

Abstract
AIM: To study the effect of gelatinases (especially MMP-9) on migration of tissue inhibitor of metalloproteinase (TIMP-1) overexpressing hepatoma cells.
METHODS: Wild type HepG2 cells, cells stably transfected with TIMP-1 and TIMP-1 antagonist (MMP-9-H401A, a catalytically inactive matrix metalloproteinase (MMP) which still binds and neutralizes TIMP-1) were incubated in Boyden chambers either with or without Galardin (a synthetic inhibitor of MMP-1, -2, -3, -8, -9) or a specific inhibitor of gelatinases.
RESULTS: Compared to wild type HepG2 cells, the cells overexpressing TIMP-1 showed 115% migration (P<0.05) and the cells overexpressing MMP-9-H401A showed 62% migration (P<0.01). Galardin reduced cell migration dose dependently in all cases. The gelatinase inhibitor reduced migration in TIMP-1 overexpressing cells predominantly. Furthermore, we examined intracellular signal transduction pathways of TIMP-1-dependent HepG2 cells. TIMP-1 deactivates cell signaling pathways of MMP-2 and MMP-9 involving p38 mitogen-activated protein kinase. Specific blockade of the ERK pathway suppresses gelatinase expression either in the presence or absence of TIMP-1.
CONCLUSION: Overexpressing functional TIMP-1- enhanced migration of HepG2-TIMP-1 cells depends on enhanced MMP-activity, especially MMP-9.
Keywords: HepG2, MMP-2, MMP-9, p38MAPK, Galardin, Genistein
INTRODUCTION
Proteins of the matrix metalloproteinases (MMPs), a family of structurally related zinc-dependent enzymes, are involved in the breakdown of extracellular matrix (ECM) in normal physiological processes, such as embryonic development, reproduction, and tissue remodeling as well as in disease processes, such as arthritis, tumor cell invasion, migration, and metastasis[1-3]. Most MMPs are secreted as inactive proteins, which are activated when cleaved by extracellular proteinases. MMPs may be expressed on the surface of cells, thus allowing a precise and localized proteolysis[4]. The gelatinases MMP-2 and MMP-9 are necessary for the migration of Langerhans cells and dermal dendritic cells from human and murine skin[5]. MMP-2 (gene ID 4313 on chromosome 16q13-q21) e.g., is involved in cell migration of human lung carcinoma cells[6]. MMP-9 (gene ID 4318, chromosome 20, q11.2-q13.1) is a major secretion product of macrophages and a component of cytoplasmic granules of neutrophils[7]. The enzyme is also secreted by lymphocytes and stromal cells upon stimulation by inflammatory cytokines, or upon delivery of bidirectional activation signals following integrin-mediated cell-cell or cell-matrix contacts[8]. MMP-9 plays an important role in tumor invasion and angiogenesis. Secretion has been reported in various cancer types including lung, colon, and breast cancer. The activity of MMPs is tightly controlled by endogenous specific inhibitors, the tissue inhibitors of metalloproteinases (TIMPs). The balance between activated MMPs and their free inhibitors determines the net MMP activity and seems to be of great importance for matrix protein turnover associated with tumor cell invasion and metastasis. A striking phenotypic characteristic of fibrotic liver is a dramatic up-regulation of TIMP-1 (GenBank number NM 003254.1, chromosome X, Xp11.3-p11.23), which seems to be a functional promotor of hepatic fibrosis[9,10]. But there is also evidence that TIMPs are associated with suppression of programed cell death, tissue remodeling during tumor progression and mitogenic activity[11,12]. TIMP-1 has been shown to suppress apoptosis in a series of Burkitt’s lymphoma cell lines[13] and to act as a survival factor blocking apoptosis in B cells[14,15]. TIMP-1 must, therefore, be regarded as a multifunctional protein[16,17]. Recently, evidence has been provided that TIMP-1 induces specific signaling pathways[18]. TIMP-1 is able to activate Ras involving the Tyk/mitogen-activated protein kinase (MAPK) pathway[19]. A specific blockade of the ERK pathway suppresses the expression of MMP-9 and inhibits markedly the invasiveness of tumor cells[20]. The PKC-dependent NFK kappa B activation is absolute for MMP-9 induction and PKC-dependent ERC activation devotes to increase the expression level of MMP-9 in hepatocellular carcinoma cells[21,22]. St-Pierre et al[8] have given an overview of recent developments in the field of MMP-9 gene expression in different cell types, from the triggering of cell-surface receptors to the activation of cytoplasmic mediators and transcription factors responsible for the activation of MMP-9 promotor. Fibronectin up-regulates MMP-9 and induces coordinated expression of MMP-2 and its activator, the membrane-type MT1-MMP (MMP-14) by human T-lymphocyte cell lines, a process repressed through Ras/MAP kinase signaling pathways[23]. Accordingly, transfection of T-cell lines with dominant negative mutant of Ha-Ras strongly increases fibronectin-induced expression of MMP-2 and MMP-9[23]. In human glioma cells, angiopoietin-2 induces tumor cell infiltration by activation of MMP-2. MMP-2 is associated with tumor size, invasiveness, and survival in breast cancer[24]. SB203580, a p38 MAP kinase inhibitor, reduces MMP-2 expression in melanoma cells[25]. Non-steroidal anti-inflammatory drugs inhibit MMP-2 via suppression of ERK/Sp1-mediated transcription[26].
Recently, we demonstrated an increased expression of MMP-2 and MMP-9 protein as a result of increased TIMP-1 expression in human hepatoma cells[27]. Beyond that TIMP-1 overexpressing HepG2 cells could grow in nests and keep in contact with each other[27, 28]. This striking phenotype could be abrogated by a TIMP-1 antagonist, the mutant MMP-9-H401A[28]. Three histidine residues constitute the active site of MMP-9 by binding to the catalytic zinc. A MMP-9 mutant has been generated in which the histidine residue at position 401 is replaced by alanine. The mutant is enzymatically inactive but is still able to bind to TIMP-1. Furthermore, MMP-9-H401A is able to neutralize the ability of TIMP-1 to inhibit MMP-9 activity[28]. With the help of this TIMP-1 antagonist the influence of TIMP-1 e.g., on cell migration and invasion might be demonstrated much more clearly. Recent discoveries regarding the main functions of TIMP-1 (inhibition of tumor growth, invasion, angiogenesis, and metastasis; exhibition of growth factor-like activity and suppression of programmed cell death independent of the MMP inhibitory activity) have complicated the understanding of this protease inhibitor. Presently TIMP-1 can no longer be regarded uniquely as a tissue inhibitor of metalloproteinases. We decided to study in more detail the role of TIMP-1 and MMPs in the migration and signal transduction of human hepatoma cells by using an established cell motility assay with clear and defined conditions. We showed here that TIMP-1 overexpressing cells exhibit increased migration by chemotaxis, which could be reduced by MMP inhibition, especially inhibition of gelatinases. Following this discovery, we further investigated the signal transduction pathways of MMP-2 and MMP-9 in the event of TIMP-1 overexpression and demonstrated that TIMP-1 deactivates specific cell signaling pathways of MMPs.
