

Abstract
AIM: To evaluate the genetic and epigenetic inactivation mechanism of the RASSF1A tumor suppressor gene at 3p21.3 in extrahepatic cholangiocarcinoma.
METHODS: RT-PCR was used to investigate the transcriptional expressing and re-expression of RASSF1A. RASSF1A mutation was analyzed with SSCP and selective sequencing. PCR was performed to detect the loss of heterozygosity (LOH) at the region of chromosome 3p21.3. Genomic DNA were modificated bisulfite and the frequency of methylation of CpG islands in RASSF1A promoter were evaluated by methylation specific PCR (MS-PCR).
RESULTS: In all 48 samples and one cell lines of extrahepatic cholangiocarcinoma, the RASSF1A mutation is rare (6.12%, 3/49), 33 samples (68.75%) and QBC-939 cell lines (χ2 = 14.270, P = 0.001<0.01) showed RASSF1A express inactivation with LOH at microsatellite loci D3S4604. Among these 33 samples and QBC-939, 28 of 33 (84.85%) tumor samples and 1 cell lines were methylated for majority of 16 CpGs, the average frequency is 73.42%.
CONCLUSION: The data we present suggest that RASSF1A which we have been searching for at 3p21.3 may be one of the key tumor suppressor gene and play an important role in the pathogenesis of extrahepatic cholangiocarcinoma, and the promoter methylation and allelic loss are the major mechanism for inactivation of RASSF1A.
Keywords: RASSF1A, Tumor suppressor gene, Cholangiocarcinoma
INTRODUCTION
Extrahepatic cholangiocarcinoma accounts for about 1% of adult malignant tumor[1], its mortality has been increased in a global worldwide over the past few decades[2] and the prognosis is poor[3,4], but its pathogenic mechanisms remains unknown. Recently, molecular and cytogenetic studies have been contributed significantly to identify the genetic and epigenetic changes associated with this cancer[5,6]. Genome-wide studies by alleotyping and CGH have demonstrated high frequencies of genetic abnormalities on 1q, 3q, 8q, 15q, 17q, and on 3p, 9p, 17p, 21p, loss of heterozygosity (LOH) of 3p was displayed in 60-70% and in 45% of precancerous lesions in extrahepatic cholangiocarcinoma[7]. These findings suggested that inactivation of tumor suppressor gene(s) on 3p is critical in the development of extrahepatic cholangiocarcinoma.
Deletion mapping and functional studies have targeted the extrahepatic cholangiocarcinoma-related tumor suppressor gene to a region at 3p21.3[8]. Dammann et al[9] have isolated a RAS association domain family protein, RASSF1, which is located in the 120-kb region of minimal homozygous deletion at 3p21.3 in lung cancer in 2000. They demonstrated that one of the major transcript of this gene, RASSF1A, is frequently inactivated by the promoter hypermethylation. Re-expression of the gene in lung cancer cell lines suppressed the malignant phenotype. These results indicated that the LOH from chromosome 3p21.3 and CpG island methylation of RASSF1A is one of the common and earliest identified events in the pathogenesis of carcinoma which first be found in lung cancer, but limited to lung tumors, other studies showed which occurred in nasopharyngeal[10], RCC[11], prostate cancer[12] and so on. In this study, we investigated the methylation status, expression, and mutation of RASSF1A in extrahepatic cholangiocarcinoma.
MATERIALS AND METHODS
Tissues and cell line
A total of 48 samples of tumor (34 adenocarcinoma, 9 squamous and 5 adenosquamous) and 12 adjacent non-cancerous tissues were collected from extrahepatic cholangiocarcinoma patients during surgery at Wuhan Tongji Hospital and Cancer Hospital of Shandong province from January to December of 2003. Pathological classification was carried out and the stage of TNM was determined according to criteria outlined by the WHO (TNM stages: 20 samples in I-II stage and 28 in III-IV stage; 29 with metastasis and 19 without metastasis). The cholangiocarcinoma cell lines QBC-939 were presented by professor Wang Shuguang of The Third Military University.
