

Abstract
AIM: To define the infection status of Helicobacter pylori in 109 patients with gastric cancers and H pylori localization in gastric carcinoma tissues in South China.
METHODS: The incidence of H pylori infection in gastric carcinomas was estimated by polymerase chain reaction (PCR), simultaneously; both morphological features and the localization of H pylori in gastric carcinomas were demonstrated by Warthin-Starry (WS) staining. The relationships between H pylori infection and the clinical-pathologic factors of gastric carcinomas were analyzed by software SPSS10.0.
RESULTS: H pylori was found in 42 (39.03%) and 58 (53.21%) cases of 109 patients with gastric carcinomas by PCR and WS, respectively. H pylori infection rate detected in gastric carcinomas by WS was higher than that by PCR (χ2 = 9.735, P<0.005<0.01). WS stain showed that H pylori existed in the gastric antrum mucus, mucosal gland of normal tissues adjacent to gastric carcinomas and the gland, mucus pool of cancer tissues. The positive rate of H pylori in normal tissues adjacent to carcinomas was higher than that in cancer tissues (χ2 = 15.750, P<0.005<0.01). No significant differences in age, sex, site, histological types and lymph node metastasis were found between H pylori-positive gastric carcinomas and H pylori-negative cases by both methods, but there were statistically significant differences of H pylori positive rate between early and advanced stage of gastric carcinomas (χ2 = 4.548 or 5.922, P = 0.033 or 0.015<0.05).
CONCLUSION: These results suggested that H pylori infection might play a certain role in the early stage of carcinogenesis of human gastric mucosa epithelia.
Keywords: Gastric carcinoma, H pylori, Polymerase chain reaction, Warthin-Starry staining
INTRODUCTION
Gastric carcinoma is one of the most common human malignant cancers in the world. Although the etiopathological mechanism of human gastric cancer remains unclear until now, most researchers think that the pathogenesis of human gastric cancer is a multifactorial, multistage and multistep process[1-3]. The epidemiological and histopathological studies have shown that Helicobacter pylori (H pylori) infection is closely associated with gastric carcinogenesis[4,5] and H pylori infection is also one of the factors of gastric carcinogenesis studied at present[6,7]. Although there were discrepancies among epidemiological studies[8-12], some meta-analyses indicated that the magnitude of the association of H pylori infection and risk of gastric cancer was ORs = 2-6[13-17]. However, the location of H pylori in the gastric cancer tissues, the mechanism and the stages in which H pylori participates in the process of gastric carcinogenesis are largely unknown. In the present study, we have detected H pylori DNA by polymerase chain reaction (PCR) and observed H pylori location in 109 cases of gastric carcinoma by Warthin-Starry (WS) stain and analyzed relationships between H pylori infection and the clinical-pathologic characteristics of gastric carcinomas.
MATERIALS AND METHODS
Patients
All of the 109 patients had not accepted radiotherapy and chemotherapy; their histological features were diagnosed in the Pathology Department of the First Affiliated Hospital and Cancer Research Institute of Nanhua University.
Seventy-seven specimens of paraffin-embedded gastric carcinoma tissues and 32 cases of resection specimens with gastric carcinomas were collected from First Affiliated Hospital of Nanhua University in 2000-2002. The fresh tissues were cut into 300-500 mg blocks suitable for sectioning, either frozen in -70 °C deepfreezed refigerabo, or fixed in formalin and embedded in paraffin.
Main reagents and dispensing
The primers for ureA gene of H pylori were synthesized in Shanghai Sangon Biological Engineering Company. Phenol: chloroform:isoamyl alcohol (25:24:1), Protease K, RNAse, DNA Marker, Taq DNA polymerase and dNTP were bought from Shanghai Songon Biological Engineering Company. 1×SSC (150 mmol/L sodium chloride, 15 mmol/L citromalic acid sodium, pH 7.0), paraffin digestion buffer (150 mmol/L sodium chloride, 15 mmol/L citromalic acid sodium, 1% sodium dodecyl sulfate (SDS)), TNE (0.1 mol/L Tris, 10 mmol/L EDTA, 2.0 mol/L NaCl, 10% SDS).