MATERIALS AND METHODS
Materials
Culture reagents were from Sigma (Steinheim, Germany), Gibco (Eggenstein, Germany), or Sarstedt (Berlin, Germany). All chemicals were purchased from Sigma (Germany), Pharmacia (Freiburg, Germany), or ICN (Meckenheim, Germany). Galardin (GM 6001), a broad spectrum inhibitor of MMPs, genistein (4,5,7-trihydroxyisoflavone), an inhibitor of tyrosine kinases, SB 202190 (FHPI; 4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazol), an inhibitor of p38 MAP kinase, UO126 (1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio) butadien), an inhibitor of MEK1/2, and MMP-2/MMP-9 inhibitor I (2R,2,4-biphenylylsulfonylamino-3-phenylpropionic acid) were obtained from CalBiochem (Bad Soden, Germany). Preparation of a rabbit polyclonal anti-murine-TIMP-1 antiserum was described elsewhere[29]. Boyden chambers were purchased from Neuroprobe (Gaithersburg, US) and membranes from Merck (Darmstadt, Germany). Recombinant murine TIMP-1 was obtained from R&D Systems (Wiesbaden-Nordenstadt, Germany). A semi-quantitative PCR detection kit was purchased from BioSource International (Camarillo, CA).
Construction of a TIMP-1 antagonist
The MMP-9 mutant MMP-9-H401A was cloned as previously described[28]. To introduce a point mutation at amino acid 401 of murine MMP-9 (MMP-9-H401A), PCR-aided mutagenesis was performed using the following primers: 5’-TGGCAGCGGCCGAGTTCGGCCATG-3’ sense and 5’-CGAACTCGGCCGCTGCCACCAGG-3’ antisense.
Cells and cell treatment
Human hepatoma cells (HepG2) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 50 mL/L fetal calf serum (FCS), 100 mg/L streptomycin and 60 mg/L penicillin. Cells were grown at 37 °C in a water-saturated atmosphere in air/CO2 (19:1). Cells were assessed for the expression of TIMP-1 by Western blotting after incubation under serum-free conditions for 24 h. Selection of HepG2 clones stably transfected with murine TIMP-1 cDNA or murine MMP-9-H401A cDNA was previously described[27,28]. For stable expression, HepG2 cells were transfected with the expression vector pcDNA3.1-mTIMP-1 or pcDNA3.1-MMP-9-mut containing the neomycin resistance gene. Colonies resistant to 500 µg/mL neomycin were isolated, several clones were established and assessed for the expression of murine TIMP-1 or mutant MMP-9 by Northern and Western blotting. Expression by these cells was maintained for more than 20 passages. Three positive clones for each plasmid were used for further analyses.
Western blot
Serum-free cell supernatants of HepG2 cells and HepG2 cell clones were separated by SDS-polyacrylamide gel electrophoresis separating gels of 12.5% polyacrylamide and stacking gels of 3% polyacrylamide. Lanes were loaded with 2 μg of total protein each. Following electrophoresis at 30 V the proteins from serum-free cell supernatants were transferred to nitrocellulose membrane. Protein bands were localized by staining Ponceau S. Blots were blocked with TBS-N (pH 7.6) containing 10% BSA, 20 mmol/L Tris, 137 mmol/L NaCl, 0.1% Nonidet P40, washed and incubated with antibodies against TIMP-1 (dilution 1:1000). Signals were visualized by enhanced chemiluminescence according to the manufacturer’s instructions (Amersham). Experiments were done in triplicate.
Boyden chamber experiments
Transwell 0.64-cm2 filter inserts (8-μm pores) coated with a thin layer of gelatin (125 μg/cm2) were re-hydrated according to the customer’s instructions essentially as described previously[30]. Prior to seeding into the transwell inserts, cells were released using trypsin-EDTA and sequentially rinsed with DMEM containing 10% FCS and serum-free DMEM. The rinsed cells were re-suspended in serum-free DMEM (2.5×105 cells/mL) and 800 μL was added to the upper chambers. Galardin (2 μmol/L), MMP-2/MMP-9 inhibitor I (20 μmol/L) or 0.1% DMSO as control were added. In the lower compartment, fetal calf serum was used as chemoattractant. Chambers were incubated in a humified tissue incubator, containing 50 mL/L CO2 for 4 h at 37 °C. The cells on the upper surface of the filters were then removed using a cotton swab and those remaining on the lower surface of the filter were fixed with 10% methanol and stained with hematoxylin and eosin. The filters were rinsed with deionized water, dried and examined using light microscopy. The number of cells in five random optical fields (×400 magnification) from triplicate filters was averaged to obtain the number of migrating cells.
Attachment assay
Attachment assays were performed in 96-well plates. The wells were coated with gelatin solutions at a concentration of 10 µg/mL at 4 °C for 18 h. Unspecific binding of cells was blocked by pre-incubation of the wells with 3% BSA/PBS for 2 h. Hepatoma cell clones were harvested by trypsin incubation for 2 min, resuspended in DMEM without FCS at a density of 2×105 cells/mL and placed in the respective wells. For 4 h cells remaining in suspension were counted every 15 min. The assays were carried out in triplicate. Numerical data were analyzed by analysis of variance and Tukey’s multiple range tests at 5 and 1% levels. Data were presented as mean±SD.
