

Abstract
AIM: To study the role of gastric mucosal ascorbic acid (AA) in the progression of acute gastric mucosal lesions induced by compound 48/80 (C48/80), a mast cell degranulator, in rats.
METHODS: C48/80 (0.75 mg/kg) was intraperitoneally injected to fasted Wistar rats. Oral administration of AA (10, 50 or 100 mg/kg) was performed 0.5 h after C48/80 treatment. Determinations for gastric mucosal lesion severity and blood flow, and assays for gastric mucosal total AA, reduced AA, oxidized AA, vitamin E, thiobarbituric acid reactive substances (TBARS), adherent mucus, nitrite/nitrate (NOx), non-protein SH (NPSH), and myeloperoxidase (MPO), and serum total AA, reduced AA, oxidized AA, and NOx were conducted 0.5 and 3 h after C48/80 treatment.
RESULTS: Gastric mucosal lesions occurred 0.5 h after C48/80 treatment and progressed at 3 h. Gastric mucosal blood flow decreased 0.5 h after C48/80 treatment but the decrease was recovered at 3 h. Gastric mucosal total AA, reduced AA, vitamin E, and adherent mucus concentrations decreased 3 h after C48/80 treatment. Gastric mucosal oxidized AA concentration remained unchanged after C48/80 treatment. Gastric mucosal NPSH concentration decreased 0.5 h after C48/80 treatment, but the decrease was recovered at 3 h. Gastric mucosal TBARS concentration and MPO activity increased 0.5 h after C48/80 treatment and further increased at 3 h. Serum total AA and reduced AA concentrations increased 0.5 h after C48/80 treatment and further increased at 3 h, while serum oxidized AA concentration increased at 0.5 h. Serum and gastric mucosal NOx concentrations increased 3 h after C48/80 treatment. AA administration to C48/80-treated rats at 0.5 h after the treatment prevented the gastric mucosal lesion progression and the changes in gastric mucosal total AA, reduced AA, vitamin E, adherent mucus, NOx, and TBARS concentrations and MPO activity and serum NOx concentration found at 3 h after the treatment dose-dependently. The AA administration to C48/80-treated rats caused further increases in serum total AA and reduced AA concentrations at 3 h after the treatment dose-dependently.
CONCLUSION: Gastric mucosal AA plays a critical role in the progression of C48/80-induced acute gastric mucosal lesions in rats.
Keywords: Compound 48/80, Ascorbic acid, Non-protein SH, Vitamin E, Nitric oxide, Lipid peroxidation, Inflammation
INTRODUCTION
Ascorbic acid (AA), also known as vitamin C, exerts an antioxidant action both by itself and by interacting with reduced glutathione (GSH) or vitamin E[1,2]. Ascorbic acid scavenges reactive oxygen species (ROS) such as superoxide radical (O2-), hydrogen peroxide (H2O2), hydroxyl radical (OH·), singlet oxygen, and hypochlorous acid (HOCl), which is produced from H2O2 and Cl- via myeloperoxidase (MPO) present in neutrophils, in vitro[3-7]. In addition, AA scavenges peroxynitrite (ONOO-), a highly reactive nitrogen oxide species, which is produced by the reaction of O2- and nitric oxide (NO) in vitro[8-10]. It is known that AA scavenges O2- generated by activated NADPH oxidase in neutrophils and in the hypoxanthine-xanthine oxidase (XO) system in vitro[11-13]. It is also known that AA prevents neutrophil adherence to endothelium by scavenging ROS derived from activated neutrophils in vitro[14].
Ekman et al[15, 16] have reported that a single intraarterial pretreatment with AA reduces gastric bleeding after hemorrhagic shock and retransfusion in rats and that the same AA pretreatment prevents gastric mucosal energy depletion after hemorrhagic shock and gastric mucosal vessel injury after retransfusion in rats. Huang and Neu[17] have shown in 5-d starved rats with HCl treatment that the oral AA administration during starvation and HCl treatment protects against gastric damage induced by starvation plus HCl through attenuation of decreased gastric GSH and mucus levels and increased gastric lipid peroxide level and acid back-diffusion. In addition, Pohle et al[18] have shown that human volunteers who were given aspirin for 3 d of oral AA administration during aspirin treatment protect themselves against aspirin-induced gastric damage by preventing increases in neutrophil infiltration and lipid peroxidation induced by ROS, the major source of which is probably activated neutrophils, and decreases in blood flow and antioxidant defense systems mediated by superoxide dismutase, glutathione peroxidase, and AA in the gastric tissue. Thus, administration of AA protects against acute gastric damage in experimental animals and humans. However, there is little information on the role of gastric mucosal AA in the development of acute gastric damage.
Compound 48/80 (C48/80) is known to cause degranulation of connective tissue mast cells, but not mucosal mast cells, with release of serotonin and histamine from the cells[19,20]. We have shown in rats with a single C48/80 treatment that gastric mucosal lesions develop with decreases in Se-glutathione peroxidase activity and vitamin E content and increases in neutrophil infiltration, XO activity, and lipid peroxide level in the gastric mucosal tissue, and that gastric mucosal blood flow is reduced with gastric mucosal lesion formation, while the decreased blood flow is recovered with the lesion progression and recovery[21]. We have also shown in rats treated once with C48/80 that neutrophils infiltrating into the gastric mucosal tissue participate in gastric mucosal lesion formation and progression, while the xanthine-XO system in the gastric mucosal tissue takes part mainly in the lesion progression[22]. Our recent report has shown in rats treated once with C48/80 that an increase in the gastric mucosal level of nitrite/nitrate (NOx), an index of NO production, which is closely associated with induction of nitric oxide synthase in neutrophils infiltrating into the gastric mucosa, contributes to the progression of acute gastric mucosal lesions[23]. Furthermore, our recent report has shown that the gastric mucosal levels of total AA and reduced AA decrease during the progression of C48/80-induced acute gastric mucosal lesions in rats although no change in the gastric mucosal level of oxidized AA, i.e., dehydroascorbic acid, occurs during the lesion progression[24]. Thus, gastric mucosal AA is depleted with the progression of C48/80-induced acute gastric mucosal lesions in rats. However, there is no information on whether gastric mucosal AA plays a critical role in the progression of C48/80-induced acute gastric mucosal lesions.
In the present study, therefore, we attempted to clarify the role of gastric mucosal AA in the progression of C48/80-induced acute gastric mucosal lesions in rats, namely, we investigated the effect of AA administered orally after the occurrence of gastric mucosal lesions on the progression of the lesions and the changes in the gastric mucosal levels of total AA, reduced AA, oxidized AA, vitamin E, non-protein SH (NPSH), adherent mucus, thiobarbituric acid reactive substances (TBARS), an index of lipid peroxidation, NOx and the activity of MPO, an index of tissue neutrophil infiltration[25], with gastric mucosal lesion progression in C48/80-treated rats. In addition, the changes in the serum levels of total AA, reduced AA, oxidized AA, and NOx with gastric mucosal lesion progression were examined in the C48/80-treated rats with and without post-AA administration.