DNA extraction
DNA of H pylori was extracted from control cells, resection tissue and serial section of the paraffin wax embedded material as described previously[18]. PCR amplification of 411-bp fragment from the ureA gene of H pylori was carried out as described by Monstein et al[19].
With 1 mL saline, 400 mg frozen fresh tissue blocks for each case of 32 human gastric carcinomas were washed thrics, shearsed to microparticles in an Eppendorf tube and then suspended in 400 μL TNE buffer containing 1 mg/mL protease K, incubated at 55 °C overnight (12-24 h).
Twenty 6-μm-thick sections of gastric carcinoma tissues were cut from each case of 77 paraffin wax embedded block and placed in an Eppendorf tube and then thoroughly dewaxed in warm xylene four times and re-hydrated by passage through graded alcohols (100, 95, 75, 50% for 30 min, respectively). The supernatant was decanted with each tube vortexed again and centrifugated at 14000 g for 3 min. The last precipitations were re-suspended and vortexed in 700 μL 1×SSC buffer. The supernatant was decanted again after another 3-min centrifugation, and the remaining ethanol was removed with a micro capillary pipette. The precipitations were dried, and then resuspended in 400 μL paraffin wax digestion buffer containing 1 mg/mL protease K, incubated for 120 h at 55 °C in a swing bed. On the fourth d, 10 μL×20 mg/mL protease K was added to each tube.
DNA of each specimen was purified by an organic extraction step. An equal volume of phenol:chloroform:isoamyl alcohol (25:24:1) was added to each sample after digestion. After 3-min centrifugation at 14000 g, it was mixed thoroughly and centrifuged to separate the aqueous and organic layers. The upper aqueous layer containing DNA was collected and the extraction was repeated. DNA samples were precipitated with two volumes of ethanol precooled (-20 °C) and one-tenth volume of 3 mol/L sodium acetate (pH 5.2) at -20 °C for 1 h, centrifuged for 10 min of 14000 g, dried and redissolved in 50 μL Tris-EDTA buffer containing 20 μg/μL RNAse. DNA samples extracted were assayed quantitatively with ultraviolet spectrophotometer with the value of A260/A280 from 1.7 to 2.0, finally stored at -20 °C.
PCR amplification
Two primers for the detection of H pylori were designed according to DNA sequences described by Monstein et al[19], and synthesized in Shanghai Sangon Biological Engineering Company. The ureA-H pylori primers used were the upstream: 5’GCCAATGGTAAATTAGTT; and the downstream: 5’CTCCTTAATTGTTTTTAC, to amplify a 411 bp fragment of ureA gene region of H pylori DNA. The PCR reaction mixture contained 2.5 μL 10×Taq polymerase buffer (500 mmol/L KCl, 100 mmol/L Tris-HCl, 1.5 mmol/L MgCl2 and 0.1% gelatin), 200 μmol/L dNTPs, 0.2 μmol/L primers, 1.25 u Taq polymerase and 0.25 μg DNA samples purified in a final reaction volume of 25 μL. After an initial 5-min incubation at 94 °C in the Biometra Thermal Cycler to fully denature the template DNA, the reaction mixtures were processed through 35 PCR cycles of 1-min denaturation at 94 °C, 1-min annealing at 47 °C, and 1-min extension at 72 °C, followed by 10 min at 72 °C to ensure that all the products were fully extended. Each PCR experiment included a positive control of DNA from H pylori NCTC 11 637 and a negative control of TE in place of DNA. Ten microliters of reaction mixtures were loaded to 1.5% agarose gel for electrophoresis. Being stained with ethidium bromide in a DNA subcell at 100 V for 30 min, the gel was then observed under ultraviolet ray. The amplified products of ureA gene were at the band of 411 bp. If an orange band appeared on the band of 411 bp, which was identical to the product from positive control well, the result was thought to be positive or the sample was thought to have been infected with H pylori.