RT-PCR
RNA was isolated from whole cells by UltraspecTM phenol chloroform extraction procedure (Biotecx Laboratories Inc., Houston, TX) according to the instruction manual, quantified spectrophotometrically at 260 nm and stored at -70 °C. Five micrograms of RNA were heated to 65 °C for 10 min in 50% formamide, 20 mmol/L 3-[N-morpholino]propane sulfonic acid (MOPS), 5 mmol/L sodium acetate, 1 mmol/L EDTA, and 2.2 mol/L formaldehyde before gel electrophoresis in 1% agarose containing 2.2 mol/L formaldehyde, 20 mmol/L MOPS, 5 mmol/L sodium acetate, and 1 mmol/L EDTA. After electrophoresis, integrity of the RNA was controlled by ethidium bromide staining of 18S and 28S ribosomal RNA. First-strand complementary DNA (cDNA) was synthesized from 1 µg of DNA-free total RNA with a T-primed first-strand kit (Pharmacia Biotech, Freiburg, Germany) according to the manufacturer’s instructions. A 1:100 final dilution of the reaction was used as template for PCR. In preliminary experiments, we determined the optimal number of cycles within the linearity of reactions for each PCR product. The cycle number was 35 cycles for MMP-9, MMP-14, and human TIMP-1, and 25 cycles for MMP-2 and GAPDH. The primers for PCR were as follows: human MMP-2 (756-1682 nt, GenBankTM accession no. AL832088), forward (5’-CCACGCTGCGGCAACCCAGATGTGGCCAAC-3’) and backward (5’-GGCGTGGCCAAACTCGTGGGCTGCCAC-3’); human GAPDH (292-885 nt, GenBankTM J04038), forward (5’-CCCATCACCATCTTCCAG) and backward (5’-CCTGCTTCACCACCTTCT); human MMP-9 (1512-2120 nt, GenBankTM BC006093), forward (5’-ACGGCCACTACTGTGCCTTTG-3’) and backward (5’-AGGGCACTGCAGGATGTCATAGG-3’); human MMP-14 (1247–1766 nt, GenBankTM NM 031056), forward (5’-GCCCATTGGCCAGTTCTGGCGGG-3’) and backward (5’-CCTCGTCCACCTCAATGATGATC-3’); human TIMP-1 (169-494 nt, GenBankTM NM003254), forward (5’-GCAATTCCGACCTCGTCATC-3’) and backward (5’-AGTGTAGGTCTTGGTGAAGC-3’). Using these primers, PCR was performed by the expand high fidelity PCR system (Roche Diagnostics) at 94 °C for 4 min followed by individual cycles at 94 °C for 30 s, at 58 °C for 30 s, and at 72 °C for 1 min with an extension step at 72 °C for 7 min at the end of the last cycle. The products were resolved in agarose gel (1.8%), followed by staining with ethidium bromide, and recorded by digital camera. The relative density of the products was quantitated by the Bio-Rad Gel-Doc system. The predicted PCR product size for MMP-2 and MMP-9 was 926 and 608 bp, respectively. For glyceraldehyde 3-phosphate dehydrogenase (GAPDH) the product size is 593 bp, for MMP-14 519 bp, and for human TIMP-1, 325 bp. Identity of the PCR product for MMP-2, MMP-9, MMP-14, and human TIMP-1 was confirmed by DNA sequencing. The values were depicted as the ratio to those of GAPDH.
Semi-quantitative multiplexPCR detection
The expression of human MMP-1, -3, -7, -2, -9, and GAPDH were detected by a multiplexPCR kit (BioSource International) according to the supplier’s instructions. The primers were selected with similar Tm such that highly stringent conditions for all primers could be utilized for RT-multiplexPCR. Buffers used were formulated to decrease competition among amplicons and to enhance the amplification of longer DNA fragments during multiplexPCR. Following PCR, the amplicons were analyzed by gel electrophoresis. Since the amplification efficiency of each primer pair was equivalent, the intensity of each amplicon band was proportional to their relative concentration in the cDNA sample. Agarose gel electrophoresis bands were scanned by computer analysis, and the optical density was plotted by histogram. The Bio-Rad Gel-Doc system was used to quantify PCR data. Data from MMP-cDNA were normalized to the respective content of GAPDH.
RESULTS
Effect of TIMP-1 and TIMP-1 antagonist on TIMP-1 expression
HepG2 wild type cells were compared with HepG2 cell clones overexpressing murine TIMP-1 (#T1, #T2, #T3) and HepG2 cells overexpressing the TIMP-1 antagonist MMP-9-H401A (#M1, #M2, #M3). Western blot analysis was performed for serum-free supernatants from HepG2 cells, HepG2-TIMP-1, (Figure 1, Lane 3) and HepG2-MMP-9-H401A. Using a specific polyclonal antiserum against murine TIMP-1 that did not detect human TIMP-1, amounts of murine TIMP-1 could be detected in HepG2-TIMP-1 cells. The supernatants of three different clones for HepG2-TIMP-1 (Figure 1, lane 2) and three different clones for MMP-9-H401A (Figure 1, lane 1), respectively, were pooled.
Figure 1.
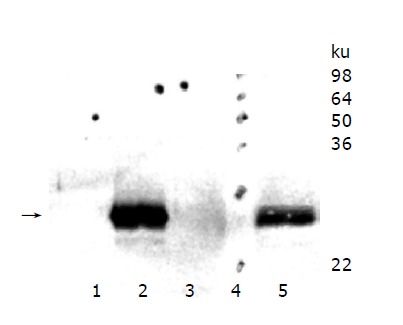
TIMP-1 protein expression of hepatoma cells overexpressing murine TIMP-1 and those overexpressing the TIMP-1 antagonist MMP-9-H401A. The arrow indicates the position of murine TIMP-1. TIMP-1 protein could be detected in HepG2-TIMP-1 cells only.
Effect of TIMP-1 and TIMP-1 antagonist on expression of MMP-1, -3, -7, -2, -9, and MMP-13
Semi-quantitative multiplexPCR was used to investigate the expression of MMP-1, -3, -2, -9, and -14 simultaneously in HepG2 cells, HepG2-TIMP-1 cells and HepG2 cells overexpressing MMP-9-H401A. The average values of three different clones for each cell line are indicated in Figure 2. Expression values of HepG2 cells were set to 100%. There were no significant changes in the expression levels of MMP-1, MMP-3, and MMP-14 between wild type HepG2 (white columns), TIMP-1 overexpressing HepG2 cells (black columns), and HepG2 overexpressing MMP-9-H401A (halftone columns). We observed a significant increase in the expression of MMP-9 (HepG2-TIMP-1: P = 0.003; HepG2-MMP-9-H401A: P = 0.05) in both HepG2-TIMP-1 and HepG2-MMP-9-H401A cells (Figure 2). MMP-9 primers did not differ between endogenous human MMP-9 and the transgene MMP-9-H401A with a single point mutation on DNA level. MMP-2 expression increased in HepG2-TIMP-1 in comparison to HepG2 cells but values did not reach the level of significance. MMP-2 expression levels in HepG2-MMP-9-H401A, however, decreased (Figure 2). There were no changes in the expression of the endogenous TIMP-1 between all cell clones examined (Figure 2, right panel).
Figure 2.
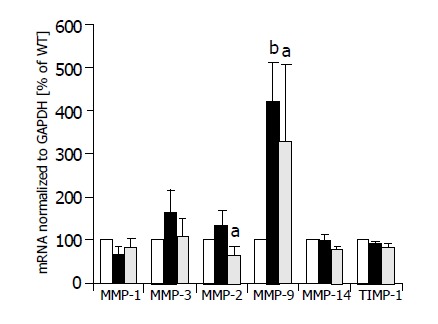
MMP mRNA expression of HepG2 (white columns), HepG2-TIMP-1 clones (black columns), and HepG2 cells overexpressing the TIMP-1 antagonist MMP-9-H401A (halftone columns). aP<0.05; bP<0.01.
