dr. Costenbader). The funders had no role in the preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.van Steenbergen HW, Huizinga TW, van der Helm-van Mil AH. The preclinical phase of rheumatoid arthritis: what is acknowledged and what needs to be assessed? Arthritis Rheum. 2013;65(9):2219–32. doi: 10.1002/art.38013. [DOI] [PubMed] [Google Scholar]
- 2.Klareskog L, Alfredsson L, Rantapaa-Dahlqvist S, Berglin E, Stolt P, Padyukov L. What precedes development of rheumatoid arthritis? Ann Rheum Dis. 2004;63(Suppl 2):ii28–ii31. doi: 10.1136/ard.2004.028225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lahiri M, Morgan C, Symmons DP, Bruce IN. Modifiable risk factors for RA: prevention, better than cure? Rheumatology (Oxford) 2012;51(3):499–512. doi: 10.1093/rheumatology/ker299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506(7488):376–81. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi HK, Nguyen US, Niu J, Danaei G, Zhang Y. Selection bias in rheumatic disease research. Nat Rev Rheumatol. 2014 doi: 10.1038/nrrheum.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karlson EW, Deane K. Environmental and gene-environment interactions and risk of rheumatoid arthritis. Rheum Dis Clin North Am. 2012;38(2):405–26. doi: 10.1016/j.rdc.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chernajovsky Y, Winyard PG, Kabouridis PS. Advances in understanding the genetic basis of rheumatoid arthritis and osteoarthritis: implications for therapy. Am J Pharmacogenomics. 2002;2(4):223–34. doi: 10.2165/00129785-200202040-00002. [DOI] [PubMed] [Google Scholar]
- 8.Neale BM, Purcell S. The positives, protocols, and perils of genome-wide association. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(7):1288–94. doi: 10.1002/ajmg.b.30747. [DOI] [PubMed] [Google Scholar]
- 9.Brookes AJ. Rethinking genetic strategies to study complex diseases. Trends Mol Med. 2001;7(11):512–6. doi: 10.1016/s1471-4914(01)02163-3. [DOI] [PubMed] [Google Scholar]
- 10.Raychaudhuri S, Remmers EF, Lee AT, Hackett R, Guiducci C, Burtt NP, et al. Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat Genet. 2008;40(10):1216–23. doi: 10.1038/ng.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42(6):508–14. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plenge RM, Seielstad M, Padyukov L, Lee AT, Remmers EF, Ding B, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis--a genomewide study. N Engl J Med. 2007;357(12):1199–209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Webb R, Kelly JA, Somers EC, Hughes T, Kaufman KM, Sanchez E, et al. Early disease onset is predicted by a higher genetic risk for lupus and is associated with a more severe phenotype in lupus patients. Ann Rheum Dis. 2011;70(1):151–6. doi: 10.1136/ard.2010.141697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott IC, Seegobin SD, Steer S, Tan R, Forabosco P, Hinks A, et al. Predicting the Risk of Rheumatoid Arthritis and Its Age of Onset through Modelling Genetic Risk Variants with Smoking. PLoS Genet. 2013;9(9):e1003808. doi: 10.1371/journal.pgen.1003808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110–21. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 16.Hayes B. Overview of Statistical Methods for Genome-Wide Association Studies (GWAS) Methods Mol Biol. 2013;1019:149–69. doi: 10.1007/978-1-62703-447-0_6. [DOI] [PubMed] [Google Scholar]
- 17.Viatte S, Plant D, Raychaudhuri S. Genetics and epigenetics of rheumatoid arthritis. Nat Rev Rheumatol. 2013;9(3):141–53. doi: 10.1038/nrrheum.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horne BD, Anderson JL, Carlquist JF, Muhlestein JB, Renlund DG, Bair TL, et al. Generating genetic risk scores from intermediate phenotypes for use in association studies of clinically significant endpoints. Ann Hum Genet. 2005;69(Pt 2):176–86. doi: 10.1046/j.1529-8817.2005.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karlson EW, Chibnik LB, Kraft P, Cui J, Keenan BT, Ding B, et al. Cumulative association of 22 genetic variants with seropositive rheumatoid arthritis risk. Ann Rheum Dis. 2010;69(6):1077–85. doi: 10.1136/ard.2009.120170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sparks JA, Chen C, Jiang X, Hiraki LT, Klareskog L, Alfredsson L, et al. Improved performance of epidemiologic and genetic risk models for rheumatoid arthritis serologic phenotypes using family history. Ann Rheum Dis. 2014 doi: 10.1136/annrheumdis-2013-205009. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karlson EW, Ding B, Keenan BT, Liao K, Costenbader KH, Klareskog L, et al. Association of environmental and genetic factors and gene-environment interactions with risk of developing rheumatoid arthritis. Arthritis Care Res (Hoboken) 2013;65(7):1147–56. doi: 10.1002/acr.22005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor KE, Chung SA, Graham RR, Ortmann WA, Lee AT, Langefeld CD, et al. Risk alleles for systemic lupus erythematosus in a large case-control collection and associations with clinical subphenotypes. PLoS Genet. 2011;7(2):e1001311. doi: 10.1371/journal.pgen.1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kraft P, Hunter D. Integrating epidemiology and genetic association: the challenge of gene-environment interaction. Philos Trans R Soc Lond B Biol Sci. 2005;360(1460):1609–16. doi: 10.1098/rstb.2005.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karlson EW, Costenbader KH. Epidemiology: Interpreting studies of interactions between RA risk factors. Nat Rev Rheumatol. 2010;6(2):72–3. doi: 10.1038/nrrheum.2009.276. [DOI] [PubMed] [Google Scholar]
- 25.Aschard H, Chen J, Cornelis MC, Chibnik LB, Karlson EW, Kraft P. Inclusion of gene-gene and gene-environment interactions unlikely to dramatically improve risk prediction for complex diseases. Am J Hum Genet. 2012;90(6):962–72. doi: 10.1016/j.ajhg.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milne RL, Gaudet MM, Spurdle AB, Fasching PA, Couch FJ, Benitez J, et al. Assessing interactions between the associations of common genetic susceptibility variants, reproductive history and body mass index with breast cancer risk in the breast cancer association consortium: a combined case-control study. Breast Cancer Res. 2010;12(6):R110. doi: 10.1186/bcr2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knol MJ, van der Tweel I, Grobbee DE, Numans ME, Geerlings MI. Estimating interaction on an additive scale between continuous determinants in a logistic regression model. Int J Epidemiol. 2007;36(5):1111–8. doi: 10.1093/ije/dym157. [DOI] [PubMed] [Google Scholar]
- 28.Crowson CS, Matteson EL, Myasoedova E, Michet CJ, Ernste FC, Warrington KJ, et al. The lifetime risk of adult-onset rheumatoid arthritis and other inflammatory autoimmune rheumatic diseases. Arthritis Rheum. 2011;63(3):633–9. doi: 10.1002/art.30155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gabriel SE, Crowson CS, O’Fallon WM. The epidemiology of rheumatoid arthritis in Rochester, Minnesota, 1955–1985. Arthritis Rheum. 1999;42(3):415–20. doi: 10.1002/1529-0131(199904)42:3<415::AID-ANR4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 30.Tighe H, Carson DA. Rheumatoid factor. In: Ruddy S, Harris ED, Sledge C, editors. Kelley’s textbook of rheumatology. W.B. Saunders Company; Philadelphia: 2001. pp. 151–61. [Google Scholar]
- 31.Schellekens GA, Visser H, de Jong BA, van den Hoogen FH, Hazes JM, Breedveld FC, et al. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000;43(1):155–63. doi: 10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 32.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, 3rd, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9):2569–81. doi: 10.1002/art.27584. [DOI] [PubMed] [Google Scholar]
- 33.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31(3):315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 34.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30(11):1205–13. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 35.Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44(3):291–6. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernando MM, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4(4):e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eyre S, Bowes J, Diogo D, Lee A, Barton A, Martin P, et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet. 2012;44(12):1336–40. doi: 10.1038/ng.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plenge RM, Cotsapas C, Davies L, Price AL, de Bakker PI, Maller J, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39(12):1477–82. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han B, Diogo D, Eyre S, Kallberg H, Zhernakova A, Bowes J, et al. Fine Mapping Seronegative and Seropositive Rheumatoid Arthritis to Shared and Distinct HLA Alleles by Adjusting for the Effects of Heterogeneity. Am J Hum Genet. 2014;94(4):522–32. doi: 10.1016/j.ajhg.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurreeman F, Liao K, Chibnik L, Hickey B, Stahl E, Gainer V, et al. Genetic basis of autoantibody positive and negative rheumatoid arthritis risk in a multi-ethnic cohort derived from electronic health records. Am J Hum Genet. 2011;88(1):57–69. doi: 10.1016/j.ajhg.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morgan AW, Thomson W, Martin SG, Carter AM, Erlich HA, Barton A, et al. Reevaluation of the interaction between HLA-DRB1 shared epitope alleles, PTPN22, and smoking in determining susceptibility to autoantibody-positive and autoantibody-negative rheumatoid arthritis in a large UK Caucasian population. Arthritis Rheum. 2009;60(9):2565–76. doi: 10.1002/art.24752. [DOI] [PubMed] [Google Scholar]
- 42.Kallberg H, Padyukov L, Plenge RM, Ronnelid J, Gregersen PK, van der Helm-van Mil AH, et al. Gene-gene and gene-environment interactions involving HLA-DRB1, PTPN22, and smoking in two subsets of rheumatoid arthritis. Am J Hum Genet. 2007;80(5):867–75. doi: 10.1086/516736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haj Hensvold A, Magnusson PK, Joshua V, Hansson M, Israelsson L, Ferreira R, et al. Environmental and genetic factors in the development of anticitrullinated protein antibodies (ACPAs) and ACPA-positive rheumatoid arthritis: an epidemiological investigation in twins. Ann Rheum Dis. 2013 doi: 10.1136/annrheumdis-2013-203947. [DOI] [PubMed] [Google Scholar]
- 44.Sugiyama D, Nishimura K, Tamaki K, Tsuji G, Nakazawa T, Morinobu A, et al. Impact of smoking as a risk factor for developing rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis. 2010;69(1):70–81. doi: 10.1136/ard.2008.096487. [DOI] [PubMed] [Google Scholar]
- 45.Criswell LA, Merlino LA, Cerhan JR, Mikuls TR, Mudano AS, Burma M, et al. Cigarette smoking and the risk of rheumatoid arthritis among postmenopausal women: results from the Iowa Women’s Health Study. Am J Med. 2002;112(6):465–71. doi: 10.1016/s0002-9343(02)01051-3. [DOI] [PubMed] [Google Scholar]
- 46.Costenbader KH, Feskanich D, Mandl LA, Karlson EW. Smoking intensity, duration, and cessation, and the risk of rheumatoid arthritis in women. Am J Med. 2006;119(6):503 e1–9. doi: 10.1016/j.amjmed.2005.09.053. [DOI] [PubMed] [Google Scholar]
- 47.Yahya A, Bengtsson C, Lai TC, Larsson PT, Mustafa AN, Abdullah NA, et al. Smoking is associated with an increased risk of developing ACPA-positive but not ACPA-negative rheumatoid arthritis in Asian populations: evidence from the Malaysian MyEIRA case-control study. Mod Rheumatol. 2012;22(4):524–31. doi: 10.1007/s10165-011-0544-2. [DOI] [PubMed] [Google Scholar]
- 48.Vessey MP, Villard-Mackintosh L, Yeates D. Oral contraceptives, cigarette smoking and other factors in relation to arthritis. Contraception. 1987;35(5):457–64. doi: 10.1016/0010-7824(87)90082-5. [DOI] [PubMed] [Google Scholar]
- 49.Hernandez Avila M, Liang MH, Willett WC, Stampfer MJ, Colditz GA, Rosner B, et al. Reproductive factors, smoking, and the risk for rheumatoid arthritis. Epidemiology. 1990;1(4):285–91. doi: 10.1097/00001648-199007000-00005. [DOI] [PubMed] [Google Scholar]
- 50.Hazes JM, Dijkmans BA, Vandenbroucke JP, de Vries RR, Cats A. Lifestyle and the risk of rheumatoid arthritis: cigarette smoking and alcohol consumption. Ann Rheum Dis. 1990;49(12):980–2. doi: 10.1136/ard.49.12.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heliovaara M, Aho K, Aromaa A, Knekt P, Reunanen A. Smoking and risk of rheumatoid arthritis. J Rheumatol. 1993;20(11):1830–5. [PubMed] [Google Scholar]
- 52.Karlson EW, Lee IM, Cook NR, Manson JE, Buring JE, Hennekens CH. A retrospective cohort study of cigarette smoking and risk of rheumatoid arthritis in female health professionals. Arthritis Rheum. 1999;42(5):910–7. doi: 10.1002/1529-0131(199905)42:5<910::AID-ANR9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 53.Uhlig T, Hagen KB, Kvien TK. Current tobacco smoking, formal education, and the risk of rheumatoid arthritis. J Rheumatol. 1999;26(1):47–54. [PubMed] [Google Scholar]