MATERIALS AND METHODS
Materials
Compound 48/80 and 3, 3’,5, 5’-tetramethylbenzidine were purchased from Sigma Chemicals (St. Louis, MO). L-AA (reduced form), α,α’-dipyridyl, disodium octyl sulfosuccinate (DSS), 5,5’-dithiobis (2-nitobenzoic acid) (DTNB), dithiothreitol (DTT), EDTA, N-ethylmaleimide (NEM), GSH, sodium dodecyl sulfate, RRR-α-tocopherol (α-Toc.), RRR-δ-tocopherol, trichloroacetic acid (TCA), and other chemicals were purchased from Wako Pure Chemical Ind., Ltd (Osaka, Japan). All chemicals were used without further purification.
Animals
Male Wistar rats aged 6 wk were purchased from Japan SLC Co. (Hamamatsu, Japan). The animals were housed in cages in a ventilated animal room with controlled temperature (23±2 °C) and relative humidity (55±5%) with 12 h of light (7:00 to 19:00). The animals were maintained with free access to rat chow, Oriental MF (Oriental Yeast Co., Tokyo, Japan) and tap water ad libitum for one week. All animals received humane care in compliance with Guidelines for the Management of Laboratory Animals in Fujita Health University.
Induction and observation of gastric mucosal lesions
Rats (7 wk old) fasted for 24 h received a single intraperitoneal injection of compound 48/80 (0.75 mg/kg body weight), dissolved in distilled water, as described previously[21-24]. The control rats received an intraperitoneal injection of an equal volume of distilled water. All animals were maintained with free access to water and without food during the experiment. The animals were sacrificed under ether anesthesia 0.5 or 3 h after compound 48/80 injection. The stomachs were removed, inflated with 10 mL of 0.9% NaCl, and put into 40 g/L formaldehyde formalin for 10 min. The stomachs were then opened along the greater curvature and examined for lesions in the glandular part under a dissecting microscope (×10). The severity of gastric mucosal lesions was estimated using the index of the following eight grades of lesions as described in our previous reports[21-24]: grade 0, no lesion (normal); grade I, edema only; grade II, damaged area of 1-10 mm2; grade III, damaged area of 11-20 mm2; grade IV, damaged area of 21-30 mm2; grade V, damaged area of 31-40 mm2; grade VI, damaged area of 41-50 mm2; grade VII, damaged area of >51 mm2.
Administration of AA
Rats treated with and without C48/80 received a single oral administration of AA dissolved in distilled water (10, 50 or 100 mg/kg body weight) or distilled water at a dose of 1 mL/100 g body weight at 0.5 h after the treatment.
Sample collection and biochemical determinations
Rats with and without C48/80 treatment were sacrificed under ether anesthesia at 0.5 or 3 h after the treatment at which time blood was collected from the inferior vena cava. Serum was obtained from the collected blood by centrifugation. Immediately after sacrifice, stomachs were isolated and gastric mucosal tissues were collected from the isolated stomachs. The collected gastric mucosal tissues and serum were stored at -80 °C until use. Gastric mucosal tissue was homogenized in 9 volumes of ice-cold 0.15 mol/L KCl containing 1 mmol/L EDTA on ice using a micro-homogenizer M-100 (Tokai-Irika Co., Tokyo, Japan). This homogenate was used for the assays of AA, NPSH, and TBARS. Total AA, reduced AA, and oxidized AA in gastric mucosal tissues and serum were assayed by the method of Zannoni et al[26]. and Okamura[27] as follows. For the determination of total AA, 0.3 mL of 10% gastric mucosal homogenate or serum was incubated with 0.1 mL of 10 mmol/L DTT at 37 °C for 30 min to convert all AA in an oxidized form in the homogenate or serum to its reduced form and then the excess DTT was removed with 0.1 mL of 0.5% NEM. An aliquot of the supernatant obtained after deproteinization with 0.5 mL of ice-cold 10% TCA was used for the assay of the resultant reduced AA plus the original reduced AA. For the determination of reduced AA, 0.3 mL of 10% gastric mucosal homogenate or serum was mixed with 0.2 mL of a solution of 10 mmol/L DTT-0.5% NEM. An aliquot of the supernatant obtained after deproteinization with 0.5 mL of ice-cold 10% TCA was used for the assay of reduced AA. Reduced AA in each sample was measured by the α,α’-dipyridyl method. The concentration of reduced AA was determined using the standard curve of authentic L-AA in a reduced form. The concentration of oxidized AA in gastric mucosal homogenates or serum was estimated from the difference between the concentrations of total AA and reduced AA determined. Gastric mucosal NPSH was assayed by the DTNB method of Sedlak and Lindsay[28] using GSH as a standard. Gastric mucosal TBARS was spectrophotometrically assayed by the method of Ohkawa et al[29] using the thiobarbituric acid reaction except that 1.0 mmol/L EDTA was added to the reaction mixture. The amount of gastric mucosal TBARS was expressed as that of malondialdehyde (MDA) equivalents. Gastric mucosal vitamin E was assayed by the method of Mitton and Trevithick[30] using high-performance liquid chromatography (HPLC) with electrochemical detection with some modifications as follows. Gastric mucosal tissue (50 mg) was mixed with 500 μL of ice cold phosphate-buffered saline containing 1 mmol/L EDTA (pH 7.0), 50 μL of butylated hydroxytoluene dissolved in ethanol, and 500 μL of 0.1 mol/L sodium dodecyl sulfate, and then homogenized on ice using a micro-homogenizer M-100. The homogenate was mixed with 2.0 mL of a mixture of acetonitrile and acetone (1:1 v/v) and 100 μL of 10 μmol/L RRR-δ-tocopherol used as an internal standard in a glass test tube with a cup and then shaken vigorously for 5 min using a shaker model SA-31 (Yamato Scientific Co., Tokyo, Japan). After shaking, 4.0 mL of hexane was added to the mixture. Once stirred on Vortex mixer vigorously, the mixture containing hexane was centrifuged at 2000 r/min for 5 min. The layer of hexane was carefully collected in a small glass test tube and then evaporated at 50 °C under nitrogen gas stream. After evaporation, the residue was dissolved in 500 μL of a mixture of ethanol and methanol (1:1 v/v). The HPLC system consisted of an EiCOM Model EP-300 HPLC pump system (EiCOM Co., Kyoto, Japan), an EiCOM ECD-300 electrochemical detector with grassy carbon electrode, an EiCOM ATC-300 column oven, an EiCOM SC-5ODS column (3.0 mm ID 150 cm), and a Chromatocorder 21 data processor (System Instrument Co., Hachioji, Japan). The analytical condition was as follows: the mobile phase was a mixture of methanol, acetic acid, and H2O (95:3.4:1.6 v/v) (pH 4.95) and it was continuously degassed with an EiCOM DG-300 degasser (EiCOM Co., Kyoto, Japan). The temperature of the column oven was 30 °C. The flow rate was 0.75 mL/min. The electrode potential was set at 800 mV (vs Ag/AgCl reference electrode). The injection volume of each sample was 10 μL. The amount of vitamin E in gastric mucosal tissues was expressed as that of α-Toc. Gastric adherent mucus was assayed by the method of Kitagawa et al[31] as follows. The removed stomach was cut open along with the greater curvature and rinsed with 10 mL of ice-cold 0.25 mol/L sucrose. Then, 50 mm2 (approx. 