Total genomic DNA was extracted from tumor cell lines QBC-939 and frozen tissue specimens (50-100 mg) according to standard protocol with some modifications, which are briefly described below. Frozen pulverized powders of the specimens were suspended with 2 mL warmed lysis buffer: 50 mmol/L Tris-HCl pH 8.0, 50 mmol/L EDTA, 1% SDS, 10 mmol/L NaCl plus 100 μg/mL boiling-treated RNase A. Following 1 h of incubation at 37 °C, Proteinase K was added to the cellular lysates for a final concentration of 100 μg/mL and the digestion was carried out at 55 °C for 2 h. Organic extractions with a half volume of Phenol/Chloroform/Isoamyl alcohol (1:1:0.04) were repeatedly carried out until no visible interphase remained after centrifugation. DNA was precipitated from the aqueous phase in the presence of 0.3 mol/L NaOAc pH 7.0 and two and a half volumes of ethanol. The DNA pellet was washed once with 70% ethanol and dissolved at 65 °C for 30 min with 0.2-0.4 mL TE (10 mmol/L Tris-HCl pH 7.4 and 1 mmol/L EDTA), followed by storage at 4 °C until further use. The DNA concentrations were calculated according to their A260nm readings.
RASSF1A expression analysis
Total RNAs from 48 tumor samples, QBC-939 cell lines and 2 normal bile duct samples were extracted using the TriZOL Reagent (Invitrogen). Prior to RT-PCR, the RNA was treated with 10 units of DNA and incubated at 37 °C for 10 min. After the reverse transcription reaction, RT-PCR was essentially performed as following: 0.5 µg of RNA were pipetted into tubes containing PCR master mix use primer RT-1 (all primers were composed by sbs Biotech Corporation, Shanghai), PCR conditions were 95 °C for 5 min, then 95 °C for 1 min, 60 °C for 1 min, and 74 °C for 1 min, 35 cycles. PCR products were separated on 2% Tris-borate EDTA agarose gels, and visualized by autoradiography.
As a control, we also assayed levels of the RASSF1C, which has been shown previously[13] to be widely expressed and not susceptible to methylation, primer RT-2 was used. Condition were 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s, 40 cycles.
RASSF1A mutation analysis and LOH studies
Mutation screening in extrahepatic cholangiocarcinoma DNA was performed using the PCR-single strand conformation polymorphism (SSCP) method with intronic primer as described in Table 1, process as Ref. 14. DNA with aberrantly migrating bands were re-collected and sequenced on an ABI automated sequencer. LOH analysis of 4 microsatellite markers targeted at chromosome 3p21.3 region was undertook by PCR with 4 pairs of primers (Table 1) labeled with fluorescence FAM, HEX and NED respectively, methods refer to Ref. 15 with some modifications.
Table 1.
PCR primer sequences for RT-PCR, SSCP, MS-PCR.