Warthin-Starry staining
Five-micrometers thick paraffin sections from each case of 109 gastric carcinoma tissues were mounted on slides and backed for 1 h at 60 °C. After routine dewaxing and re-hydration with deionized water, tissue sections were washed twice with 0.2 mol/L acetic acid buffer, incubated at 56 °C for 1 h in 1% silver nitrate buffer in the dark box, then dipped in developer solution and stained for 3-8 min. After dipping into distilled water at 56 °C and washed for 2 min, sections were washed once more with distilled water, then dehydrated with 100% alcohol, cleared with xylene, and mounted with neutral gum. Under microscope observation, H pylori was stained into buffy or black color, and the background was light yellow.
Statistical analysis
Results of H pylori detection and the clinical-pathologic parameters of 109 cases of gastric carcinomas were statistically analyzed with software SPSS10.0.
RESULTS
PCR detection of H pylori in gastric carcinomas
PCR amplification products of H pylori DNA were presented in 42 (39.03%) cases of gastric carcinomas. Only one positive orange band was presented on the band of 411 bp from positive control and positive samples, but no amplification band was found from negative control and negative samples. PCR products were evaluated by 1.5% agarose gel electrophoresis, as shown in Figure 1. Positive bands were presented in 17 (53.12%) from frozen tissues with gastric carcinomas and in 25 (32.47%) from paraffin-section tissues with gastric carcinomas. It is quite obvious that there was a statistically significant difference between frozen tissues and paraffin sections (0.01<P<0.05).
Figure 1.
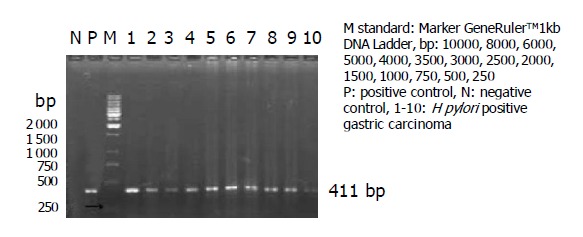
1.5% agarose gel electrophoresis of PCR products, showing amplification of 411-bp fragment of ureA gene from DNA extracted.
Detection of H pylori by WS stain
WS stain showed 58 out of 109 cases (53.21%) of tissue sections carrying bacterial bodies of H pylori from 109 gastric carcinomas, and the positive rate was higher than that detected by PCR method. Simultaneously, shape and location of H pylori could be clearly seen (Figure 2), H pylori of different sizes showed different forms such as curve, S-shape, arc or stem. In non-tumor tissues adjacent to gastric cancers, some bacteria gathered together just like shoals of fish. However, in tumor tissues, they diffused in every direction. In 25 of 58 cases with H pylori-positive gastric carcinomas, bacterial bodies of H pylori were observed in the mucus, mucosal gland cavum, epithelial cell plasmas of normal tissues adjacent to cancers. Bacterial bodies of H pylori in 3 of 58 cases were observed in the gland, mucous pool and plasma of cancer cells. H pylori was observed in both tumor and non-tumor sites in 30 of 109 cases with gastric carcinomas (Table 1). By χ2 test (χ2 = 15.750, P<0.005<0.01), H pylori positive rate in non-tumor sites was higher than that in tumor sites.
Figure 2.
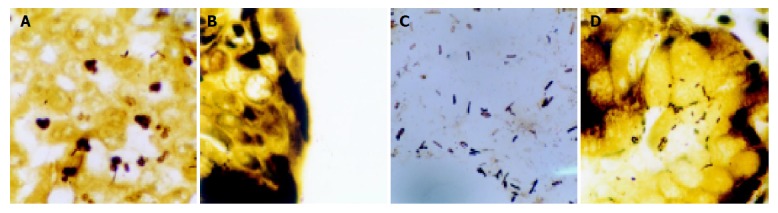
H pylori in different sites. A: H pylori in cancer cell plasmas and cancer tissues; B: H pylori in mucous pool of mucous adenocarcinoma; C: H pylori in the mucus of gastric cavum; D: H pylori in mucosal gland cavum, epithelial cell plasmas of normal tissues adjacent to cancer. WS stain ×1000.
Table 1.
Numbers of H pylori positive cases in different histological sites.
| Tumor site |
No tumor site |
Total | |
| + | - | ||
| + | 30 | 3 | 33 |
| - | 25 | 51 | 76 |
| Total | 55 | 54 | 109 |
The results of H pylori in 109 cases with gastric carcinomas were statistically analyzed by Pearson Chi-Square of SPSS10.0, which were χ2 = 9.735, P<0.005<0.01 (Table 2). There was significant difference in H pylori positive rates detected by PCR and WS methods, respectively.