Migration of human hepatoma cells was influenced by endogenous TIMP-1
To investigate whether TIMP-1 or TIMP-1 antagonism contributed to differences in cell migration, HepG2-TIMP-1 and HepG2-MMP-9-H401A cells were incubated in Boyden chambers. Fetal calf serum was used as chemoattractant. After incubated for 4 h, migrated cells were numbered on the undersite of the permeable membrane coated with gelatin. Data were normalized by setting the number of HepG2 wild type cells to 100% (Figure 3, white column, lane 1) and were presented as mean±SD. Compared with wild type HepG2 cells, the cells overexpressing TIMP-1 showed 114.6±10.7% (P = 0.039) migration (Figure 3, black column, lane 2) and the cells overexpressing the TIMP-1 antagonist showed 62.2±16% (P = 0.0196) migration (Figure 3, halftone column, lane 3). The difference between TIMP-1 clones and clones overexpressing the TIMP-1 antagonist was highly significant (P = 0.0057). Thus, TIMP-1 overexpression associated with increased expression of MMP-9 and MMP-2 favors migration in contrast to TIMP-1 inhibition by MMP-9H401A.
Figure 3.
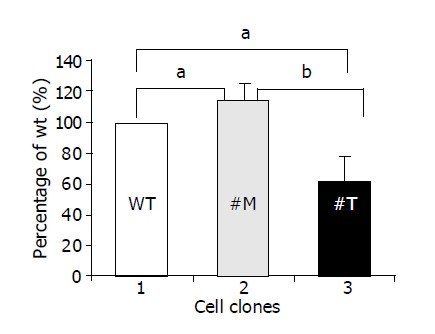
TIMP-1 and TIMP-1 antagonist contribute to differences in cell migration. aP<0.05; bP<0.01.
Migration of hepatoma cells overexpressing TIMP-1 was inhibited by a broad spectrum inhibitor of MMPs and a specific inhibitor of MMP-2 and MMP-9
To investigate whether MMPs were responsible for the different migration rates of HepG2 cells overexpressing TIMP-1 or MMP-9-H401A, cells were incubated with the broad spectrum MMP-inhibitor Galardin (Figure 4, lanes 2, 5, 8, 11, 14, 17, 20; few points) and a specific inhibitor of MMP-2/MMP-9 (Figure 4, lanes 3, 6, 9, 12, 15, 18, 21; many points). HepG2, HepG2-TIMP-1, and HepG2-MMP-9-H401A cells were incubated in Boyden chambers either with or without 2 µmol/L Galardin (GM 6001, inhibitor of MMP-1, -2, -3, -8, -9) and 20 µmol/L MMP-2/MMP-9 inhibitor. Fetal calf serum was used as chemoattractant. After 4 h of incubation migrated cells were numbered on the bottom of the permeable membrane coated with gelatin. Incubation with 2 µmol/L Galardin and 20 µmol/L gelatinase inhibitor reduced cell migration from 27.7±15.6 to 22.2±10.3 (P = 0.038) and 25.1±15.7 in HepG2 wild type cells (Figure 4, column 1-3). In the case of HepG2-MMP-9-H401A, incubation with Galardin reduced cell migration significantly from 21.5 to 16.1 (P = 0.02, #M1), 17.6 to 4.8 (P = 0.0016, #M2), and 12.6 to 6.9 (P = 0.03, #M3) for all clones analyzed separately. Gelatinase inhibitor was not able to reduce cell migration significantly in wild type cells and two of three cell clones overexpressing the TIMP-1 antagonist MMP-9-H401A (Figure 4, lanes 3, 6, and 12; many points). In the case of HepG2-TIMP-1, incubation with Galardin reduced cell migration from 33.9 to 25.8 (P = 0.02, #T1), 30.8 to 24.6 (P = 0.018, #T2), and 28.1 to 14.5 (P<0.0001, #T3). In the presence of MMP-2/-9 inhibitor a reduced migration was observed in all TIMP-1 overexpressing cell clones (Figure 4, lane 15, 18, and 21: many points), although reduction did not achieve significance in all cases (Figure 4, lane 18). Incubation of HepG2-TIMP-1 (#T1) with the specific MMP-2/-9 inhibitor reduced cell migration from 33.9±18.9 to 15.4±10.7 (P = 0.000003) beyond the number achieved by incubation with Galardin.
Figure 4.
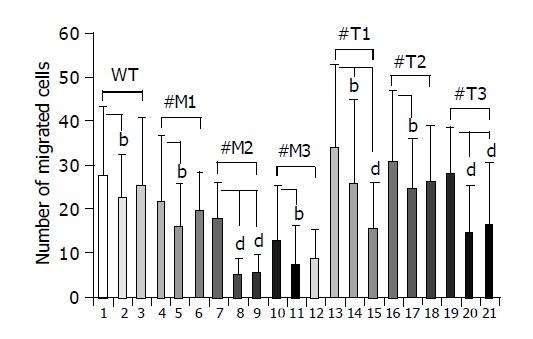
Effect of a broad spectrum MMP inhibitor and a specific MMP-2/MMP-9 inhibitor on the migration of HepG2, HepG2-TIMP-1 and HepG2-MMP-9-H401A cells. bP<0.01; dP<0.001.
Effect of TIMP-1 overexpression and TIMP-1 antagonism on cell attachment
We examined the attachment of all cell clones used to gelatin-coated membranes as shown in Figure 5. Three different clones of HepG2-TIMP-1 (#T1, #T2, #T3) and HepG2-MMP-9-H401A (#M1, #M2, #M3) behaved similarly to wild type HepG2 cells and showed a comparable attachment (Figure 5).
Figure 5.
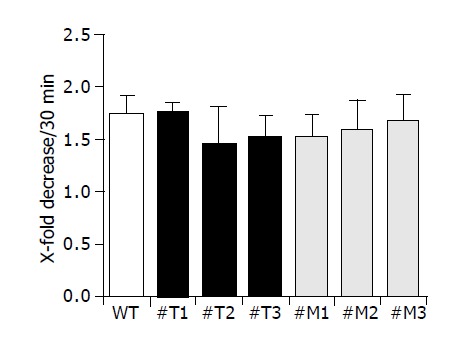
TIMP-1 and TIMP-1 antagonist do not influence cell attachment.