- 54.Di Giuseppe D, Orsini N, Alfredsson L, Askling J, Wolk A. Cigarette smoking and smoking cessation in relation to risk of rheumatoid arthritis in women. Arthritis Res Ther. 2013;15(2):R56. doi: 10.1186/ar4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okamoto S, Adachi M, Chujo S, Yamada K, Akita K, Itoh S, et al. Etiological role of cigarette smoking in rheumatoid arthritis: Nasal exposure to cigarette smoke condensate extracts augments the development of collagen-induced arthritis in mice. Biochem Biophys Res Commun. 2011;404(4):1088–92. doi: 10.1016/j.bbrc.2010.12.118. [DOI] [PubMed] [Google Scholar]
- 56.Kallberg H, Ding B, Padyukov L, Bengtsson C, Ronnelid J, Klareskog L, et al. Smoking is a major preventable risk factor for rheumatoid arthritis: estimations of risks after various exposures to cigarette smoke. Ann Rheum Dis. 2011;70(3):508–11. doi: 10.1136/ard.2009.120899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wesley A, Bengtsson C, Elkan AC, Klareskog L, Alfredsson L, Wedren S. Association between body mass index and anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis: results from a population-based case-control study. Arthritis Care Res (Hoboken) 2013;65(1):107–12. doi: 10.1002/acr.21749. [DOI] [PubMed] [Google Scholar]
- 58.Shapiro JA, Koepsell TD, Voigt LF, Dugowson CE, Kestin M, Nelson JL. Diet and rheumatoid arthritis in women: a possible protective effect of fish consumption. Epidemiology. 1996;7(3):256–63. doi: 10.1097/00001648-199605000-00007. [DOI] [PubMed] [Google Scholar]
- 59.Symmons DP, Bankhead CR, Harrison BJ, Brennan P, Barrett EM, Scott DG, et al. Blood transfusion, smoking, and obesity as risk factors for the development of rheumatoid arthritis: results from a primary care-based incident case-control study in Norfolk, England. Arthritis Rheum. 1997;40(11):1955–61. doi: 10.1002/art.1780401106. [DOI] [PubMed] [Google Scholar]
- 60.Karlson EW, Mandl LA, Hankinson SE, Grodstein F. Do breast-feeding and other reproductive factors influence future risk of rheumatoid arthritis? Results from the Nurses’ Health Study. Arthritis Rheum. 2004;50(11):3458–67. doi: 10.1002/art.20621. [DOI] [PubMed] [Google Scholar]
- 61.Orellana C, Wedren S, Kallberg H, Holmqvist M, Karlson EW, Alfredsson L, et al. Parity and the risk of developing rheumatoid arthritis: results from the Swedish Epidemiological Investigation of Rheumatoid Arthritis study. Ann Rheum Dis. 2013 doi: 10.1136/annrheumdis-2013-203567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stolt P, Kallberg H, Lundberg I, Sjogren B, Klareskog L, Alfredsson L. Silica exposure is associated with increased risk of developing rheumatoid arthritis: results from the Swedish EIRA study. Ann Rheum Dis. 2005;64(4):582–6. doi: 10.1136/ard.2004.022053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stolt P, Yahya A, Bengtsson C, Kallberg H, Ronnelid J, Lundberg I, et al. Silica exposure among male current smokers is associated with a high risk of developing ACPA-positive rheumatoid arthritis. Ann Rheum Dis. 2010;69(6):1072–6. doi: 10.1136/ard.2009.114694. [DOI] [PubMed] [Google Scholar]
- 64.Rosell M, Wesley AM, Rydin K, Klareskog L, Alfredsson L. Dietary fish and fish oil and the risk of rheumatoid arthritis. Epidemiology. 2009;20(6):896–901. doi: 10.1097/EDE.0b013e3181b5f0ce. [DOI] [PubMed] [Google Scholar]
- 65.Di Giuseppe D, Wallin A, Bottai M, Askling J, Wolk A. Long-term intake of dietary long-chain n-3 polyunsaturated fatty acids and risk of rheumatoid arthritis: a prospective cohort study of women. Ann Rheum Dis. 2013 doi: 10.1136/annrheumdis-2013-203338. [DOI] [PubMed] [Google Scholar]
- 66.Demoruelle MK, Deane KD, Holers VM. When and where does inflammation begin in rheumatoid arthritis? Curr Opin Rheumatol. 2014;26(1):64–71. doi: 10.1097/BOR.0000000000000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Makrygiannakis D, Hermansson M, Ulfgren AK, Nicholas AP, Zendman AJ, Eklund A, et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis. 2008;67(10):1488–92. doi: 10.1136/ard.2007.075192. [DOI] [PubMed] [Google Scholar]