Table 2.
Detection of H pylori by WS stain and PCR.
| PCR |
WS stain |
Total | |
| + | - | ||
| + | 38 | 4 | 42 |
| - | 20 | 47 | 67 |
| Total | 58 | 51 | 109 |
Relationships between H pylori infection and the clinical-pathologic characteristics of human gastric carcinomas
On the basis of data in Table 3, the results were statistically analyzed by Pearson Chi-Square of SPSS10.0. No statistically significant differences in age, sex, site and lymph node metastasis were found between H pylori-positive cases and H pylori-negative cases of gastric carcinomas by both methods of PCR and WS.
Table 3.
Relationships between H pylori infection and gastric carcinomas in age, sex, site and lymph node metastasis.
| Factors | Total cases |
H pylori positive |
χ2 | P | |
| PCR | WS | ||||
| Age (yr) | |||||
| ≥60 | 29 | 7 | 11 | 1.999P | 0.157P |
| <60 | 80 | 35 | 47 | 3.705W | 0.054W |
| Sex | |||||
| Male | 64 | 23 | 33 | 1.131P | 0.288P |
| Female | 45 | 19 | 25 | 2.500W | 0.114W |
| Locus | |||||
| Gastric antrum | 55 | 22 | 29 | 1.221P | 0.269P |
| Gastric fundus and gastric corpus | 54 | 20 | 29 | 0.079W | 0.778W |
| Lymph node metastasis | |||||
| LN+ | 66 | 26 | 36 | 0.052P | 0.819P |
| LN- | 43 | 16 | 22 | 0.120W | 0.440W |
P: PCR results, W: WS results.
On the basis of data in Table 4, the results were statistically analyzed by Pearson Chi-Square of SPSS10.0, and no significant differences of H pylori detection were shown among four types of gastric carcinoma according to histological morphology by both PCR and WS stain.
Table 4.
H pylori infection comparison with histological types of gastric carcinoma.
| Histological types | Total cases |
PCR |
WS |
||
| P | N | P | N | ||
| Well-differentiated adenocarcinoma | 23 | 9 | 14 | 12 | 11 |
| Poorly-differentiated adenocarcinoma | 49 | 16 | 33 | 24 | 25 |
| Signet-ring cell carcinoma | 17 | 7 | 10 | 11 | 6 |
| Mucous adenocarcinoma | 20 | 10 | 10 | 11 | 9 |
The positive rates of H pylori DNA were 62.5 and 34.4% in 16 cases of early gastric carcinomas and 93 cases of advanced gastric carcinomas by PCR detecting H pylori, respectively. Whereas, the positive rates of H pylori were 75.0 and 49.5% in early gastric carcinomas and advanced gastric carcinomas by WS, respectively. The results showed that statistically significant differences between H pylori-positive gastric carcinomas and H pylori-negative cases were found in clinical stages of gastric carcinomas (χ2 = 4.548, 5.922, P = 0.033 or P = 0.015<0.05), and H pylori positive rate of early gastric carcinomas was higher than that of the advanced gastric carcinomas (Table 5).
Table 5.
Relationships between H pylori infection and clinical stages of gastric carcinomas.
| Clinical stage | Total cases |
Positive |
Negative |
χ2 | P | ||
| PCR | WS | PCR | WS | ||||
| Early | 16 | 10 | 12 | 6 | 4 | 4.548P | 0.033P |
| Advanced | 93 | 32 | 46 | 61 | 47 | 5.922W | 0.015W |
P: PCR results, W: WS results.