Signaling pathways required for expression of MMP-2 and MMP-9 in TIMP-1 overexpressing cells
To test which signaling pathways were involved in the expression of MMP-2 and MMP-9 in wild type HepG2 and TIMP-1 overexpressing HepG2, cells were incubated with different kinase inhibitors. Ras proteins are key molecular switches that mediate transmembrane signaling through the activation of multiple downstream pathways, which include the MAPK kinase pathways[31]. A series of pharmacological inhibitors were used to test the potential contribution of one or more signaling pathways that might regulate the steady-state transcription of the MMP-2 and the MMP-9 gene in wild-type or TIMP-1 overexpressing HepG2 cells: genistein (25 and 50 μmol/L; tyrosine kinase inhibitor), SB 202190 (2.5 μmol/L; potent inhibitor of p38 MAP kinase), and UO126 (1 μmol/L; potent and specific inhibitor of MEK1 and MEK2).
UO126 and genistein treatment at a dose of 50 μmol/L inhibited the secretion of MMP-2 in wild type and TIMP-1 overexpressing hepatoma cells (Figures 6A, B and columns 3, and 5). There was also a less pronounced but reproducible effect using the general tyrosin kinase inhibitor genistein at a dose of 25 μmol/L (Figure 6A, B and column 2). The specific p38 MAP kinase inhibitor SB 202190 was effective in blocking MMP-2 expression in HepG2 wild-type cells only, suggesting that the MMP-2 expression was not regulated through p38 MAPK in the case of TIMP-1 overexpression. These data all supported the hypothesis that constitutive MMP-2 expression is regulated through a steady-state signaling event, passing through Ras, MEK1/2, and p38 MAP kinases (Figure 6A). All TIMP-1 overexpressing cell clones (#T1, #T2, #T3) behaved similar (Figure 6B).
Figure 6.
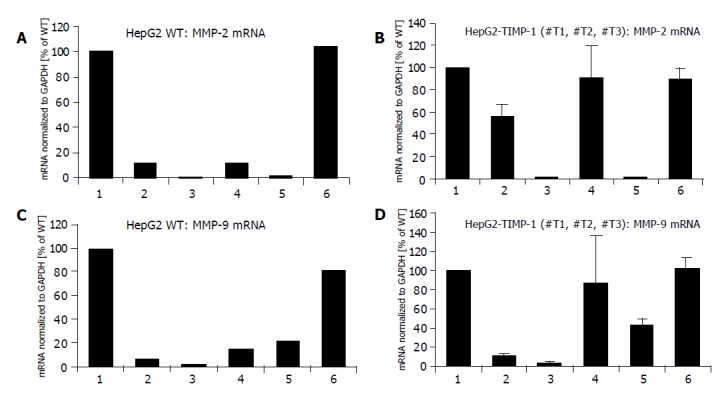
Effect of genistein, p38 MAPK inhibitor (SB 202190), and UO126 on the induction of MMP-2 and MMP-9 mRNA expression in HepG2 (A and C) and HepG2 TIMP-1 overexpressing cell clones (B and D). Cells were treated with Genistein 25 µM (2), Genistein 50 µM (3), SB202190 (4), and UO126 (5). mRNA expression of untreated cells (1) were set to 100%. Incubation with DMSO served as control (6).
Genistein at a dose of 25 and 50 µmol/L inhibited the secretion of MMP-9 in wild-type and TIMP-1 overexpressing hepatoma cells (Figure 6C, D and columns 2, 3). There was also a less-pronounced but reproducible effect using the specific MEK1/2 inhibitor UO126 at a dose of 1 μmol/L (Figure 6C, D and column 5). The specific p38 MAP kinase inhibitor SB 202190 was effective in blocking MMP-9 expression in HepG2 wild-type cells and less pronounced in one (#T1) of the TIMP-1 overexpressing clones (individual data not shown) whereas MMP-9 expression could not be influenced by p38 MAK inhibition in other TIMP-1 overexpressing clones (#T2, #T3) (individual data not shown). These data suggested that TIMP-1 overexpression attenuates MMP-9 expression through p38 MAPK. Similar to MMP-2 constitutive MMP-9 expression is regulated through a steady-state signaling event passing through Ras, MEK1/2, and p38 MAP kinases (Figure 6C).
DISCUSSION
A critical involvement of TIMP-1 and MMPs, in the extent of migration of human hepatoma cells, is shown here. It has previously been reported that TIMP-1 may play an important role in growth and migration, especially of hepatocellular and cholangiocellular carcinomas[32]. In this report, we extend these data in several ways. The migration of human hepatoma cells is characterized by the presence of TIMP-1 and a novel TIMP-1 antagonist. The influence of MMPs in the presence of high TIMP-1 levels can be determined by the application of a broad spectrum MMP inhibitor (Galardin GM6001) and a specific inhibitor of MMP-2, and MMP-9. MMP-2 and MMP-9 were critical for the migration of hepatoma cells in the presence of TIMP-1 (Figure 4). The action of TIMP-1 on the migration of hepatoma cells is functionally different from the MMP-inhibitors indicating other functions of TIMP-1 besides its MMP inhibitory capacity. The influence of cell attachment on the extent of migration is excluded by defining the attachment of all cell clones used. Signal transduction pathways responsible for the gene expression of MMP-2 and MMP-9 are analyzed either in the presence or absence of TIMP-1 and differences are elaborated concerning p38 MAPK.
TIMP-1 overexpression leads to increased migration of hepatoma cells (Figure 2). This phenomenon linked to TIMP-1 is confirmed by the use of the novel TIMP-1 antagonist MMP-9-H401-A[28]. This MMP-9 mutant has the opposite effect of TIMP-1 and reduces migration in our system. Although both TIMP-1 and the synthetic MMP-inhibitor GM6001 are able to inhibit MMPs, TIMP-1 increases and GM6001 decreases migration. Thus, the action of TIMP-1 on migration of hepatoma cells is functionally different from GM6001. Since TIMP-1 enhances migration in hepatoma cells, the influence of TIMP-1 on migration seems to be at least partly independent of its MMP inhibitory activity and indicates other functions of TIMP-1 besides its MMP inhibitory capacity. Recent studies showed that apoptotic effects of TIMP-1, especially the inhibition of apoptosis in tumor cells, are independent of MMP inhibition[11,33]. However, apoptosis would hardly be the reason for increased migration in our experiments because we measured migration over 4 h, too short for significant effects of programed cell death. Furthermore, we measured apoptosis in all cell clones used vs HepG2 wild-type cells by a cell death detection (nuclear matrix protein) but ELISA and apoptotic DNA Ladder kit could not detect any significant differences (data not shown). Further biological functions of TIMP-1, mitogenic activity, and cell-growth promoting activity, can be mainly excluded for the short period of time selected[11,34-36]. The effect of TIMP-1 on hepatoma cell migration might be cell tissue specific because in B16-F10 melanoma cells a specific up-regulation of murine TIMP-1 expression directly suppresses the invasive ability in a matrigel transwell invasion assay[37]. Recently the modulation of cell morphology has been reported to have a function for TIMP-1[11,38]. In smooth muscle cells a minimum expression of the contractile proteins and a maximal proliferation rate are correlated with the highest levels of both MMP-1 and TIMP-1[38]. TIMP-1 overexpressing HepG2 cells actually have a characteristic phenotype; they grow in nests and lie on top of each other[27]. It might be possible that this specific phenotype enables the cells to enhance migration through the gelatin-coated transwell filters. Whether the modulation of cell morphology by TIMP-1 takes place by changing the expression pattern of contractile proteins is the subject of our current investigation.