- 68.de Pablo P, Dietrich T, McAlindon TE. Association of periodontal disease and tooth loss with rheumatoid arthritis in the US population. J Rheumatol. 2008;35(1):70–6. [PubMed] [Google Scholar]
- 69.Chen HH, Huang N, Chen YM, Chen TJ, Chou P, Lee YL, et al. Association between a history of periodontitis and the risk of rheumatoid arthritis: a nationwide, population-based, case-control study. Ann Rheum Dis. 2013;72(7):1206–11. doi: 10.1136/annrheumdis-2012-201593. [DOI] [PubMed] [Google Scholar]
- 70.Mangat P, Wegner N, Venables PJ, Potempa J. Bacterial and human peptidylarginine deiminases: targets for inhibiting the autoimmune response in rheumatoid arthritis? Arthritis Res Ther. 2010;12(3):209. doi: 10.1186/ar3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mikuls TR, Thiele GM, Deane KD, Payne JB, O’Dell JR, Yu F, et al. Porphyromonas gingivalis and disease-related autoantibodies in individuals at increased risk of rheumatoid arthritis. Arthritis Rheum. 2012;64(11):3522–30. doi: 10.1002/art.34595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Too CL, Yahya A, Murad S, Dhaliwal JS, Larsson PT, Muhamad NA, et al. Smoking interacts with HLA-DRB1 shared epitope in the development of anti-citrullinated protein antibody-positive rheumatoid arthritis: results from the Malaysian Epidemiological Investigation of Rheumatoid Arthritis (MyEIRA) Arthritis Res Ther. 2012;14(2):R89. doi: 10.1186/ar3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Karlson EW, Chang SC, Cui J, Chibnik LB, Fraser PA, De Vivo I, et al. Gene-environment interaction between HLA-DRB1 shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis. Ann Rheum Dis. 2010;69(1):54–60. doi: 10.1136/ard.2008.102962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Klareskog L, Stolt P, Lundberg K, Kallberg H, Bengtsson C, Grunewald J, et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006;54(1):38–46. doi: 10.1002/art.21575. [DOI] [PubMed] [Google Scholar]
- 75.Lundstrom E, Kallberg H, Alfredsson L, Klareskog L, Padyukov L. Gene-environment interaction between the DRB1 shared epitope and smoking in the risk of anti-citrullinated protein antibody-positive rheumatoid arthritis: all alleles are important. Arthritis Rheum. 2009;60(6):1597–603. doi: 10.1002/art.24572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Klareskog L, Padyukov L, Ronnelid J, Alfredsson L. Genes, environment and immunity in the development of rheumatoid arthritis. Curr Opin Immunol. 2006;18(6):650–5. doi: 10.1016/j.coi.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 77.Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E. Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol. 2003;171(2):538–41. doi: 10.4049/jimmunol.171.2.538. [DOI] [PubMed] [Google Scholar]
- 78.Klareskog L, Malmstrom V, Lundberg K, Padyukov L, Alfredsson L. Smoking, citrullination and genetic variability in the immunopathogenesis of rheumatoid arthritis. Semin Immunol. 2011;23(2):92–8. doi: 10.1016/j.smim.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 79.Mahdi H, Fisher BA, Kallberg H, Plant D, Malmstrom V, Ronnelid J, et al. Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat Genet. 2009;41(12):1319–24. doi: 10.1038/ng.480. [DOI] [PubMed] [Google Scholar]
- 80.Lundberg K, Bengtsson C, Kharlamova N, Reed E, Jiang X, Kallberg H, et al. Genetic and environmental determinants for disease risk in subsets of rheumatoid arthritis defined by the anticitrullinated protein/peptide antibody fine specificity profile. Ann Rheum Dis. 2013;72(5):652–8. doi: 10.1136/annrheumdis-2012-201484. [DOI] [PubMed] [Google Scholar]
- 81.Keenan BT, Chibnik LB, Cui J, Ding B, Padyukov L, Kallberg H, et al. Effect of interactions of glutathione S-transferase T1, M1, and P1 and HMOX1 gene promoter polymorphisms with heavy smoking on the risk of rheumatoid arthritis. Arthritis Rheum. 2010;62(11):3196–210. doi: 10.1002/art.27639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Urowitz MB, Gladman DD, Tom BD, Ibanez D, Farewell VT. Changing patterns in mortality and disease outcomes for patients with systemic lupus erythematosus. J Rheumatol. 2008;35(11):2152–8. doi: 10.3899/jrheum.080214. [DOI] [PubMed] [Google Scholar]