8 mm in diameter) of the glandular portion of the stomach was excised with a scalpel and the excised part was weighed. The excised stomach was soaked in 2 mL of 0.1% Alcian blue, which was dissolved in 0.16 mol/L sucrose buffered with 0.05 mol/L sodium acetate (pH 5.8), for 2 h. Uncomplexed dye was removed by two successive washes in 2 mL of 0.25 mol/L sucrose for 15 and 45 min, and then dye complex with mucus was extracted with 30% DSS for 2 h. After centrifugation (3000 r/min for 10 min), the optical density of the solution of Alcian blue extracted with DSS was read at 620 nm and the concentration of the extracted Alcian blue was calculated in comparison with a calibration curve obtained from known concentrations of Alcian blue solutions. The concentration of gastric mucosal adherent mucus was expressed as that of Alcian blue adhered to the gastric mucosal surface (μg/g tissue). NOx (nitrite/nitrate) in gastric mucosal tissues or serum was assayed by the Griess reaction-dependent method of Green et al[32]. For this assay, gastric mucosal tissue was homogenized in 9 volumes of ice-cold 50 mmol/L Tris-HCl buffer (pH 7.5) using a micro-homogenizer M-100. The homogenate was sonicated two times on ice for 30 s using a Handy Sonic model UR-20P (Tomy Seiko Co., Tokyo, Japan). The sonicated homogenate was centrifuged at 10 000 g for 20 min at 4 °C and the resultant supernatant was filtrated at 4 °C under centrifugation using a membrane filter Ultrafree-MC (Millipore Co., Bedford, MA, USA). Serum was also filtrated using an Ultrafree-MC. NOx in the filtrate obtained from gastric mucosal tissues or serum was determined using a nitric oxide assay kit (Roche Diagnostics Co., Tokyo, Japan). Gastric mucosal MPO was assayed by the method of Suzuki et al[33]. For this enzyme assay, the supernatant fraction of the sonicated gastric mucosal homogenate, which was prepared for the assay of gastric mucosal NOx, was dialyzed against 100 volumes of 50 mmol/L Tris-HCl buffer (pH 7.5) at 4 °C for 24 h. MPO activity in the dialyzed supernatant was assessed by measuring the H2O2-dependent oxidation of tetramethylbenzidine at 37 °C. One unit (U) of this enzyme was defined as the amount of enzyme causing a change in absorbance of 1.0 per min at 655 nm.
Measurement of gastric mucosal blood flow
Gastric mucosal blood flow was measured using a laser Doppler flowmeter, Laser Flow BRL-100 (Bio Research Center Co., Nagoya), as described in our previous reports[21-24]. Rats used for this measurement were anesthetized with pentobarbital sodium 10 min before the onset of the measurement and the abdomen was opened on an operation mat. The mat was heated at 37 °C during the operation and blood flow measurement. The laser probe was attached to the serosal side of the corpus mucosa by aid of a cyanoacrylate-typed instantaneous adhesive, Aron Alpha (Toagosei Co., Tokyo), and the blood flow changes were monitored on a recorder for at least 5 min after the onset of the measurement. Gastric mucosal blood flow in C48/80-treated rats was expressed as a relative percentage toward the mean value of gastric mucosal blood flow determined in control rats without C48/80 treatment. The values of gastric mucosal blood flow measured in C48/80-untreated rats were constant within at least 5% of standard deviation.
Statistical analysis
Results of biochemical determinations in the gastric mucosal tissue and serum were expressed as mean±SD. The results were analyzed by computerized statistical package (StatView). Each mean value was compared by one-way analysis of variance (one-way ANOVA) and Fisher’s protected least significance difference (PLSD) for multiple comparisons as the post hoc test. Statistical analyses of the severity of mucosal lesions were carried out using the Kruskal-Wallis test. Values of significance were set at P<0.05 for both tests.
RESULTS
Effect of post-AA administration on gastric mucosal lesion development in C48/80-treated rats
As shown in Table 1, C48/80-treated rats showed apparent gastric mucosal lesions at 0.5 h after the treatment and progressed gastric mucosal lesions at 3 h when the severity of gastric mucosal lesions was estimated using the lesion gradation. Untreated control rats showed no gastric mucosal lesion during the experiment period (data not shown). When AA (10, 50 or 100 mg/kg) was orally administered to C48/80-treated rats at 0.5 h after the treatment, the severity of gastric mucosal lesions found at 3 h after the treatment was significantly attenuated by AA administered at a dose of 50 or 100 mg/kg and this attenuating effect was dose-dependent (Table 1). In addition, the severity of gastric mucosal lesions in the C48/80-treated group given AA at a dose of 100 mg/kg was not significantly different from that found at 0.5 h after the treatment (Table 1).
Table 1.
Effect of post-AA administration on gastric mucosal lesion development in rats with a single C48/80 treatment.
| Time after C48/80 treatment and groups |
Lesion index (%) |
P (vsC48/80 at 3 h) | P (vsC48/80 at 0.5 h) | |||||||
| 0 | I | II | III | IV | V | VI | VII | |||
| 0.5 h C48/80 | 0 | 20 | 50 | 30 | 0 | 0 | 0 | 0 | ¯ | ¯ |
| 3 h C48/80 | 0 | 0 | 0 | 0 | 0 | 20 | 40 | 40 | ¯ | 0.05 |
| + AA (10 mg/kg) | 0 | 0 | 0 | 0 | 10 | 30 | 50 | 10 | NS | 0.05 |
| + AA (50 mg/kg) | 0 | 0 | 0 | 30 | 40 | 30 | 0 | 0 | 0.05 | 0.05 |
| + AA (100 mg/kg) | 0 | 0 | 40 | 50 | 10 | 0 | 0 | 0 | 0.05 | NS |
Rats received oral administration of AA (10, 50 or 100 mg/kg) or vehicle at 0.5 h after a single intraperitoneal injection of compound 48/80 80.75 mg/kg) and the severity of gastric mucosal lesions was estimated at 0.5 and 3 h after the compound 48/80 injection. The number of rats used in each group was 10. NS indicates not significant.