MMP-2 and -9 are necessary for the migration of many cell types and tumor cells, e.g., Langerhans cells and dermal dendritic cells from human and murine skin[5]. The migration of the latter cells, however, is inhibited by TIMP-1 and TIMP-2. We also demonstrated that MMPs are critical for the migration of hepatoma cells either in the presence or absence of TIMP-1 (Figure 4). Overexpressing functional TIMP-1 seems to depend on enhanced MMP-activity. Since Galardin is able to inhibit MMP-1, -2, -3, -8 and -9, one or more of these MMPs might be responsible for migration in HepG2 cells in our system. Overexpression of MMP-9 in HepG2-TIMP-1 cells has been demonstrated earlier[27]. Actually, we observed a reduced migration of all cell clones used in the presence of MMP-2/MMP-9 inhibitor. In some cases (Figure 4), however, reduction of migration did not achieve significance.
Migration of cells through a coated membrane involves not only ECM degradation but also requires the ability of adhesive interactions between cells and matrix. Cell migration can be viewed as a process regulated by counter-balanced forces or activities. Strength of cell adhesion and inhibitory degrees of cell adhesion can regulate the speed of motility[6,39]. In the present study, however, decreased and increased migration did not correlate with different adhesion (Figure 5).
MAPK activities exert a crucial role in cellular migration, and their implication in this process has been highlighted by the use of specific inhibitors (indicated in Figure 7).
Figure 7.
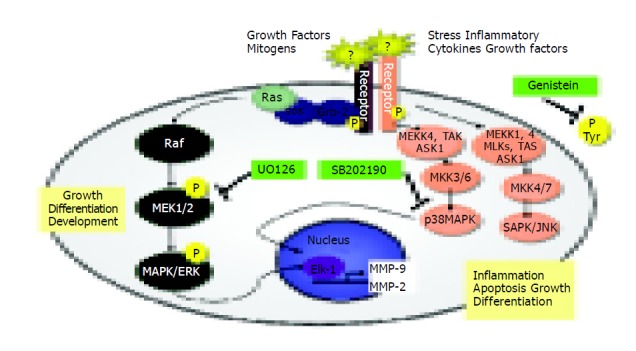
Signal transduction pathways and MAP kinase signaling cascades in mammalian cells shown in schematic form.
Several reports have shown that SAPK2/p38 is involved in chemokine-induced chemotaxis of cells[40-42]. For this reason, we examined the migration potential of TIMP-1 overexpressing cells in the presence of specific signal transduction inhibitors (Figure 7) and our study presents evidence that p38 but not ERK, intervenes in the control of hepatoma cell migration. Signal transduction has traditionally been examined by short-term stimulation of cells with agents that induce mitogenesis, differentiation, or apoptosis[43]. Recent work showed that maintenance of basal cell function also requires “steady-state” signal transduction[44]. In this specific instance, the steady-state production of MMP-2 and MMP-9 involved in cell attachment, detachment, and migration was examined in our study. Constitutive MMP-2 and MMP-9 expression is regulated through a steady-state signaling event passing through tyrosine kinases, MEK1/2, and p38 MAP kinases (Figures 6, Figures 7). Our results are in accordance with others[42-44,48]. For head and neck cancer cells, it has already been reported that the general tyrosine kinase inhibitor genistein induces several specific molecular changes, such as down-regulation of c-erbB-2 expression, down-regulation of MMP-2 and MMP-9 secretion, inhibition of tumor cell invasion and down-regulation of nuclear factor-kappaB DNA binding activity[45]. In human keratinocytes, the inhibition of MAPK pathway is correlated with down-regulation of MMP-9 secretion induced by TNF-alpha[46]. In brain cells, ERK and p38 MAP kinases are up-regulated after mechanical injury, and mediate the secretion of MMP-9[47]. Recently, it has been shown that inhibition of monocyte migration occurs via inhibiting crucial signaling pathways, like SAPK2/p38 (Figure 7) and MMP-2 activities[48].
Given the data presented in this manuscript, we could now predict that expression of MMP-2 and MMP-9 in liver tumors containing high TIMP-1 activity cannot be influenced by inhibition of p38 MAK (Figure 7). This should result in a higher metastatic potential and probably a poorer overall prognosis.
Our data add new insights in the complex function of TIMP-1 and control of MMP activity in human liver cells. Thus, the suggested role of TIMP-1 in tumor cell migration has substantially changed from the initial focus on MMP inhibition to a broader focus including modulation of MMP secretion, cell morphology, and cell migration. With regard to the treatment of inflammatory disorders and metastasis, it might be useful to target the signaling pathways regulating these MMPs in more detail.
ACKNOWLEDGEMENTSq
A part of the data has been presented as a plenary talk on the Single Topic Conference Liver Fibrosis of the EASL in Florence, 2001.