- 83.Von Feldt JM. Systemic lupus erythematosus. Recognizing its various presentations. Postgrad Med. 1995;97(4):79, 83. 6 passim. [PubMed] [Google Scholar]
- 84.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 85.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25(11):1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 86.Rullo OJ, Tsao BP. Recent insights into the genetic basis of systemic lupus erythematosus. Ann Rheum Dis. 2013;72(Suppl 2):ii56–61. doi: 10.1136/annrheumdis-2012-202351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chung SA, Criswell LA. PTPN22: its role in SLE and autoimmunity. Autoimmunity. 2007;40(8):582–90. doi: 10.1080/08916930701510848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Suarez-Gestal M, Calaza M, Dieguez-Gonzalez R, Perez-Pampin E, Pablos JL, Navarro F, et al. Rheumatoid arthritis does not share most of the newly identified systemic lupus erythematosus genetic factors. Arthritis Rheum. 2009;60(9):2558–64. doi: 10.1002/art.24748. [DOI] [PubMed] [Google Scholar]
- 89.Hughes T, Adler A, Kelly JA, Kaufman KM, Williams AH, Langefeld CD, et al. Evidence for gene-gene epistatic interactions among susceptibility loci for systemic lupus erythematosus. Arthritis Rheum. 2012;64(2):485–92. doi: 10.1002/art.33354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Borchers AT, Naguwa SM, Shoenfeld Y, Gershwin ME. The geoepidemiology of systemic lupus erythematosus. Autoimmun Rev. 2010;9(5):A277–87. doi: 10.1016/j.autrev.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 91.Costenbader KH, Kim DJ, Peerzada J, Lockman S, Nobles-Knight D, Petri M, et al. Cigarette smoking and the risk of systemic lupus erythematosus: a meta-analysis. Arthritis Rheum. 2004;50(3):849–57. doi: 10.1002/art.20049. [DOI] [PubMed] [Google Scholar]
- 92.Bengtsson AA, Rylander L, Hagmar L, Nived O, Sturfelt G. Risk factors for developing systemic lupus erythematosus: a case-control study in southern Sweden. Rheumatology (Oxford) 2002;41(5):563–71. doi: 10.1093/rheumatology/41.5.563. [DOI] [PubMed] [Google Scholar]
- 93.Cooper GS, Wither J, Bernatsky S, Claudio JO, Clarke A, Rioux JD, et al. Occupational and environmental exposures and risk of systemic lupus erythematosus: silica, sunlight, solvents. Rheumatology (Oxford) 2010;49(11):2172–80. doi: 10.1093/rheumatology/keq214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Barbhaiya M, Costenbader KH. Ultraviolet Radiation and Systemic Lupus Erythematosus. Lupus. 2014 doi: 10.1177/0961203314530488. In press. [DOI] [PubMed] [Google Scholar]
- 95.Kang I, Quan T, Nolasco H, Park SH, Hong MS, Crouch J, et al. Defective control of latent Epstein-Barr virus infection in systemic lupus erythematosus. J Immunol. 2004;172(2):1287–94. doi: 10.4049/jimmunol.172.2.1287. [DOI] [PubMed] [Google Scholar]
- 96.Poole BD, Scofield RH, Harley JB, James JA. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity. 2006;39(1):63–70. doi: 10.1080/08916930500484849. [DOI] [PubMed] [Google Scholar]
- 97.Cooper GS, Dooley MA, Treadwell EL, St Clair EW, Gilkeson GS. Hormonal and reproductive risk factors for development of systemic lupus erythematosus: results of a population-based, case-control study. Arthritis Rheum. 2002;46(7):1830–9. doi: 10.1002/art.10365. [DOI] [PubMed] [Google Scholar]
- 98.Simard JF, Costenbader KH. What can epidemiology tell us about systemic lupus erythematosus? Int J Clin Pract. 2007;61(7):1170–80. doi: 10.1111/j.1742-1241.2007.01434.x. [DOI] [PubMed] [Google Scholar]
- 99.Costenbader KH, Feskanich D, Stampfer MJ, Karlson EW. Reproductive and menopausal factors and risk of systemic lupus erythematosus in women. Arthritis Rheum. 2007;56(4):1251–62. doi: 10.1002/art.22510. [DOI] [PubMed] [Google Scholar]
- 100.Bernier MO, Mikaeloff Y, Hudson M, Suissa S. Combined oral contraceptive use and the risk of systemic lupus erythematosus. Arthritis Rheum. 2009;61(4):476–81. doi: 10.1002/art.24398. [DOI] [PubMed] [Google Scholar]
- 101.Kiyohara C, Washio M, Horiuchi T, Tada Y, Asami T, Ide S, et al. Cigarette smoking, N-acetyltransferase 2 polymorphisms and systemic lupus erythematosus in a Japanese population. Lupus. 2009;18(7):630–8. doi: 10.1177/0961203309102809. [DOI] [PubMed] [Google Scholar]