Effect of post-AA administration on gastric mucosal AA levels in C48/80-treated rats
When gastric mucosal total AA, reduced AA, and oxidized AA concentrations in C48/80-treated and untreated control rats were determined at 0.5 and 3 h after the treatment, the results shown in Figure 1 were obtained. There were no significant differences in the gastric mucosal concentrations of total AA and reduced AA between the C48/80-treated and untreated control rats at 0.5 h after the treatment, although gastric mucosal oxidized AA concentration in the C48/80-treated group tends to be higher than that in the control group. The C48/80-treated group had significantly lower gastric mucosal total AA and reduced AA concentrations than the control group at 3 h after the treatment. Gastric mucosal total AA and reduced AA concentrations in the C48/80-treated group were 54.7 and 50.0% respectively of those in the control group. There was no difference in gastric mucosal oxidized AA concentration between the C48/80-treated and control groups at 3 h after the treatment. When AA (10, 50 or 100 mg/kg) was orally administered to C48/80-treated rats at 0.5 h after the treatment, the decreased gastric mucosal total AA and reduced AA concentrations found at 3 h after the treatment were significantly attenuated by AA administered at a dose of 50 or 100 mg/kg (but not 10 mg/kg) and this attenuating effect was dose-dependent. Gastric mucosal total AA and reduced AA concentrations in the C48/80-treated group given AA at a dose of 100 mg/kg were 73.5 and 71.4%, respectively of those in the untreated control group (Figures 1A and B). However, the post-AA administration did not affect the gastric mucosal oxidized AA concentration found at 3 h after C48/80 treatment (Figure 1C). In C48/80-untreated rats, AA administered at a dose of 50 or 100 mg/kg (but not 10 mg/kg) in the same manner caused significant increases in gastric mucosal total AA and reduced AA concentrations without affecting gastric mucosal oxidized AA concentration and these increases were dose-dependent (Figure 1).
Figure 1.
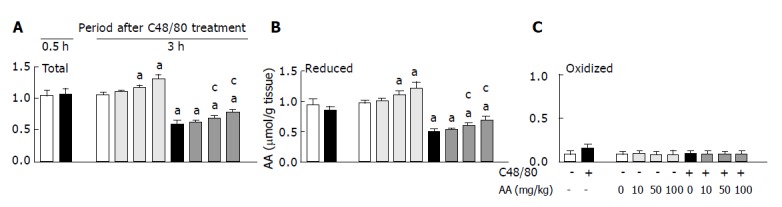
Effect of post-AA administration on gastric mucosal total AA (A), reduced AA (B), and oxidized AA (C) concentrations in C48/80-treated rats. mean±SD (n = 5 for C48/80-untreated groups; n = 10 for C48/80-treated groups). aP<0.05 vs corresponding control rats without C48/80 treatment; cP<0.05 vs rats treated with C48/80 alone.
Effect of post-AA administration on serum AA levels in 48/80-treated rats
When serum total AA, reduced AA, and oxidized AA concentrations in the C48/80-treated and untreated control groups were determined at 0.5 and 3 h after the treatment, the results shown in Figure 2 were obtained. The C48/80-treated group contained significantly higher serum total AA, reduced AA, and oxidized AA concentrations than the control group at 0.5 h after the treatment. Further increases in serum total AA and reduced AA concentrations in the C48/80-treated group occurred 3 h after the treatment. However, there was no significant difference in serum oxidized AA concentration between the C48/80-treated and untreated control groups at 3 h after the treatment. When AA (10, 50 or 100 mg/kg) was orally administered to C48/80-treated rats at 0.5 h after the treatment, the increased total AA and reduced AA concentrations found at 3 h after the treatment were further increased significantly by AA administered at a dose of 50 or 100 mg/kg (but not 10 mg/kg) and these increases were dose-dependent (Figures 2A and B). However, the post-AA administration did not affect the serum oxidized AA concentration found at 3 h after C48/80 treatment (Figure 2C). In C48/80-untreated rats, AA administered at a dose of 50 or 100 mg/kg (but not 10 mg/kg) in the same manner caused significant increases in serum total AA and reduced AA concentrations without affecting serum oxidized AA concentration and these increases were dose-dependent (Figure 2).
Figure 2.
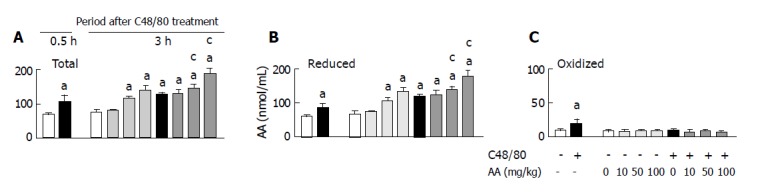
Effect of post-AA administration on serum total AA (A), reduced AA (B), and oxidized AA (C) concentrations in C48/80-treated rats. Mean±SD (n = 5 for C48/80-untreated groups; n = 10 for C48/80-treated groups). aP<0.05 vs corresponding control rats without C48/80 treatment; cP<0.05 vs rats treated with C48/80 alone.
Effect of post-AA administration on gastric mucosal vitamin E, NPSH, and adherent mucus levels in C48/80-treated rats
As shown in Figures 3A and C, there were no differences in gastric mucosal vitamin E and adherent mucus concentrations between C48/80-treated and untreated control rats at 0.5 h after the treatment, but the C48/80-treated rats had significantly lower gastric mucosal vitamin E and adherent mucus concentrations than the control rats at 3 h. When AA (10, 50 or 100 mg/kg) was orally administered to C48/80-treated rats at 0.5 h after the treatment, the decreased gastric mucosal vitamin E and adherent mucus concentrations found at 3 h after the treatment were significantly attenuated by AA administered at a dose of 50 or 100 mg/kg (but not 10 mg/kg), and these attenuating effects were dose-dependent (Figures 3A and C). As shown in Figure 3B, C48/80-treated rats had significantly lower gastric mucosal NPSH concentration than untreated control rats at 0.5 h after the treatment, but there was no difference in the concentration between the two groups at 3 h. Oral administration of AA (10, 50 or 100 mg/kg) to C48/80-treated rats at 0.5 h after the treatment did not affect the concentration of gastric mucosal NPSH found at 3 h after the treatment (Figure 3B). In C48/80-untreated rats, AA administered at a dose of 10, 50 or 100 mg/kg in the same manner had no effect on the gastric mucosal vitamin E, NPSH, and adherent mucus concentrations (Figure 3).
Figure 3.
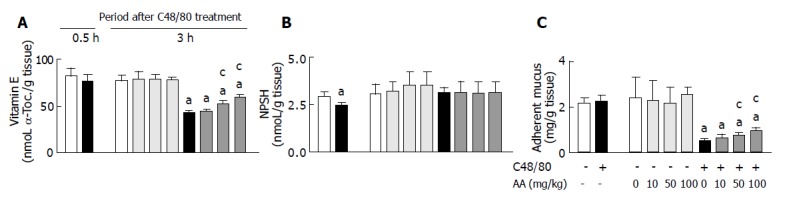
Effect of post-AA administration on gastric mucosal vitamin E (A), NPSH (B), and adherent mucus (C) concentrations in C48/80-treated rats. mean±SD (n = 5 for C48/80-untreated groups; n = 10 for C48/80-treated groups). aP<0.05 vs corresponding control rats without C48/80 treatment; cP<0.05 vs rats treated with C48/80 alone.