Footnotes
Supported by grants from the Federal Ministry of Education and Research of Germany, Deutsche Forschungsgemeinschaft (DFG), and Aachen University
References
- 1.Stetler-Stevenson WG, Aznavoorian S, Liotta LA. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu Rev Cell Biol. 1993;9:541–573. doi: 10.1146/annurev.cb.09.110193.002545. [DOI] [PubMed] [Google Scholar]
- 2.Coussens LM, Werb Z. Matrix metalloproteinases and the development of cancer. Chem Biol. 1996;3:895–904. doi: 10.1016/s1074-5521(96)90178-7. [DOI] [PubMed] [Google Scholar]
- 3.Shapiro SD. Matrix metalloproteinase degradation of extracellular matrix: biological consequences. Curr Opin Cell Biol. 1998;10:602–608. doi: 10.1016/s0955-0674(98)80035-5. [DOI] [PubMed] [Google Scholar]
- 4.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 5.Ratzinger G, Stoitzner P, Ebner S, Lutz MB, Layton GT, Rainer C, Senior RM, Shipley JM, Fritsch P, Schuler G, et al. Matrix metalloproteinases 9 and 2 are necessary for the migration of Langerhans cells and dermal dendritic cells from human and murine skin. J Immunol. 2002;168:4361–4371. doi: 10.4049/jimmunol.168.9.4361. [DOI] [PubMed] [Google Scholar]
- 6.Gu J, Nishiuchi R, Sekiguchi K. Matrix metalloproteinase-2 is involved in A549 cell migration on laminin-10/11. Biochem Biophys Res Commun. 2002;296:73–77. doi: 10.1016/s0006-291x(02)00831-8. [DOI] [PubMed] [Google Scholar]
- 7.Kang T, Yi J, Guo A, Wang X, Overall CM, Jiang W, Elde R, Borregaard N, Pei D. Subcellular distribution and cytokine- and chemokine-regulated secretion of leukolysin/MT6-MMP/MMP-25 in neutrophils. J Biol Chem. 2001;276:21960–21968. doi: 10.1074/jbc.M007997200. [DOI] [PubMed] [Google Scholar]
- 8.St-Pierre Y, Van Themsche C, Estève PO. Emerging features in the regulation of MMP-9 gene expression for the development of novel molecular targets and therapeutic strategies. Curr Drug Targets Inflamm Allergy. 2003;2:206–215. doi: 10.2174/1568010033484133. [DOI] [PubMed] [Google Scholar]
- 9.Arthur MJ. Collagenases and liver fibrosis. J Hepatol. 1995;22:43–48. [PubMed] [Google Scholar]
- 10.McCrudden R, Iredale JP. Liver fibrosis, the hepatic stellate cell and tissue inhibitors of metalloproteinases. Histol Histopathol. 2000;15:1159–1168. doi: 10.14670/HH-15.1159. [DOI] [PubMed] [Google Scholar]
- 11.Mannello F, Gazzanelli G. Tissue inhibitors of metalloproteinases and programmed cell death: conundrums, controversies and potential implications. Apoptosis. 2001;6:479–482. doi: 10.1023/a:1012493808790. [DOI] [PubMed] [Google Scholar]
- 12.Murphy FR, Issa R, Zhou X, Ratnarajah S, Nagase H, Arthur MJ, Benyon C, Iredale JP. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: implications for reversibility of liver fibrosis. J Biol Chem. 2002;277:11069–11076. doi: 10.1074/jbc.M111490200. [DOI] [PubMed] [Google Scholar]
- 13.Guedez L, Courtemanch L, Stetler-Stevenson M. Tissue inhibitor of metalloproteinase (TIMP)-1 induces differentiation and an antiapoptotic phenotype in germinal center B cells. Blood. 1998;92:1342–1349. [PubMed] [Google Scholar]
- 14.Guedez L, Stetler-Stevenson WG, Wolff L, Wang J, Fukushima P, Mansoor A, Stetler-Stevenson M. In vitro suppression of programmed cell death of B cells by tissue inhibitor of metalloproteinases-1. J Clin Invest. 1998;102:2002–2010. doi: 10.1172/JCI2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaudin P, Trocmé C, Berthier S, Kieffer S, Boutonnat J, Lamy C, Surla A, Garin J, Morel F. TIMP-1/MMP-9 imbalance in an EBV-immortalized B lymphocyte cellular model: evidence for TIMP-1 multifunctional properties. Biochim Biophys Acta. 2000;1499:19–33. doi: 10.1016/s0167-4889(00)00084-7. [DOI] [PubMed] [Google Scholar]
- 16.Lambert E, Boudot C, Kadri Z, Soula-Rothhut M, Sowa ML, Mayeux P, Hornebeck W, Haye B, Petitfrere E. Tissue inhibitor of metalloproteinases-1 signalling pathway leading to erythroid cell survival. Biochem J. 2003;372:767–774. doi: 10.1042/BJ20030187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ikenaka Y, Yoshiji H, Kuriyama S, Yoshii J, Noguchi R, Tsujinoue H, Yanase K, Namisaki T, Imazu H, Masaki T, et al. Tissue inhibitor of metalloproteinases-1 (TIMP-1) inhibits tumor growth and angiogenesis in the TIMP-1 transgenic mouse model. Int J Cancer. 2003;105:340–346. doi: 10.1002/ijc.11094. [DOI] [PubMed] [Google Scholar]
- 18.Alexander JP, Acott TS. Involvement of the Erk-MAP kinase pathway in TNFalpha regulation of trabecular matrix metalloproteinases and TIMPs. Invest Ophthalmol Vis Sci. 2003;44:164–169. doi: 10.1167/iovs.01-1201. [DOI] [PubMed] [Google Scholar]
- 19.Wang T, Yamashita K, Iwata K, Hayakawa T. Both tissue inhibitors of metalloproteinases-1 (TIMP-1) and TIMP-2 activate Ras but through different pathways. Biochem Biophys Res Commun. 2002;296:201–205. doi: 10.1016/s0006-291x(02)00741-6. [DOI] [PubMed] [Google Scholar]
- 20.Tanimura S, Asato K, Fujishiro SH, Kohno M. Specific blockade of the ERK pathway inhibits the invasiveness of tumor cells: down-regulation of matrix metalloproteinase-3/-9/-14 and CD44. Biochem Biophys Res Commun. 2003;304:801–806. doi: 10.1016/s0006-291x(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 21.Woo JH, Park JW, Lee SH, Kim YH, Lee IK, Gabrielson E, Lee SH, Lee HJ, Kho YH, Kwon TK. Dykellic acid inhibits phorbol myristate acetate-induced matrix metalloproteinase-9 expression by inhibiting nuclear factor kappa B transcriptional activity. Cancer Res. 2003;63:3430–3434. [PubMed] [Google Scholar]
- 22.Hah N, Lee ST. An absolute role of the PKC-dependent NF-kappaB activation for induction of MMP-9 in hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2003;305:428–433. doi: 10.1016/s0006-291x(03)00788-5. [DOI] [PubMed] [Google Scholar]
- 23.Esparza J, Vilardell C, Calvo J, Juan M, Vives J, Urbano-Márquez A, Yagüe J, Cid MC. Fibronectin upregulates gelatinase B (MMP-9) and induces coordinated expression of gelatinase A (MMP-2) and its activator MT1-MMP (MMP-14) by human T lymphocyte cell lines. A process repressed through RAS/MAP kinase signaling pathways. Blood. 1999;94:2754–2766. [PubMed] [Google Scholar]