| Primer | Upstream sequence | Downstream sequence |
| RT-PCR | ||
| RT-1 | 5’-ACACGTGGTGCGACCTCT-3’ | 5’-GATGAAGCCTGTGTAAGAACCGTCCT-3’ |
| Rt-2 | 5’-ACCTAGCCTTTCTCAAGCTG-3’ | 5’-CATCCTTGGGAGGTAAAAG-3’ |
| SSCP | ||
| E-1a | 5’-CCTGCTAGCGCCCAAAGC-3’ | 5’-CAGCTCCCGCAGCTCAAT-3’ |
| E-1b | 5’-ATGTCGGGGGAGCCTGAG-3’ | 5’-CACGGCCAGGGACCAG-3’ |
| E-2 | 5’-GGGACATTTCCCCGACCT-3’ | 5’-ATCCTCGCCCTTCCCATAC-3’ |
| E-3 | 5’-CCAGAGGCCATTTTTCAGAG-3’ | 5’-TAGCTGGGTACCTGCTCCTC-3’ |
| E-4 | 5’-GCCGACTTCCTTTTACCT-3’ | 5’-GCATGCGTATATACCCTCACA-3’ |
| E-5 | 5’-TGGCCCTGTCTCTGATCATT-3’ | 5’-TCCTCCTCCAAGCCTTACTG-3’ |
| E-6 | 5’-CCTGTACACTCCCCTTTTGC-3’ | 5’-ACCTGGGGGTACAAGAGGTC-3’ |
| MS-PCR | ||
| Meth- | 5’-GTGTTAACGCGTTGCGTATC-3’ | 5’-AACCCCGCGAACTAAAAACGA-3’ |
| Unmeth- | 5’-TTTGGTTGGAGTGTGTTAATGTG-3’ | 5’-CAAACCCCACAAACTAAAAACAAA-3’ |
| LOH | ||
| D3S4597 | 5’-CCTCACTACTCCTGGAATTG-3’ | 5’-CCAAGGAAGGGTTTTACTTA-3’ |
| D3S4604 | 5’-ACACGTGGTGCGACCTCT- 3’ | 5’-GATGAAGCCTGTGTAAGAACCGTCCT-3’ |
| D3S1568 | 5’-TGGCCCTGTCTCTGATCATT-3’ | 5’-TCCTCCTCCAAGCCTTTACTG-3’ |
| D3S1621 | 5’-ACTGACATCTCACCTGGAACC-3’ | 5’-CACATGCAGGTGCTTGACA-3’ |
Bisulfite modification
Bisulfite modification of genomic DNA was carried out by a bisulfite genomic sequencing protocol (Sigma). Briefly, 1 µg of genomic DNA was denatured by incubation with 0.3 mol/L sodium hydroxide for 15 min at 37 °C. Then sulfonated in 3.12 mol/L sodium bisulfite and 5 mmol/L hydroquinone (pH 5) (Sigma) in a thermocycler for 16 h at 55 °C. The DNA samples were recovered using the Wizard DNA Clean-Up System (Promega) in accordance with the manufacture instruction. The DNA is then desulfonated in 0.3 mmol/L NaOH for 10 min at room temperature before ethanol precipitation and resuspension in water.
Methylated-specific PCR
To amplicate the bisulfite-converted RASSF1A promoter sequences, we had designed methylated and unmethylated primers (Table 1). One hundred microliters of reaction buffer containing 5 µg bisulfite-treated DNA, 200 µmol/L of each dNTP, 10 units of Taq polymerase, 10× MS buffer 10 µL and 25 mmol/L MgCl2. Reaction proceeded as following: pre-denatured for 7 min at 95 °C, then at 95 °C for 30 s, 55-65 °C (according to the annealing temperature of every pair of primer) for 30 s, and at 74 °C for 30 s for 35 cycles and finally a 5 min extension at 72 °C. To identify the methylation, PCR products were purified using a QIAquick PCR purification kit (Qiagen), 50-100 ng purified PCR products (204-bp) were digested by restriction enzyme with 20 unit of TaqI for 2 h at 65 °C. The possible sizes of the TaqI restriction enzyme digestion products are 173 and 31 bp or 112 and 81 bp. The restriction enzyme digestion products were then visualized by electrophoresis in a 2% agarose gel, and the presence of methylation was verified in a limited number of samples by direct sequencing of PCR product. Other more, to determine the precise pattern of CpG methylation within the RASSF1A CpG island, we directly sequenced the 28 samples with RASSF1A methylation from nucleotides -110 to +41 bp after sodium bisulfite modification.
Re-expression of RASSF1A after 5-Aza-CdR treatment
QBC-939 cell lines were treated with 5-Aza-CdR (Sigma). Cells (2×106 cfu) each grown for upto 9 d with the different concentration of 0.25, 0.5, 1, 3, 5, 7, and 10 µmol/L. Isol ated RNA from the third day and detected the RNA expression using RT-PCR as described above.