DISCUSSION
UreA gene ordinarily exists in the genome of H pylori, so amplification of 411-bp fragment nucleotides is used to detect the presence of H pylori. However, its sensibility is determined by the primers selected, the source of DNA and quantity of bacterial presented in tissues. The fragment of 411 bp of ureA gene was amplified by PCR to detect the presence of H pylori genome in this study. The positive rate detected by PCR method was lower than that by WS stain. The possible explanation: (1) The order of magnitude of bacterial detected by PCR was over one hundred, whereas H pylori existing in the section could be easily detected by WS stain when it was carefully observed under light microscope by an experienced observer; (2) When the presence of H pylori genome was detected by PCR, its sensibility was closely related to the size of fragment amplified by PCR. Because the fragment amplified by PCR was longer, DNA degradation or fragmentation might take place in treatment process of tissue samples, consequently, the amplification efficiency was lower. Since major samples of gastric carcinomas in the present study were paraffin wax embedded blocks (77/109 cases), DNA degradation or fragmentation might take place in fixation and section of tissue samples and amplification efficiency was lower. The positive rate of H pylori was 53.12% from fresh tissues, whereas 32.47% from paraffin wax embedded blocks. The difference might be avoided by small fragment primers to amplify DNA fragments, Fabre et al[20] detected the presence of H pylori genome by using fresh tissues and primers to amplify 210 bp fragments, and their result showed that the positive rate by PCR was identical to that by Giemsa stain.
By WS stain technology H pylori were observed under light microscope, which lied in the mucus, mucosal gland cavum, epithelial cell plasma of normal tissues adjacent to cancer and the gland, mucous pool, cancer cell plasmas of cancer tissues. But the H pylori positive rate of normal tissues adjacent to cancer was higher than that of cancer tissues. In non-tumor tissues, bacteria gathered together just like shoals of fish. However, H pylori diffused in every direction in tumor tissues. The reason was regarded that acidity environments in stomach were suitable for H pylori breeding, but with the formation of stomach cancer, constitution structure and microenvironments of gastric mucosa were changed correspondently, which were not suitable for H pylori surviving and led to loss of H pylori[21].
In addition, H pylori positive rate at early gastric cancer was higher than that at advanced gastric cancer, which suggested H pylori might be partly involved in the occurrence and development process of early gastric cancer. Accumulative infection and movement of H pylori might play an important role in the development of gastric cancer. In vitro experiments showed that H pylori at low inocula stimulated cell proliferation. But at higher inocula (bacteria to cell ratio >100), it caused a time- and concentration-dependent reduction of cell cycle, which would be arrested at G1 phase, inhibit gastric cancer cell proliferation and induce apoptosis.
H pylori infection can lead to excretion of gastric acid and decrease in ascorbic acid in the gastric gland, cause gastric epithelial cell proliferation by long-term urgent and chronic damage to gastric mucosa and induce long-term tolerant inflammation response, so as to increase carcinogenesis of carcinogen. In a human model of gastric carcinogenesis, Correa thought that H pylori might play a role of precursor in gastric cancer, colonic metaplasia was a high-risk -factor of gastric cancer and H pylori positive colonic metaplasia along, but should also be considered as a high risk factor of gastric cancer. Cahill divided 151 patients into normal mucosa and H pylori negative group, chronic active gastritis and H pylori positive group, chronic atrophic gastritis group, intestinal metaplasia group and gastric carcinoma group. Gastric antral epithelial cell proliferation was assessed as the labeling index percent. The results showed that from chronic active gastritis group to gastric carcinoma one, epithelial cell proliferation increased when compared with that of normal mucosa, which was associated with H pylori infection. The increase in gastric epithelial cell proliferation associated with H pylori infection was not significantly different from that associated with the gastric precancerous lesions. H pylori infection, however, did not seem to influence the changed gastric epithelial cell proliferation in subjects with precancerous lesions or gastric cancer, which suggested that H pylori played a certain role in early gastric carcinogenesis, although it might not have so strong an influence in the later stages of the disease as that in early ones[22]. Watanabe et al[23] first reported that 5-wk-old Mongolian gerbils were orally inoculated with H pylori and infected alone with H pylori and induced gastric carcinomas, that could be located in the pyloric region. After the 26th wk, severe active chronic gastritis, ulcers, and intestinal metaplasia could be observed in the infected animals. After the 62nd wk, adenocarcinoma had developed in the pyloric region of 37% (10/27) of the infected animals. It was found that adenocarcinoma development seemed to be closely related to intestinal metaplasia. After this, 5-wk-old Mongolian gerbils were infected with H pylori ATCC-43504 strain by Honda et al[24]. It was reported that atrophic gastritis and intestinal metaplasia also appeared in the lesser curvature of the ventral mucosa 6 mo after inoculation. Eighteen months after H pylori inoculation, 40% (2/5) infected Mongolian gerbils showed three well-differentiated gastric cancer. Because of the use of 5-wk-old Mongolian gerbils for study, it suggested H pylori infection of early stage might be one of the risk-factors increasing carcinogenesis of gastric cancer. Both of the two groups showed the pathway of “H pylori→atrophic gastritis→intestinal metaplasia→atypical hyperplasia→intestinal-type gastric cancer”.