- 24.Nakopoulou L, Tsirmpa I, Alexandrou P, Louvrou A, Ampela C, Markaki S, Davaris PS. MMP-2 protein in invasive breast cancer and the impact of MMP-2/TIMP-2 phenotype on overall survival. Breast Cancer Res Treat. 2003;77:145–155. doi: 10.1023/a:1021371028777. [DOI] [PubMed] [Google Scholar]
- 25.Denkert C, Siegert A, Leclere A, Turzynski A, Hauptmann S. An inhibitor of stress-activated MAP-kinases reduces invasion and MMP-2 expression of malignant melanoma cells. Clin Exp Metastasis. 2002;19:79–85. doi: 10.1023/a:1013857325012. [DOI] [PubMed] [Google Scholar]
- 26.Pan MR, Hung WC. Nonsteroidal anti-inflammatory drugs inhibit matrix metalloproteinase-2 via suppression of the ERK/Sp1-mediated transcription. J Biol Chem. 2002;277:32775–32780. doi: 10.1074/jbc.M202334200. [DOI] [PubMed] [Google Scholar]
- 27.Roeb E, Winograd R, Breuer B, Nguyen H, Matern S. Increased TIMP-1 activity results in increased expression of gelatinases and altered cell motility. J Cell Biochem. 1999;75:346–355. [PubMed] [Google Scholar]
- 28.Roeb E, Behrmann I, Grötzinger J, Breuer B, Matern S. An MMP-9 mutant without gelatinolytic activity as a novel TIMP-1-antagonist. FASEB J. 2000;14:1671–1673. doi: 10.1096/fj.99-0947fje. [DOI] [PubMed] [Google Scholar]
- 29.Roeb E, Graeve L, Müllberg J, Matern S, Rose-John S. TIMP-1 protein expression is stimulated by IL-1 beta and IL-6 in primary rat hepatocytes. FEBS Lett. 1994;349:45–49. doi: 10.1016/0014-5793(94)00636-9. [DOI] [PubMed] [Google Scholar]
- 30.Wach F, Eyrich AM, Wustrow T, Krieg T, Hein R. Comparison of migration and invasiveness of epithelial tumor and melanoma cells in vitro. J Dermatol Sci. 1996;12:118–126. doi: 10.1016/0923-1811(95)00470-x. [DOI] [PubMed] [Google Scholar]
- 31.Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene. 1998;17:1395–1413. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- 32.Nakatsukasa H, Ashida K, Higashi T, Ohguchi S, Tsuboi S, Hino N, Nouso K, Urabe Y, Kinugasa N, Yoshida K, et al. Cellular distribution of transcripts for tissue inhibitor of metalloproteinases 1 and 2 in human hepatocellular carcinomas. Hepatology. 1996;24:82–88. doi: 10.1053/jhep.1996.v24.pm0008707287. [DOI] [PubMed] [Google Scholar]
- 33.Li G, Fridman R, Kim HR. Tissue inhibitor of metalloproteinase-1 inhibits apoptosis of human breast epithelial cells. Cancer Res. 1999;59:6267–6275. [PubMed] [Google Scholar]
- 34.Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP. Tissue inhibitors of metalloproteinases: structure, regulation and biological functions. Eur J Cell Biol. 1997;74:111–122. [PubMed] [Google Scholar]
- 35.Murphy G, Willenbrock F. Tissue inhibitors of matrix metalloendopeptidases. Methods Enzymol. 1995;248:496–510. doi: 10.1016/0076-6879(95)48032-3. [DOI] [PubMed] [Google Scholar]
- 36.Nagase H, Woessner JF. Matrix metalloproteinases. J Biol Chem. 1999;274:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 37.Khokha R, Zimmer MJ, Graham CH, Lala PK, Waterhouse P. Suppression of invasion by inducible expression of tissue inhibitor of metalloproteinase-1 (TIMP-1) in B16-F10 melanoma cells. J Natl Cancer Inst. 1992;84:1017–1022. doi: 10.1093/jnci/84.13.1017. [DOI] [PubMed] [Google Scholar]
- 38.Kato S, Yasukawa H, Fujii T, Yamaguchi M, Miyagi N, Okamoto K, Wada Y, Miyamoto T, Morimatsu M, Fox JC. Coordinate regulation of matrix metalloproteinase-1 and tissue inhibitor of metalloproteinase-1 expression in human vascular smooth muscle cells. Connect Tissue Res. 2000;41:143–153. doi: 10.3109/03008200009067666. [DOI] [PubMed] [Google Scholar]
- 39.Palecek SP, Loftus JC, Ginsberg MH, Lauffenburger DA, Horwitz AF. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 1997;385:537–540. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
- 40.Ashida N, Arai H, Yamasaki M, Kita T. Distinct signaling pathways for MCP-1-dependent integrin activation and chemotaxis. J Biol Chem. 2001;276:16555–16560. doi: 10.1074/jbc.M009068200. [DOI] [PubMed] [Google Scholar]
- 41.Sun Y, Cheng Z, Ma L, Pei G. Beta-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem. 2002;277:49212–49219. doi: 10.1074/jbc.M207294200. [DOI] [PubMed] [Google Scholar]
- 42.Cara DC, Kaur J, Forster M, McCafferty DM, Kubes P. Role of p38 mitogen-activated protein kinase in chemokine-induced emigration and chemotaxis in vivo. J Immunol. 2001;167:6552–6558. doi: 10.4049/jimmunol.167.11.6552. [DOI] [PubMed] [Google Scholar]
- 43.Kerkhoff E, Rapp UR. Cell cycle targets of Ras/Raf signalling. Oncogene. 1998;17:1457–1462. doi: 10.1038/sj.onc.1202185. [DOI] [PubMed] [Google Scholar]
- 44.Liao J, Wolfman JC, Wolfman A. K-ras regulates the steady-state expression of matrix metalloproteinase 2 in fibroblasts. J Biol Chem. 2003;278:31871–31878. doi: 10.1074/jbc.M301931200. [DOI] [PubMed] [Google Scholar]
- 45.Alhasan SA, Aranha O, Sarkar FH. Genistein elicits pleiotropic molecular effects on head and neck cancer cells. Clin Cancer Res. 2001;7:4174–4181. [PubMed] [Google Scholar]
- 46.Holvoet S, Vincent C, Schmitt D, Serres M. The inhibition of MAPK pathway is correlated with down-regulation of MMP-9 secretion induced by TNF-alpha in human keratinocytes. Exp Cell Res. 2003;290:108–119. doi: 10.1016/s0014-4827(03)00293-3. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, Mori T, Jung JC, Fini ME, Lo EH. Secretion of matrix metalloproteinase-2 and -9 after mechanical trauma injury in rat cortical cultures and involvement of MAP kinase. J Neurotrauma. 2002;19:615–625. doi: 10.1089/089771502753754082. [DOI] [PubMed] [Google Scholar]
- 48.Vitale S, Schmid-Alliana A, Breuil V, Pomeranz M, Millet MA, Rossi B, Schmid-Antomarchi H. Soluble fractalkine prevents monocyte chemoattractant protein-1-induced monocyte migration via inhibition of stress-activated protein kinase 2/p38 and matrix metalloproteinase activities. J Immunol. 2004;172:585–592. doi: 10.4049/jimmunol.172.1.585. [DOI] [PubMed] [Google Scholar]