RESULTS
Methylation analysis of a CpG island in promoter region of RASSF1A
Twenty-eight of 48 (58.33%) tumor samples and 1 cell line demonstrated that methylated for majority of 16 CpGs (Figure 1), and the other 20 samples were not methylated for any CpG island. After being purified and digested by restriction enzyme TaqI, every PCR band showed to be detached two bands in accordance with the same sample (Figure 2). These data displayed that bisulfite modification relieved the protection of CpG island so it can be digested by restriction enzyme TaqI.
Figure 1.
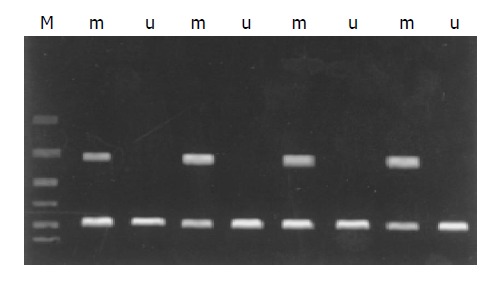
Methylation specific PCR (MSP) analysis of RASSF1A in extrahepatic cholangiocarcinoma with methylated DNA-specific primers (m) and unmethylated DNA-specific primers (u).
Figure 2.
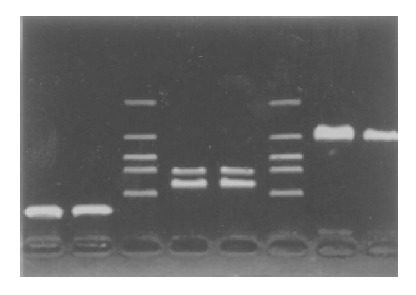
Restriction enzyme digestion of the promoter CpG island of bisulfite-treated in 4 tumors using Restriction enzyme TaqI.
Direct sequencing of the 28 samples is carried out with RASSF1A methylation from nucleotides -110 to +41 bp after sodium bisulfite modification. Four tumor samples demonstrated methylation of all 16 CpGs, and 6 had methylation at 14 of 16 CpGs, 3 at 13 of 16 CpGs, 5 at 11 of 16 CpGs, methylation at 10, 9, 8 and 7 of 16 CpGs every 2, and the last 4 samples had methylation at 4 of 16 CpGs, the average frequency of methylation of all 16 CpGs were 72.77%. Figure 3 summarizes the sequencing data of the frequency of methylation of each CpG in RASSF1A promoter region in extrahepatic cholangiocarcinoma.
Figure 3.
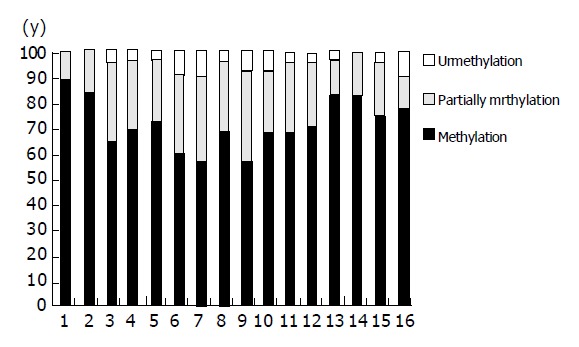
Frequency of methylation each CpG in RASSF1A promote in extrahepatic cholangiocarcinoma.
We also analyzed the methylation status of the corresponding normal extrahepatic bile duct tissues DNA, in 2 normal samples, the RASSF1A methylation was detected by the TaqI restriction digest assay, we proceeded to sequence 16 clones from each of the 2 normal samples from nucleotide -110 to +41 bp after sodium bisulfite modification. Both samples demonstrated that methylation at 10 of 16 CpGs, including those constituting one of the TaqI recognition sites. This explains the presence of TaqI digestion products, although only partial methylation of the fragment had occurred.