Effect of post-AA administration on gastric mucosal TBARS level and MPO activity in C48/80-treated rats
As shown in Figure 4, gastric mucosal TBARS concentration and MPO activity in C48/80-treated rats were significantly higher than those in untreated control rats at 0.5 h after the treatment and the C48/80-treated rats showed further increases in gastric mucosal TBARS concentration and MPO activity at 3 h after the treatment. When AA (10, 50 or 100 mg/kg) was orally administered to C48/80-treated rats at 0.5 h after the treatment, the increased gastric mucosal TBARS concentration and MPO activity found at 3 h after the treatment were significantly attenuated by AA administered at a dose of 50 or 100 mg/kg (but not 10 mg/kg), and these attenuating effects were dose-dependent (Figure 4). In C48/80-untreated rats, AA administered at a dose of 10, 50 or 100 mg/kg in the same manner had no effect on the gastric mucosal MPO activity, but AA administered at a dose of 100 mg/kg (but not 10 or 50 mg/kg) reduced the gastric mucosal TBARS concentration significantly (Figure 4).
Figure 4.
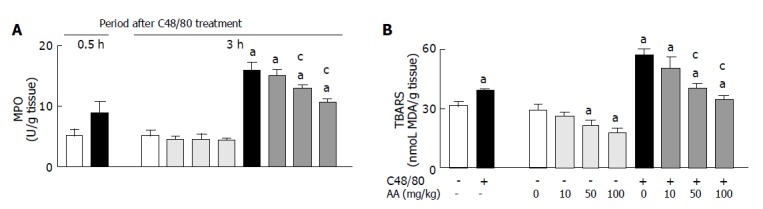
Effect of post-AA administration on gastric mucosal TBARS concentration (A) and MPO activity (B) in C48/80-treated rats. mean±SD (n = 5 for C48/80-untreated groups; n = 10 for C48/80-treated groups). aP<0.05 vs corresponding control rats without C48/80 treatment; cP<0.05 vs rats treated with C48/80 alone.
Effect of post-AA administration on gastric mucosal and serum NOx levels in C48/80-treated rats
As shown in Figure 5, there were no significant differences in gastric mucosal and serum NOx concentrations between C48/80-treated and untreated control rats at 0.5 h after the treatment, but the C48/80-treated group had significantly higher gastric mucosal and serum NOx concentrations than the control group at 3 h. When AA (10, 50 or 100 mg/kg) was orally administered to C48/80-treated rats at 0.5 h after the treatment, the increased gastric mucosal NOx concentration found at 3 h after the treatment was significantly attenuated by AA administered at a dose of 50 or 100 mg/kg (but not 10 mg/kg), and this attenuating effect was dose-dependent (Figure 5A). The post-administration of AA at a dose of 100 mg/kg (but not 10 or 50 mg/kg) attenuated the increased serum NOx concentration found at 3 h after C48/80 treatment significantly (Figure 5B). In C48/80-untreated rats, AA administered at a dose of 10, 50 or 100 mg/kg in the same manner had no effect on the gastric mucosal and serum NOx concentrations (Figure 5).
Figure 5.
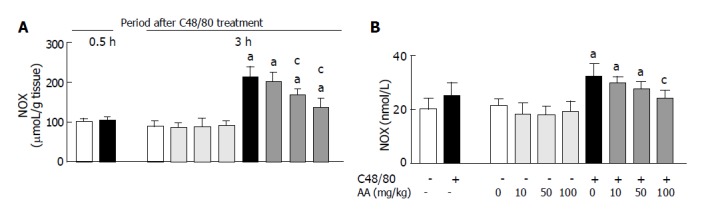
Effect of post-AA administration on NOx concentrations in the gastric mucosal tissue (A) and serum (B) of C48/80-treated rats. mean±SD (n = 5 for C48/80-untreated groups; n = 10 for C48/80-treated groups). aP<0.05 vs corresponding control rats without C48/80 treatment; cP<0.05 vs rats treated with C48/80 alone.
Effect of post-AA administration on gastric mucosal blood flow in C48/80-treated rats
As shown in Figure 6, C48/80-treated rats had about 25% of gastric blood flow in untreated control rats at 0.5 h after the treatment, but the decreased gastric blood flow in the C48/80-treated group was returned to about 75% of gastric mucosal blood flow in the control group at 3 h. Oral administration of AA (10, 50 or 100 mg/kg) to C48/80-treated rats at 0.5 h after the treatment did not affect the gastric mucosal blood flow found at 3 h after the treatment (Figure 6). Oral administration of AA to C48/80-untreated rats in the same manner did not change the gastric mucosal blood flow (Figure 6).
Figure 6.
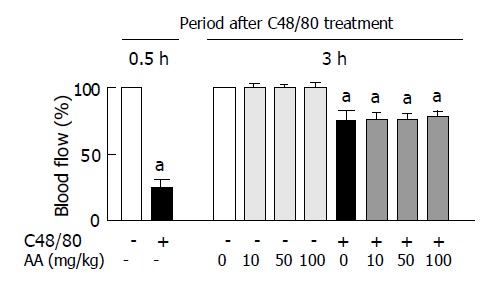
Effect of post-AA administration on gastric mucosal blood flow in C48/80-treated rats. Mean±SD (n = 5 for C48/80-untreated groups; n = 10 for C48/80-treated groups). aP<0.05 vs corresponding control rats without C48/80 treatment.
DISCUSSION
The model of acute gastric mucosal lesions in rats treated once with C48/80, a mast cell degranulator, is important for clarifying the roles of ischemia-reperfusion, oxidative stress, and inflammation in the pathogenesis of gastritis in humans[21-24].
In the present study, decreases in gastric mucosal total AA and reduced AA concentrations occurred at a progressed stage of acute gastric mucosal lesions in rats with a single C48/80 treatment, although increases in the serum total AA and reduced AA concentrations were observed at the same time point, as reported previously[24]. Oral administration of AA (10, 50 or 100 mg/kg) to C48/80-treated rats at an early stage of acute gastric mucosal lesions was found to attenuate the decreases in gastric mucosal total AA and reduced AA concentrations found at a progressed stage of the lesions by causing further increases in serum total AA and reduced AA concentrations in a dose-dependent manner. This oral administration of AA to C48/80-treated rats at an early stage of acute gastric mucosal lesions was found to reduce the severity of gastric mucosal lesions found at a progressed stage of the lesions in a dose-dependent manner. Thus, replenishment of depleted AA in the gastric mucosal tissue of rats treated once with C48/80 by oral administration of AA resulted in the prevention of the progression of acute gastric mucosal lesions. This finding suggests that gastric mucosal AA plays a critical role in the progression of C48/80-induced acute gastric mucosal lesions.