These results suggested that H pylori infection might play a certain role in the early stage of carcinogenesis of gastric mucosa epithelia.
Footnotes
Co-correspondents: Run-Liang Gan
Assistant Editor Li WZ Edited by Gabbe M
References
- 1.Hiyama T, Haruma K, Kitadai Y, Masuda H, Miyamoto M, Tanaka S, Yoshihara M, Shimamoto F, Chayama K. K-ras mutation in helicobacter pylori-associated chronic gastritis in patients with and without gastric cancer. Int J Cancer. 2002;97:562–566. doi: 10.1002/ijc.1644. [DOI] [PubMed] [Google Scholar]
- 2.Menaker RJ, Sharaf AA, Jones NL. Helicobacter pylori infection and gastric cancer: host, bug, environment, or all three? Curr Gastroenterol Rep. 2004;6:429–435. doi: 10.1007/s11894-004-0063-9. [DOI] [PubMed] [Google Scholar]
- 3.Lee SG, Kim B, Yook JH, Oh ST, Lee I, Song K. TNF/LTA polymorphisms and risk for gastric cancer/duodenal ulcer in the Korean population. Cytokine. 2004;28:75–82. doi: 10.1016/j.cyto.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Fujioka T, Murakami K, Kodama M, Kagawa J, Okimoto T, Sato R. Helicobacter pylori and gastric carcinoma--from the view point of animal model. Keio J Med. 2002;51 Suppl 2:69–73. doi: 10.2302/kjm.51.supplement2_69. [DOI] [PubMed] [Google Scholar]
- 5.Wang J, Chi DS, Kalin GB, Sosinski C, Miller LE, Burja I, Thomas E. Helicobacter pylori infection and oncogene expressions in gastric carcinoma and its precursor lesions. Dig Dis Sci. 2002;47:107–113. doi: 10.1023/a:1013223722331. [DOI] [PubMed] [Google Scholar]
- 6.Leung WK, Lin SR, Ching JY, To KF, Ng EK, Chan FK, Lau JY, Sung JJ. Factors predicting progression of gastric intestinal metaplasia: results of a randomised trial on Helicobacter pylori eradication. Gut. 2004;53:1244–1249. doi: 10.1136/gut.2003.034629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nardone G, Morgner A. Helicobacter pylori and gastric malignancies. Helicobacter. 2003;8 Suppl 1:44–52. doi: 10.1046/j.1523-5378.2003.00160.x. [DOI] [PubMed] [Google Scholar]
- 8.Hofman P, Waidner B, Hofman V, Bereswill S, Brest P, Kist M. Pathogenesis of Helicobacter pylori infection. Helicobacter. 2004;9 Suppl 1:15–22. doi: 10.1111/j.1083-4389.2004.00246.x. [DOI] [PubMed] [Google Scholar]
- 9.Inoue M, Tajima K, Matsuura A, Suzuki T, Nakamura T, Ohashi K, Nakamura S, Tominaga S. Severity of chronic atrophic gastritis and subsequent gastric cancer occurrence: a 10-year prospective cohort study in Japan. Cancer Lett. 2000;161:105–112. doi: 10.1016/s0304-3835(00)00603-0. [DOI] [PubMed] [Google Scholar]
- 10.Hwang IR, Kodama T, Kikuchi S, Sakai K, Peterson LE, Graham DY, Yamaoka Y. Effect of interleukin 1 polymorphisms on gastric mucosal interleukin 1beta production in Helicobacter pylori infection. Gastroenterology. 2002;123:1793–1803. doi: 10.1053/gast.2002.37043. [DOI] [PubMed] [Google Scholar]
- 11.Ishizuka J, Sugiyama T, Aoyama T, Hirayama F, Tada M, Kato M, Moriuchi T, Asaka M. Molecular cloning of p53 cDNA of Mongolian gerbil and establishment of yeast p53 functional assay system. Helicobacter. 2003;8:81–89. doi: 10.1046/j.1523-5378.2003.00127.x. [DOI] [PubMed] [Google Scholar]