Mutation of RASSF1A in extrahepatic cholangiocarcinoma
We have screened all six exons of RASSF1A in 48 primary tumors and QBC-939 cell lines for mutation by SSCP analysis (Figure 4). SSCP analysis detected identical mobility shifts of exon 1α in QBC-939, DNA sequencing confirmed multiple base substitutions at codons 53, 56, 57, and 60. The sequence change in codon 60 (GCC to ACC) leads to an amino acid change from Ala to Thr, whereas those in codon 53 (CGC to CGT), 56 (CCC to CCT), and 57 (GCG to GCA) do not. Mobility shifts were also detected in 3 of 48 primary tumors. Abnormal bands on SSCP gels for exon 3, exon 4, and exon 5 were found. DNA sequencing detected the sequence changes in exon 3, a missense mutation (GCT to TCT/G to T) at nt 435 was observed. The mutation leads to an amino acid change (Ala to Ser) at codon 133. The sequencing analysis also confirmed a missense mutation of exon 4 at nt 640 (G to A) and an amino acid change (Arg to His) at codon 201. We also detected a frame shift mutation at exon 5, a single base deletion at nt 829 (A). No sequence change was found in the corresponding normal control sample of those 4 patients.
Figure 4.
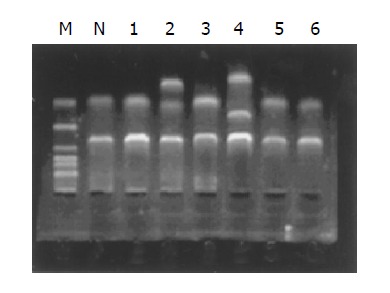
RASSF1A mutation analysis using SSCP in normal bile duct and 6 extrahepatic cholangiocarcinoma samples. It displayed that tumor 2 has a extra band, tumor 3 has a extra band with shifting. We consider that there are mutation in those two tumor samples.
LOH analysis
Allele loss was considered to be present in tumor samples with a 50% or greater reduction in signal intensity of an allele in tumor DNA compared with normal DNA[16] (Figure 5). All 4 microsatellite sites of D3S4604, D3S4597,D3S1568 and D3S16214 in chromosome 3p21.3 region, the average rate of LOH is 22.45%, very similar to the common rate of population. The highest LOH was detected at loci D3S4604 (68.75%, 33/48). The frequency of LOH at D3S4597, D3S1568, D3S1621 loci are 6.25% (3/48), 8.33% (4/48) and 4.17% (2/48), respectively.
Figure 5.
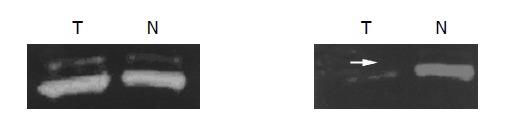
LOH analysis of microsatellite loci D3S1568 and D3S4604 at the region of chromosome 3p21.3 using the primers labeled with FAM and HEX.
Expression and re-expression of RASSF1A
In all 48 samples, there were 28 samples which RASSF1A express silencing, and QBC-939 cell lines express silencing too, which was consistent with the methylation of the RASSF1A gene at the region of promoter, but in the normal matching tissues they were all expressed. With the treatment of 5-Aza-CdR, QBC-939 cell lines RNA re-expressed in fifth-day and strongest at the concentration of 7 µmol/L. Regrettably, we have only one extrahepatic cholangiocarcinoma cell lines with methylation-positive, and have no tumor cell lines with methylation-negative, so we could not observe the influence of 5-Aza-CdR to them.
RASSF1A methylation and clinicopathological information
To determine whether the presence of RASSF1A was associated with significant differences in tumor grade and Tumor-Node-Metastasis stage, we have compared 48 samples (28 with methylation and 20 with unmethylation). However, there were no significant differences (P>0.05) between methylated and unmethylated tumor for any of these parameters. In addition, we compared RASSF1A methylated and unmethylated tumor samples for age, sex tumor size and grade, there were no significant differences too (P>0.05).
DISCUSSION
Deletion of the short arm of chromosome 3 is one of the most common genetic alteration in human cancer[17], Wong et al[7] had investigated that inactivation of the tumor suppressor genes on this region may be critical and early events for the development of this cancer. Although, multiple tumor suppressor loci have been examined, results of deletion mapping and functional studies have target the extrahepatic cholangiocarcinoma associated gene to the 3p21.3 region. Frequent allelic loss and the presence of homozygous deletions in this region have been detected in several human cancers, including carcinomas of lung, breast, ovary, head and neck cancers[10-12]. Recently, studies have focused on the candidate TSGs located within the 630-kb homozygous deletion region.