It has been shown that rats treated once with C48/80 have a marked decrease in gastric mucosal blood flow at an early stage of acute gastric mucosal lesions and a partial recovery of the decreased gastric mucosal blood flow at a progressed stage of the lesions[21,22,34]. This ischemia-reperfusion-like change of gastric mucosal blood flow is caused by serotonin and histamine released from the degranulated connective tissue mast cells of C48/80-treated rats[21,34]. In the present study, no dose of AA administered to C48/80-treated rats at an early stage of gastric mucosal lesions affected the recovery of gastric mucosal blood flow found at a progressed stage of the lesions. In addition, there was no change in gastric mucosal blood flow in normal rats given AA. It is known that AA does not inhibit C48/80-induced histamine release from isolated rat peritoneal mast cells[35]. It is also known that the release of serotonin and histamine from degranulated mast cells in rats treated once with C48/80 (0.75 mg/kg) reaches a peak around 0.5 h after the treatment[21,36]. Therefore, these findings indicate that gastric mucosal AA has no effect on the change in gastric mucosal blood flow associated with serotonin and histamine released from degranulated mast cells in rats treated once with C48/80.
We have shown in rats treated once with C48/80 that gastric mucosal Se-glutathione peroxidase activity and vitamin E level decrease at a progressed stage of gastric mucosal lesions, while gastric mucosal neutrophil infiltration, XO activity, and lipid peroxide level begin to increase at an early stage of the lesions and further increase at a progressed stage of the lesions[21,22]. Vitamin E functions as a chain-breaking antioxidant for lipid peroxidation in cell membranes and also as a scavenger of ROS[37]. Lipid peroxidation occurs via ROS generated not only by the xanthine-XO system but also by activated NADPH oxidase in neutrophils[38,39]. MPO present in neutrophils mediates lipid peroxidation in the presence of H2O2 and halide ions[40]. AA not only scavenges ROS such as O2-, H2O2, OH, singlet oxygen, and HOCl, but also supports the chain-breaking antioxidant action of vitamin E by reducing vitamin E radical to vitamin E at the lipid/aqueous interface[2-7]. In the present study, oral administration of AA (10, 50 or 100 mg/kg) to C48/80-treated rats at an early stage of acute gastric mucosal lesions could attenuate the increased gastric mucosal TBARS concentration, an index of lipid peroxidation, and the decreased gastric mucosal vitamin E concentration found at a progressed stage of the lesions in a dose-dependent manner. Rat gastric mucosal tissues contain a large amount of NPSH, a large part of which is GSH[41]. GSH converts oxidized AA to reduced AA in a non-enzymatic manner or via the enzyme-dependent AA recycling system, which contributes to the antioxidant action of AA[1]. In the present study, gastric mucosal NPSH concentration in rats treated once with C48/80 decreased at an early stage of acute gastric mucosal lesions, but the decreased NPSH concentration returned to the level in C48/80-untreated rats at a progressed stage of the lesions, as reported previously[21]. Oral administration of AA (10, 50 or 100 mg/kg) to the C48/80-treated rats at an early stage of acute gastric mucosal lesions did not affect the gastric mucosal NPSH concentration found at a progressed stage of the lesions. In addition, the same AA administration had no effect on gastric mucosal NPSH level in C48/80-untreated rats. Thus, there is little possibility that GSH-dependent AA recycling in the gastric mucosal tissue of C48/80-treated rats is impaired at a progressed stage of acute gastric mucosal lesions. Gastric mucus plays a critical role in the primary defense of the gastric mucosa and provides a protective barrier in the gastric epithelium[42]. It is known that gastric mucin interacts with ROS, especially OH, in vitro[43]. In the present study, C48/80-treated rats showed a marked decrease in gastric adherent mucus concentration at a progressed stage of acute gastric mucosal lesions, as reported previously[44]. Oral administration of AA (10, 50 or 100 mg/kg) at an early stage of gastric mucosal lesions attenuated this decrease in gastric adherent mucus concentration in a dose-dependent manner. These findings indicate that gastric mucosal AA play a very important role in gastric mucosal antioxidant defense system in rats treated once with C48/80.
AA can prevent the adherence of neutrophils to endothelial cells by scavenging ROS derived from activated neutrophils in vitro[14]. Kearns et al[45,46] have shown that orally administered vitamin C attenuates acute ischemia-reperfusion injury in the lung or skeletal muscle of rats by inhibiting neutrophil infiltration into the respective tissues and neutrophil respiratory burst activity. In the present study, MPO activity, an index of tissue neutrophil infiltration[25], in the gastric mucosal tissue of rats treated once with C48/80 increased at an early stage of acute gastric mucosal lesions and further increased at a progressed stage of the lesions, as reported previously[21-23,44]. Oral administration of AA (10, 50 or 100 mg/kg) to the C48/80-treated rats at an early stage of acute gastric mucosal lesions could attenuate the increased gastric mucosal MPO activity found at a progressed stage of the lesions in a dose-dependent manner. The C48/80-treated rats with oral AA administration showed further increases in serum total AA and reduced AA concentrations, as described above. AA does not inhibit intracellular MPO activity in neutrophils[11]. Therefore, it is suggested that orally administered AA prevents neutrophil infiltration into the gastric mucosal tissue of C48/80-treated rats by replenishment of depleted gastric mucosal AA, rather than by enhancement of serum AA level. This may allow us to think that gastric mucosal AA itself plays an important role in neutrophil infiltration into the gastric mucosal tissue of rats treated once with C48/80.
We have shown in rats treated once with C48/80 that increase in NOx level in gastric mucosal tissue occurs with a concomitant increase in NOx level in serum and that this NO production in gastric mucosal tissue is closely associated with induction of nitric oxide synthase in neutrophils infiltrating into the gastric mucosal tissue and contributes to the progression of acute gastric mucosal lesions[23]. In the present study, oral administration of AA (10, 50 or 100 mg/kg) to rats with a single C48/80 treatment at an early stage of acute gastric mucosal lesions attenuated the increases in gastric mucosal and serum NOx concentrations in a dose-dependent manner. These results further show that gastric mucosal AA plays an important role in neutrophil infiltration into the gastric mucosal tissue of rats treated once with C48/80. AA is not known to scavenge NO itself. However, AA scavenges ONOO-, which is produced by the reaction of O2- and NO in vitro[8-10]. It was reported that ONOO- participates in lipid peroxidation induced by activated neutrophils[47] and is involved in lipid peroxidation induced by NO in the gastric mucosal tissue of rats with local intra-arterial infusion of NO donors[48]. Therefore, these findings suggest that gastric mucosal AA plays an important role in NO-induced lipid peroxidation in the gastric mucosal tissue of rats treated once with C48/80.
In conclusion, the results of the present study indicate that gastric mucosal AA plays a critical role in the progression of compound 48/80-induced acute gastric mucosal lesions in rats through its antioxidant and anti-inflammatory actions. However, further investigation is needed to clarify the exact role of gastric mucosal AA in the progression of C48/80-induced acute gastric mucosal lesions.