- 12.Yamagata H, Kiyohara Y, Aoyagi K, Kato I, Iwamoto H, Nakayama K, Shimizu H, Tanizaki Y, Arima H, Shinohara N, et al. Impact of Helicobacter pylori infection on gastric cancer incidence in a general Japanese population: the Hisayama study. Arch Intern Med. 2000;160:1962–1968. doi: 10.1001/archinte.160.13.1962. [DOI] [PubMed] [Google Scholar]
- 13.Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology. 1998;114:1169–1179. doi: 10.1016/s0016-5085(98)70422-6. [DOI] [PubMed] [Google Scholar]
- 14.Danesh J. Helicobacter pylori infection and gastric cancer: systematic review of the epidemiological studies. Aliment Pharmacol Ther. 1999;13:851–856. doi: 10.1046/j.1365-2036.1999.00546.x. [DOI] [PubMed] [Google Scholar]
- 15.Eslick GD, Lim LL, Byles JE, Xia HH, Talley NJ. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol. 1999;94:2373–2379. doi: 10.1111/j.1572-0241.1999.01360.x. [DOI] [PubMed] [Google Scholar]
- 16.Gastric cancer and Helicobacter pylori: a combined analysis of 12 case control studies nested within prospective cohorts. Gut. 2001;49:347–353. doi: 10.1136/gut.49.3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue FB, Xu YY, Wan Y, Pan BR, Ren J, Fan DM. Association of H. pylori infection with gastric carcinoma: a Meta analysis. World J Gastroenterol. 2001;7:801–804. doi: 10.3748/wjg.v7.i6.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Satoh Y, Takasaka N, Hoshikawa Y, Osaki M, Ohfuji S, Ito H, Kaibara N, Kurata T, Sairenji T. Pretreatment with restriction enzyme or bovine serum albumin for effective PCR amplification of Epstein-Barr virus DNA in DNA extracted from paraffin-embedded gastric carcinoma tissue. J Clin Microbiol. 1998;36:3423–3425. doi: 10.1128/jcm.36.11.3423-3425.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monstein HJ, Ellnebo-Svedlund K. Molecular typing of Helicobacter pylori by virulence-gene based multiplex PCR and RT-PCR analysis. Helicobacter. 2002;7:287–296. doi: 10.1046/j.1523-5378.2002.00099.x. [DOI] [PubMed] [Google Scholar]
- 20.Fabre R, Sobhani I, Laurent-Puig P, Hedef N, Yazigi N, Vissuzaine C, Rodde I, Potet F, Mignon M, Etienne JP. Polymerase chain reaction assay for the detection of Helicobacter pylori in gastric biopsy specimens: comparison with culture, rapid urease test, and histopathological tests. Gut. 1994;35:905–908. doi: 10.1136/gut.35.7.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGuigan JE. Helicobacter pylori: the versatile pathogen. Dig Dis. 1996;14:289–303. doi: 10.1159/000171560. [DOI] [PubMed] [Google Scholar]
- 22.Cahill RJ, Kilgallen C, Beattie S, Hamilton H, O'Morain C. Gastric epithelial cell kinetics in the progression from normal mucosa to gastric carcinoma. Gut. 1996;38:177–181. doi: 10.1136/gut.38.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology. 1998;115:642–648. doi: 10.1016/s0016-5085(98)70143-x. [DOI] [PubMed] [Google Scholar]
- 24.Honda S, Fujioka T, Tokieda M, Satoh R, Nishizono A, Nasu M. Development of Helicobacter pylori-induced gastric carcinoma in Mongolian gerbils. Cancer Res. 1998;58:4255–4259. [PubMed] [Google Scholar]