In 2000, Reinhard Dammann[11] isolated a tumor suppressor gene RASSF1 in this homozygous deletion region. The RASSF1 protein was found to be interacted with the human DNA repair protein XPA and its carboxy terminus shows high homology (55% identity) to the mouse Ras effector protein Norel and Maxp1. As the amino terminus of the cDNA showed almost no homology to these proteins, screened additional cDNA library and obtained three alternatively spliced transcripts: RASSF1A, RASSF1B and RASSF1C. All three transcripts have four common exons (exon 3-6), which encode a RAS association domain[18]. Transcript A has two 5’ exons, designed 1α and 2αβ. This cDNA is 1873 bp and contain an ORE of 340 amino acids with a calculated molecular mass of 38.8 kD. The N terminals of RASSF1A has high homology to a cysteine-rich diacylglycerol/phorbol ester binding domain, also known as the protein kinase C conserved region 1 (C1) domain. Vos et al[19] have shown that RASSF1A binds RAS in a GTP-dependent manner and mediates the apoptosis effects of oncogenic RAS. It was suggested that this protein may be involved in the DNA repair system or the RAS pathway. The most striking findings in the major transcripts of this gene, RASSF1A, is altered in the cell lines and in the primary tumors of lung cancer. The tumor suppressor function of this gene has also been demonstrated by transfection of the full-length RASSF1A in lung cancer cell lines. Re-expression of RASSF1A in lung cancer cells leads to reduced colony formation, suppressed anchorage-independent growth and inhibited tumor formation in nude mice[11]. Now, we have shown a high incidence of RASSF1A alternations in extrahepatic cholangiocarcinoma, the data presented in this study support RASSF1A as the major target suppressor at 3p21.3 in human cancers.
In our studies, we have analyzed the expression of RASSF1A mRNA in 48 samples and one cell lines, the RASSF1A express silencing in 33 samples (68.75%) and QBC-939 cell lines, the difference is significant (P = 0.001-<0.01). This suggested that RASSF1A, as a TSG, play an important role in the pathogenesis of human extrahepatic cholangiocarcinoma.
Many other investigators considered the major mechanism of TSGs inactivation include base mutation, loss of heterozygosity or deletion of homozygosity, and promoter methylation through their studies previously[12], so we assumed three hypothesis on RASSF1A inactivation: (1) Base mutation, (2) Loss of heterozygosity or deletion of homozygosity and promoter methylation separatively, (3) Loss of heterozygosity or deletion of homozygosity combine with promoter methylation.
Our data displayed that the incidence of RASSF1A mutation is very low (6.25%, 3/48), it is unclear whether these mutations alter the function of the protein or represent rare polymorphisms, which is very difficult to decide. Now, we only know a common polymorphisms exists in exon 1α [AAG(Lys21)/CAG(Gln21)], but it suggested strongly that base mutation is not the major mechanism in this gene inactivation.
Homozygous deletion of 3p21.3 has been reported in several breast cancer and lung cell lines and may be one of the major mechanisms for inactivation of RASSF1A in these cancers[10-12]. In extrahepatic cholangiocarcinoma of our investigated team, no homozygous deletion of RASSF1A was detected. The allelic status of chromosome 3p21.3 in our tumor has been examined by LOH analysis, the RASSF1A gene is located near to the microsatellite loci D3S4604, the frequency of LOH at D3S4604 is 68.75%, significantly higher than the common. So, we can believe that the LOH of RASSF1A may be the critical events for extrahepatic cholangiocarcinoma tumorigenesis.