Footnotes
Edited by Wang XL and Gabbe M
References
- 1.Winkler BS, Orselli SM, Rex TS. The redox couple between glutathione and ascorbic acid: a chemical and physiological perspective. Free Radic Biol Med. 1994;17:333–349. doi: 10.1016/0891-5849(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 2.Beyer RE. The role of ascorbate in antioxidant protection of biomembranes: interaction with vitamin E and coenzyme Q. J Bioenerg Biomembr. 1994;26:349–358. doi: 10.1007/BF00762775. [DOI] [PubMed] [Google Scholar]
- 3.Som S, Raha C, Chatterjee IB. Ascorbic acid: a scavenger of superoxide radical. Acta Vitaminol Enzymol. 1983;5:243–250. [PubMed] [Google Scholar]
- 4.Deutsch JC. Ascorbic acid oxidation by hydrogen peroxide. Anal Biochem. 1998;255:1–7. doi: 10.1006/abio.1997.2293. [DOI] [PubMed] [Google Scholar]
- 5.Rose RC. Ascorbic acid metabolism in protection against free radicals: a radiation model. Biochem Biophys Res Commun. 1990;169:430–436. doi: 10.1016/0006-291x(90)90349-r. [DOI] [PubMed] [Google Scholar]
- 6.Bodannes RS, Chan PC. Ascorbic acid as a scavenger of singlet oxygen. FEBS Lett. 1979;105:195–196. doi: 10.1016/0014-5793(79)80609-2. [DOI] [PubMed] [Google Scholar]
- 7.Halliwell B, Wasil M, Grootveld M. Biologically significant scavenging of the myeloperoxidase-derived oxidant hypochlorous acid by ascorbic acid. Implications for antioxidant protection in the inflamed rheumatoid joint. FEBS Lett. 1987;213:15–17. doi: 10.1016/0014-5793(87)81456-4. [DOI] [PubMed] [Google Scholar]
- 8.Shi X, Rojanasakul Y, Gannett P, Liu K, Mao Y, Daniel LN, Ahmed N, Saffiotti U. Generation of thiyl and ascorbyl radicals in the reaction of peroxynitrite with thiols and ascorbate at physiological pH. J Inorg Biochem. 1994;56:77–86. doi: 10.1016/0162-0134(94)85039-9. [DOI] [PubMed] [Google Scholar]
- 9.Bartlett D, Church DF, Bounds PL, Koppenol WH. The kinetics of the oxidation of L-ascorbic acid by peroxynitrite. Free Radic Biol Med. 1995;18:85–92. doi: 10.1016/0891-5849(94)e0133-4. [DOI] [PubMed] [Google Scholar]
- 10.Whiteman M, Halliwell B. Protection against peroxynitrite-dependent tyrosine nitration and alpha 1-antiproteinase inactivation by ascorbic acid. A comparison with other biological antioxidants. Free Radic Res. 1996;25:275–283. doi: 10.3109/10715769609149052. [DOI] [PubMed] [Google Scholar]
- 11.Hemilä H, Roberts P, Wikström M. Activated polymorphonuclear leucocytes consume vitamin C. FEBS Lett. 1984;178:25–30. doi: 10.1016/0014-5793(84)81232-6. [DOI] [PubMed] [Google Scholar]
- 12.Anderson R, Lukey PT. A biological role for ascorbate in the selective neutralization of extracellular phagocyte-derived oxidants. Ann N Y Acad Sci. 1987;498:229–247. doi: 10.1111/j.1749-6632.1987.tb23764.x. [DOI] [PubMed] [Google Scholar]
- 13.Dwenger A, Funck M, Lueken B, Schweitzer G, Lehmann U. Effect of ascorbic acid on neutrophil functions and hypoxanthine/xanthine oxidase-generated, oxygen-derived radicals. Eur J Clin Chem Clin Biochem. 1992;30:187–191. doi: 10.1515/cclm.1992.30.4.187. [DOI] [PubMed] [Google Scholar]
- 14.Jonas E, Dwenger A, Hager A. In vitro effect of ascorbic acid on neutrophil-endothelial cell interaction. J Biolumin Chemilumin. 1993;8:15–20. doi: 10.1002/bio.1170080104. [DOI] [PubMed] [Google Scholar]
- 15.Ekman T, Risberg B, Bagge U. Ascorbate reduces gastric bleeding after hemorrhagic shock and retransfusion in rats. Eur Surg Res. 1994;26:187–193. doi: 10.1159/000129335. [DOI] [PubMed] [Google Scholar]
- 16.Ekman T, Bagge U, Risberg B, Soussi B. Ascorbate preserves gastric mucosal metabolism and microcirculation after hemorrhagic shock and retransfusion in rats. Eur Surg Res. 1995;27:39–48. doi: 10.1159/000129371. [DOI] [PubMed] [Google Scholar]
- 17.Hung CR, Neu SL. Acid-induced gastric damage in rats is aggravated by starvation and prevented by several nutrients. J Nutr. 1997;127:630–636. doi: 10.1093/jn/127.4.630. [DOI] [PubMed] [Google Scholar]
- 18.Pohle T, Brzozowski T, Becker JC, Van der Voort IR, Markmann A, Konturek SJ, Moniczewski A, Domschke W, Konturek JW. Role of reactive oxygen metabolites in aspirin-induced gastric damage in humans: gastroprotection by vitamin C. Aliment Pharmacol Ther. 2001;15:677–687. doi: 10.1046/j.1365-2036.2001.00975.x. [DOI] [PubMed] [Google Scholar]
- 19.Enerbäck L, Lundin PM. Ultrastructure of mucosal mast cells in normal and compound 48-80-treated rats. Cell Tissue Res. 1974;150:95–105. doi: 10.1007/BF00220383. [DOI] [PubMed] [Google Scholar]
- 20.Irman-Florjanc T, Erjavec F. Compound 48/80 and substance P induced release of histamine and serotonin from rat peritoneal mast cells. Agents Actions. 1983;13:138–141. doi: 10.1007/BF01967317. [DOI] [PubMed] [Google Scholar]
- 21.Ohta Y, Kobayashi T, Nishida K, Ishiguro I. Relationship between changes of active oxygen metabolism and blood flow and formation, progression, and recovery of lesions is gastric mucosa of rats with a single treatment of compound 48/80, a mast cell degranulator. Dig Dis Sci. 1997;42:1221–1232. doi: 10.1023/a:1018854107623. [DOI] [PubMed] [Google Scholar]
- 22.Ohta Y, Kobayashi T, Ishiguro I. Participation of xanthine-xanthine oxidase system and neutrophils in development of acute gastric mucosal lesions in rats with a single treatment of compound 48/80, a mast cell degranulator. Dig Dis Sci. 1999;44:1865–1874. doi: 10.1023/a:1018803025043. [DOI] [PubMed] [Google Scholar]
- 23.Kamiya Y, Ohta Y, Ogawa H, Imai Y, Arisawa Y, Nakano H. Relationship between nitric oxide and the development of compound 48/80-induced acute gastric mucosa lesions. Bull Fujita Med Soc. 2003;27:55–60. [Google Scholar]