- 102.Fraser PA, Ding WZ, Mohseni M, Treadwell EL, Dooley MA, St Clair EW, et al. Glutathione S-transferase M null homozygosity and risk of systemic lupus erythematosus associated with sun exposure: a possible gene-environment interaction for autoimmunity. J Rheumatol. 2003;30(2):276–82. [PubMed] [Google Scholar]
- 103.Shapira Y, Agmon-Levin N, Shoenfeld Y. Geoepidemiology of autoimmune rheumatic diseases. Nat Rev Rheumatol. 2010;6(8):468–76. doi: 10.1038/nrrheum.2010.86. [DOI] [PubMed] [Google Scholar]
- 104.van der Linden S, Valkenburg HA, Cats A. Evaluation of diagnostic criteria for ankylosing spondylitis. A proposal for modification of the New York criteria. Arthritis Rheum. 1984;27(4):361–8. doi: 10.1002/art.1780270401. [DOI] [PubMed] [Google Scholar]
- 105.Sieper J, Rudwaleit M, Baraliakos X, Brandt J, Braun J, Burgos-Vargas R, et al. The Assessment of SpondyloArthritis international Society (ASAS) handbook: a guide to assess spondyloarthritis. Ann Rheum Dis. 2009;68(Suppl 2):ii1–44. doi: 10.1136/ard.2008.104018. [DOI] [PubMed] [Google Scholar]
- 106.Schlosstein L, Terasaki PI, Bluestone R, Pearson CM. High association of an HL-A antigen, W27, with ankylosing spondylitis. N Engl J Med. 1973;288(14):704–6. doi: 10.1056/NEJM197304052881403. [DOI] [PubMed] [Google Scholar]
- 107.Reveille JD. Genetics of spondyloarthritis--beyond the MHC. Nat Rev Rheumatol. 2012;8(5):296–304. doi: 10.1038/nrrheum.2012.41. [DOI] [PubMed] [Google Scholar]
- 108.Cortes A, Hadler J, Pointon JP, Robinson PC, Karaderi T, Leo P, et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet. 2013;45(7):730–8. doi: 10.1038/ng.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sorrentino R, Bockmann RA, Fiorillo MT. HLA-B27 and antigen presentation: at the crossroads between immune defense and autoimmunity. Mol Immunol. 2014;57(1):22–7. doi: 10.1016/j.molimm.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 110.Payeli SK, Kollnberger S, Marroquin Belaunzaran O, Thiel M, McHugh K, Giles J, et al. Inhibiting HLA-B27 homodimer-driven immune cell inflammation in spondylarthritis. Arthritis Rheum. 2012;64(10):3139–49. doi: 10.1002/art.34538. [DOI] [PubMed] [Google Scholar]
- 111.Mear JP, Schreiber KL, Munz C, Zhu X, Stevanovic S, Rammensee HG, et al. Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J Immunol. 1999;163(12):6665–70. [PubMed] [Google Scholar]
- 112.Colbert RA, DeLay ML, Klenk EI, Layh-Schmitt G. From HLA-B27 to spondyloarthritis: a journey through the ER. Immunol Rev. 2010;233(1):181–202. doi: 10.1111/j.0105-2896.2009.00865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Carter JD, Gerard HC, Espinoza LR, Ricca LR, Valeriano J, Snelgrove J, et al. Chlamydiae as etiologic agents in chronic undifferentiated spondylarthritis. Arthritis Rheum. 2009;60(5):1311–6. doi: 10.1002/art.24431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nickerson CL, Luthra HS, David CS. Role of enterobacteria and HLA-B27 in spondyloarthropathies: studies with transgenic mice. Ann Rheum Dis. 1990;49 (Suppl 1):426–33. [PubMed] [Google Scholar]
- 115.Berthelot JM, Glemarec J, Guillot P, Laborie Y, Maugars Y. New pathogenic hypotheses for spondyloarthropathies. Joint Bone Spine. 2002;69(2):114–22. doi: 10.1016/s1297-319x(02)00371-8. [DOI] [PubMed] [Google Scholar]
- 116.Van Praet L, Van den Bosch F, Mielants H, Elewaut D. Mucosal inflammation in spondylarthritides: past, present, and future. Curr Rheumatol Rep. 2011;13(5):409–15. doi: 10.1007/s11926-011-0198-2. [DOI] [PubMed] [Google Scholar]
- 117.Wendling D, Prati C. Spondyloarthritis and smoking: towards a new insight into the disease. Expert Rev Clin Immunol. 2013;9(6):511–6. doi: 10.1586/eci.13.35. [DOI] [PubMed] [Google Scholar]
- 118.Gooren LJ, Giltay EJ, van Schaardenburg D, Dijkmans BA. Gonadal and adrenal sex steroids in ankylosing spondylitis. Rheum Dis Clin North Am. 2000;26(4):969–87. doi: 10.1016/s0889-857x(05)70179-4. [DOI] [PubMed] [Google Scholar]
- 119.Kuon W, Holzhutter HG, Appel H, Grolms M, Kollnberger S, Traeder A, et al. Identification of HLA-B27-restricted peptides from the Chlamydia trachomatis proteome with possible relevance to HLA-B27-associated diseases. J Immunol. 2001;167(8):4738–46. doi: 10.4049/jimmunol.167.8.4738. [DOI] [PubMed] [Google Scholar]