The TSGs methylation is increasingly recognized as a major contributor to the pathogenesis of human tumors, the precise etiology of epigenetic changes in cancer were enigmatic. In extrahepatic cholangiocarcinoma, 58.33% (28/48) samples and one cell lines which RASSF1A promoter CpG islands were completely or partially methylated and induced-RASSF1A transcript the silenced. Glen et al[20] indicated that a degree of methylation of more than 50% would suggest either that both alleles are methylated or that one allele is methylated and the other one is lost, our data hold out the latter. In QBC-939,treatment with 5’-aza-deoxycytidine led to demethylation of 5’CpG island of RASSF1A and re-expression of its transcripts. This strongly suggests that aberrant hypermethylation of RASSF1A promoter is directly responsible for transcriptional inactivation of its expression in our investigated tumor samples.
We divided the 28 samples in which promoters were methylation into LOH-positive and LOH-negative team and study the relationship LOH and promoter methylation. In LOH-positive team, 25 of 33 (75.76%) samples had methylation in CpG islands of RASSF1A promoter, and the contrast data is 15% (3/20) in LOH-negative team. Furthermore, the average frequence of all CpG islands with LOH-positive is significantly higher then LOH-negative team (P<0.01). Our data accord with the revised Knudson two-hit theory[21], in this new hypothesis, epigenetic mechanism of gene inactivation are included.
In summary, we have demonstrated a high frequency of RASSF1A gene in extrahepatic cholangiocarcinoma. In 58.33% of primary tumors, either promoter hypermethylation or LOH of RASSF1A were seen, these observation provide that promoter methylation and allelic loss are the major mechanism for inactivation of this tumor suppressor gene, RASSF1A may be one of the key tumor suppressor gene that we have been searching for at 3p21.3 in extrahepatic cholangiocarcinoma. Inactivation of its function, resulting in either defective DNA repair or disruption of the RAS pathway, may be closely associated with extrahepatic cholangiocarcinoma occurrence and development.
Footnotes
Supported by the National High Technology Research and Development Program of China (863 Program), No. 2002AA214061
Assistant Editor Guo SY Edited by Gabbe M
References
- 1.Liu C, Wang J, Ou QJ. Possible stem cell origin of human cholangiocarcinoma. World J Gastroenterol. 2004;10:3374–3376. doi: 10.3748/wjg.v10.i22.3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel T. Worldwide trends in mortality from biliary tract malignancies. BMC Cancer. 2002;2:10. doi: 10.1186/1471-2407-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ichikawa K, Imura J, Kawamata H, Takeda J, Fujimori T. Down-regulated p16 expression predicts poor prognosis in patients with extrahepatic biliary tract carcinomas. Int J Oncol. 2002;20:453–461. doi: 10.3892/ijo.20.3.453. [DOI] [PubMed] [Google Scholar]
- 4.Niiyama H, Mizumoto K, Kusumoto M, Ogawa T, Suehara N, Shimura H, Tanaka M. Activation of telomerase and its diagnostic application in biopsy specimens from biliary tract neoplasms. Cancer. 1999;85:2138–2143. doi: 10.1002/(sici)1097-0142(19990515)85:10<2138::aid-cncr7>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 5.Ito Y, Takeda T, Sasaki Y, Sakon M, Yamada T, Ishiguro S, Imaoka S, Tsujimoto M, Monden M, Matsuura N. Expression of p57/Kip2 protein in extrahepatic bile duct carcinoma and intrahepatic cholangiocellular carcinoma. Liver. 2002;22:145–149. doi: 10.1034/j.1600-0676.2002.01532.x. [DOI] [PubMed] [Google Scholar]