- 24.Ohta Y, Kamiya Y, Imai Y, Arisawa T, Nakano H. A change in gastric mucosal ascorbic acid status with the formation, progression, and recovery of compound 48/80-induced acute gastric mucosal lesions in rats. J Nutr Sci Vitaminol (Tokyo) 2004;50:371–376. doi: 10.3177/jnsv.50.371. [DOI] [PubMed] [Google Scholar]
- 25.Krawisz JE, Sharon P, Stenson WF. Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology. 1984;87:1344–1350. [PubMed] [Google Scholar]
- 26.Zannoni V, Lynch M, Goldstein S, Sato P. A rapid micromethod for the determination of ascorbic acid in plasma and tissues. Biochem Med. 1974;11:41–48. doi: 10.1016/0006-2944(74)90093-3. [DOI] [PubMed] [Google Scholar]
- 27.Okamura M. An improved method for determination of L-ascorbic acid and L-dehydroascorbic acid in blood plasma. Clin Chim Acta. 1980;103:259–268. doi: 10.1016/0009-8981(80)90144-8. [DOI] [PubMed] [Google Scholar]
- 28.Sedlak J, Lindsay RH. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman's reagent. Anal Biochem. 1968;25:192–205. doi: 10.1016/0003-2697(68)90092-4. [DOI] [PubMed] [Google Scholar]
- 29.Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 30.Mitton KP, Trevithick JR. High-performance liquid chromatography-electrochemical detection of antioxidants in vertebrate lens: glutathione, tocopherol, and ascorbate. Methods Enzymol. 1994;233:523–539. doi: 10.1016/s0076-6879(94)33058-1. [DOI] [PubMed] [Google Scholar]
- 31.Kitagawa H, Takeda F, Kohei H. A simple method for estimation of gastric mucus and effects of antiulcerogenic agents on the decrease in mucus during water-immersion stress in rats. Arzneimittelforschung. 1986;36:1240–1244. [PubMed] [Google Scholar]
- 32.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki K, Ota H, Sasagawa S, Sakatani T, Fujikura T. Assay method for myeloperoxidase in human polymorphonuclear leukocytes. Anal Biochem. 1983;132:345–352. doi: 10.1016/0003-2697(83)90019-2. [DOI] [PubMed] [Google Scholar]
- 34.Ohta Y, Kobayashi T, Ishiguro I. Role of endogenous serotonin and histamine in the pathogenesis of gastric mucosal lesions in unanaesthetised rats with a single treatment of compound 48/80, a mast cell degranulator. Pharmacol Res. 1999;39:261–267. doi: 10.1006/phrs.1998.0403. [DOI] [PubMed] [Google Scholar]
- 35.Alvarez RG, Mesa MG. Ascorbic acid and pyridoxine in experimental anaphylaxis. Agents Actions. 1981;11:89–93. doi: 10.1007/BF01991466. [DOI] [PubMed] [Google Scholar]
- 36.Takeuchi K, Ohtsuki H, Okabe S. Pathogenesis of compound 48/80-induced gastric lesions in rats. Dig Dis Sci. 1986;31:392–400. doi: 10.1007/BF01311675. [DOI] [PubMed] [Google Scholar]
- 37.Liebler DC. The role of metabolism in the antioxidant function of vitamin E. Crit Rev Toxicol. 1993;23:147–169. doi: 10.3109/10408449309117115. [DOI] [PubMed] [Google Scholar]
- 38.Fukuzawa K, Soumi K, Iemura M, Goto S, Tokumura A. Dynamics of xanthine oxidase- and Fe(3+)-ADP-dependent lipid peroxidation in negatively charged phospholipid vesicles. Arch Biochem Biophys. 1995;316:83–91. doi: 10.1006/abbi.1995.1013. [DOI] [PubMed] [Google Scholar]
- 39.Zimmerman JJ, Ciesielski W, Lewandoski J. Neutrophil-mediated phospholipid peroxidation assessed by gas chromatography-mass spectroscopy. Am J Physiol. 1997;273:C653–C661. doi: 10.1152/ajpcell.1997.273.2.C653. [DOI] [PubMed] [Google Scholar]
- 40.Stelmaszyńska T, Kukovetz E, Egger G, Schaur RJ. Possible involvement of myeloperoxidase in lipid peroxidation. Int J Biochem. 1992;24:121–128. doi: 10.1016/0020-711x(92)90237-u. [DOI] [PubMed] [Google Scholar]
- 41.Body SC, Sasame HA, Body MR. High concentrations of glutathione in glandular stomach: possible implications for carcinogenesis. Science. 1979;205:1010–1012. doi: 10.1126/science.572989. [DOI] [PubMed] [Google Scholar]
- 42.Kaunitz JD. Barrier function of gastric mucus. Keio J Med. 1999;48:63–68. doi: 10.2302/kjm.48.63. [DOI] [PubMed] [Google Scholar]
- 43.Grisham MB, Von Ritter C, Smith BF, Lamont JT, Granger DN. Interaction between oxygen radicals and gastric mucin. Am J Physiol. 1987;253:G93–G96. doi: 10.1152/ajpgi.1987.253.1.G93. [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi T, Ohta Y, Inui K, Yoshino J, Nakazawa S. Protective effect of omeprazole against acute gastric mucosal lesions induced by compound 48/80, a mast cell degranulator, in rats. Pharmacol Res. 2002;46:75–84. doi: 10.1016/s1043-6618(02)00034-8. [DOI] [PubMed] [Google Scholar]
- 45.Kearns SR, Kelly CJ, Barry M, Abdih H, Condron C, Leahy A, Bouchier-Hayes D. Vitamin C reduces ischaemia-reperfusion-induced acute lung injury. Eur J Vasc Endovasc Surg. 1999;17:533–536. doi: 10.1053/ejvs.1999.0833. [DOI] [PubMed] [Google Scholar]
- 46.Kearns SR, Moneley D, Murray P, Kelly C, Daly AF. Oral vitamin C attenuates acute ischaemia-reperfusion injury in skeletal muscle. J Bone Joint Surg Br. 2001;83:1202–1206. doi: 10.1302/0301-620x.83b8.11754. [DOI] [PubMed] [Google Scholar]
- 47.Ródenas J, Carbonell T, Mitjavila MT. Different roles for nitrogen monoxide and peroxynitrite in lipid peroxidation induced by activated neutrophils. Free Radic Biol Med. 2000;28:374–380. doi: 10.1016/s0891-5849(99)00250-6. [DOI] [PubMed] [Google Scholar]
- 48.Lamarque D, Whittle BJ. Involvement of peroxynitrite in the lipid peroxidation induced by nitric oxide in rat gastric mucosa. Eur J Pharmacol. 1996;313:R5–R7. doi: 10.1016/0014-2999(96)00669-3. [DOI] [PubMed] [Google Scholar]