- 120.Hjelholt A, Carlsen T, Deleuran B, Jurik AG, Schiottz-Christensen B, Christiansen G, et al. Increased levels of IgG antibodies against human HSP60 in patients with spondyloarthritis. PLoS One. 2013;8(2):e56210. doi: 10.1371/journal.pone.0056210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Feng XG, Xu XJ, Ye S, Lin YY, Chen P, Zhang XJ, et al. Recent Chlamydia pneumoniae infection is highly associated with active ankylosing spondylitis in a Chinese cohort. Scand J Rheumatol. 2011;40(4):289–91. doi: 10.3109/03009742.2011.560891. [DOI] [PubMed] [Google Scholar]
- 122.Costenbader KH, Gay S, Alarcon-Riquelme ME, Iaccarino L, Doria A. Genes, epigenetic regulation and environmental factors: which is the most relevant in developing autoimmune diseases? Autoimmun Rev. 2012;11(8):604–9. doi: 10.1016/j.autrev.2011.10.022. [DOI] [PubMed] [Google Scholar]
- 123.Brooks WH, Le Dantec C, Pers JO, Youinou P, Renaudineau Y. Epigenetics and autoimmunity. J Autoimmun. 2010;34(3):J207–19. doi: 10.1016/j.jaut.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 124.Ballestar E, Esteller M, Richardson BC. The epigenetic face of systemic lupus erythematosus. J Immunol. 2006;176(12):7143–7. doi: 10.4049/jimmunol.176.12.7143. [DOI] [PubMed] [Google Scholar]
- 125.Cortijo S, Wardenaar R, Colome-Tatche M, Gilly A, Etcheverry M, Labadie K, et al. Mapping the epigenetic basis of complex traits. Science. 2014;343(6175):1145–8. doi: 10.1126/science.1248127. [DOI] [PubMed] [Google Scholar]
- 126.Garaud S, Le Dantec C, Jousse-Joulin S, Hanrotel-Saliou C, Saraux A, Mageed RA, et al. IL-6 modulates CD5 expression in B cells from patients with lupus by regulating DNA methylation. J Immunol. 2009;182(9):5623–32. doi: 10.4049/jimmunol.0802412. [DOI] [PubMed] [Google Scholar]
- 127.Lu Q, Wu A, Richardson BC. Demethylation of the same promoter sequence increases CD70 expression in lupus T cells and T cells treated with lupus-inducing drugs. J Immunol. 2005;174(10):6212–9. doi: 10.4049/jimmunol.174.10.6212. [DOI] [PubMed] [Google Scholar]
- 128.Lu Q, Kaplan M, Ray D, Zacharek S, Gutsch D, Richardson B. Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis Rheum. 2002;46(5):1282–91. doi: 10.1002/art.10234. [DOI] [PubMed] [Google Scholar]
- 129.Teh AL, Pan H, Chen L, Ong ML, Dogra S, Wong J, et al. The effect of genotype and in utero environment on inter-individual variation in neonate DNA methylomes. Genome Res. 2014 doi: 10.1101/gr.171439.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dogan MV, Shields B, Cutrona C, Gao L, Gibbons FX, Simons R, et al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics. 2014;15:151. doi: 10.1186/1471-2164-15-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol. 2013;31(2):142–7. doi: 10.1038/nbt.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu Y, Li X, Aryee MJ, Ekstrom TJ, Padyukov L, Klareskog L, et al. GeMes, clusters of DNA methylation under genetic control, can inform genetic and epigenetic analysis of disease. Am J Hum Genet. 2014;94(4):485–95. doi: 10.1016/j.ajhg.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202. doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Scrivo R, Casadei L, Valerio M, Priori R, Valesini G, Manetti C. Metabolomics approach in allergic and rheumatic diseases. Curr Allergy Asthma Rep. 2014;14(6):445. doi: 10.1007/s11882-014-0445-5. [DOI] [PubMed] [Google Scholar]
- 135.Young SP, Kapoor SR, Viant MR, Byrne JJ, Filer A, Buckley CD, et al. The impact of inflammation on metabolomic profiles in patients with arthritis. Arthritis Rheum. 2013;65(8):2015–23. doi: 10.1002/art.38021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kapoor SR, Filer A, Fitzpatrick MA, Fisher BA, Taylor PC, Buckley CD, et al. Metabolic profiling predicts response to anti-tumor necrosis factor alpha therapy in patients with rheumatoid arthritis. Arthritis Rheum. 2013;65(6):1448–56. doi: 10.1002/art.37921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Klareskog L, Ronnelid J, Lundberg K, Padyukov L, Alfredsson L. Immunity to citrullinated proteins in rheumatoid arthritis. Annu Rev Immunol. 2008;26:651–75. doi: 10.1146/annurev.immunol.26.021607.090244. [DOI] [PubMed] [Google Scholar]