- 6.Abraham SC, Lee JH, Boitnott JK, Argani P, Furth EE, Wu TT. Microsatellite instability in intraductal papillary neoplasms of the biliary tract. Mod Pathol. 2002;15:1309–1317. doi: 10.1097/01.MP.0000038461.80167.34. [DOI] [PubMed] [Google Scholar]
- 7.Wong N, Li L, Tsang K, Lai PB, To KF, Johnson PJ. Frequent loss of chromosome 3p and hypermethylation of RASSF1A in cholangiocarcinoma. J Hepatol. 2002;37:633–639. doi: 10.1016/s0168-8278(02)00269-6. [DOI] [PubMed] [Google Scholar]
- 8.Lo KW, Teo PM, Hui AB, To KF, Tsang YS, Chan SY, Mak KF, Lee JC, Huang DP. High resolution allelotype of microdissected primary nasopharyngeal carcinoma. Cancer Res. 2000;60:3348–3353. [PubMed] [Google Scholar]
- 9.Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet. 2000;25:315–319. doi: 10.1038/77083. [DOI] [PubMed] [Google Scholar]
- 10.Lo KW, Kwong J, Hui AB, Chan SY, To KF, Chan AS, Chow LS, Teo PM, Johnson PJ, Huang DP. High frequency of promoter hypermethylation of RASSF1A in nasopharyngeal carcinoma. Cancer Res. 2001;61:3877–3881. [PubMed] [Google Scholar]
- 11.Dreijerink K, Braga E, Kuzmin I, Geil L, Duh FM, Angeloni D, Zbar B, Lerman MI, Stanbridge EJ, Minna JD, et al. The candidate tumor suppressor gene, RASSF1A, from human chromosome 3p21.3 is involved in kidney tumorigenesis. Proc Natl Acad Sci USA. 2001;98:7504–7509. doi: 10.1073/pnas.131216298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu L, Yoon JH, Dammann R, Pfeifer GP. Frequent hypermethylation of the RASSF1A gene in prostate cancer. Oncogene. 2002;21:6835–6840. doi: 10.1038/sj.onc.1205814. [DOI] [PubMed] [Google Scholar]
- 13.Burbee DG, Forgacs E, Zöchbauer-Müller S, Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S, et al. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst. 2001;93:691–699. doi: 10.1093/jnci/93.9.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laghi L, Orbetegli O, Bianchi P, Zerbi A, Di Carlo V, Boland CR, Malesci A. Common occurrence of multiple K-RAS mutations in pancreatic cancers with associated precursor lesions and in biliary cancers. Oncogene. 2002;21:4301–4306. doi: 10.1038/sj.onc.1205533. [DOI] [PubMed] [Google Scholar]
- 15.Lieberfarb ME, Lin M, Lechpammer M, Li C, Tanenbaum DM, Febbo PG, Wright RL, Shim J, Kantoff PW, Loda M, et al. Genome-wide loss of heterozygosity analysis from laser capture microdissected prostate cancer using single nucleotide polymorphic allele (SNP) arrays and a novel bioinformatics platform dChipSNP. Cancer Res. 2003;63:4781–4785. [PubMed] [Google Scholar]
- 16.Mitsudomi T, Oyama T, Nishida K, Ogami A, Osaki T, Sugio K, Yasumoto K, Sugimachi K, Gazdar AF. Loss of heterozygosity at 3p in non-small cell lung cancer and its prognostic implication. Clin Cancer Res. 1996;2:1185–1189. [PubMed] [Google Scholar]
- 17.Chan AS, To KF, Lo KW, Mak KF, Pak W, Chiu B, Tse GM, Ding M, Li X, Lee JC, et al. High frequency of chromosome 3p deletion in histologically normal nasopharyngeal epithelia from southern Chinese. Cancer Res. 2000;60:5365–5370. [PubMed] [Google Scholar]
- 18.Ponting CP, Benjamin DR. A novel family of Ras-binding domains. Trends Biochem Sci. 1996;21:422–425. doi: 10.1016/s0968-0004(96)30038-8. [DOI] [PubMed] [Google Scholar]
- 19.Vos MD, Ellis CA, Bell A, Birrer MJ, Clark GJ. Ras uses the novel tumor suppressor RASSF1 as an effector to mediate apoptosis. J Biol Chem. 2000;275:35669–35672. doi: 10.1074/jbc.C000463200. [DOI] [PubMed] [Google Scholar]
- 20.Dammann R, Yang G, Pfeifer GP. Hypermethylation of the cpG island of Ras association domain family 1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3 locus, occurs in a large percentage of human breast cancers. Cancer Res. 2001;61:3105–3109. [PubMed] [Google Scholar]
- 21.Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157–162. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]