

Abstract
AIM: Chronic hepatitis B virus (HBV) infection is predominantly treated with interferon alpha (IFN-α), which results in an efficient reduction of the viral load only in 20-40% of treated patients. Mutations at HBV precore prevail in different clinical status of HBV infection. The roles of precore mutation in the progression of chronic hepatitis and interferon sensitivity are still unknown. The aim of this study was to explore if there was any relationship between HBV precore mutation and sensitivity to interferon in vitro.
METHODS: HBV replication-competent recombinant constructs with different patterns of precore mutations were developed. Then the recombinants were transiently transfected into hepatoma cell line (Huh7) by calcium phosphate transfection method. With or without IFN, viral products in culture medium were collected and quantified 3 d after transfection.
RESULTS: We obtained 4 recombinant constructs by orientation-cloning 1.2-fold-overlength HBV genome into pUC18 vector via the EcoRI and Hind III and PCR mediated site-directed mutagenesis method. All the recombinants contained mutations within precore region. Huh7 cells transfected with recombinants secreted HBsAg and HBV particles into the cell culture medium, indicating that all the recombinants were replication-competent. By comparing the amount of HBV DNA in the medium, we found that HBV DNA in medium reflecting HBV replication efficiency was different in different recombinants. Recombinants containing precore mutation had fewer HBV DNAs in culture medium than wild type. This result showed that recombinants containing precore mutation had lower replication efficiency than wild type. HBV DNA was decreased in pUC18-HBV1.2-WT recombinants after IFN was added while others with precore mutations were not, indicating that HBV harboring precore mutation was less sensitive to IFN in cell culture system.
CONCLUSION: These data indicate that HBV harboring precore mutation may be resistant to IFN in vitro.
Keywords: Hepatitis B virus, Mutation, Interferon, Viral resistance
INTRODUCTION
Hepatitis B virus (HBV) is a small, partially double-stranded DNA (dsDNA) virus that causes acute and chronic hepatitis in humans. HBV infection is a global health problem. Current estimates are that 2 billion people are infected with HBV worldwide, 360 million of them suffer from chronic HBV infection resulting in over 520000 deaths each year (52000 from acute hepatitis B and 470000 from cirrhosis or liver cancer)[1]. Transient infections may lead to serious illness, and approximately 0.5% are terminated in fatal, fulminant hepatitis. Chronic infections may also have serious consequences, nearly 25% are terminated in untreatable liver cancer[2]. Chronic HBV infection is predominantly treated with IFN alpha, which could reduce the viral load in only 20-40% of treated patients with low-level viremia and evidence of active liver disease[3,4]. It is known that IFN alpha reduces viremia in responding patients by modulation of immunological responses and/or by direct induction of an intracellular antiviral state in infected cells. So far, drug-induced intracellular antiviral mechanisms against HBV have mostly been examined by use of hepatoma cell lines and transgenic mice with integrated HBV DNA, since cell lines permissive for HBV infection are not available. Studies have shown that treatment of various stable HBV-expressing hepatoma cell lines with IFN-α could lead to intracellular inhibition of synthesis of one or several HBV products, depending on the type of cells and systems used[5-7].
HBV sequence variability has been increasingly recognized as a factor that modulates the course and outcome of HBV infection. Variants with disturbed hepatitis B e antigen (HBeAg) synthesis, large deletions in the nucleocapsid gene, or mutations in the basic core promoter (BCP) have been found to be associated with severe liver diseases and might be related to the sensitivity of interferon therapy[8]. There have been many studies on HBV replication and IFN sensitivity, but the results are inconclusive. The significance of mutations of HBV precore/core antigen in causing persistent infection and subsequent liver diseases is debatable. Therefore the roles of precore mutations in the progression of chronic hepatitis are still unknown. G to A transition at nt1896 in the precore (preC) region terminates translation of the HBeAg precursor and results in HBeAg-minus HBV. Although A1896 mutation was reported previously to be associated with severe forms of chronic liver diseases[9], the real significance of A1896 mutation in the course of hepatitis is still controversial[10,11].
In addition, there are a few works about HBV mutation and IFN sensitivity in vitro. HBV genome is a condensed structure, the open reading frames are overlapped. Therefore mutation at one site may alter transcription or translation of more than one viral gene. Most previous studies focused on HBV subgenomic fragments, nevertheless,there are a few researches on the full length of HBV. Therefore we introduced recombinant constructs harboring more than one copy of HBV genome into hepatoma cell line by transient transfection. Since the recombinants contained different mutations at the precore region, we could study the relationship of these mutations and interferon sensitivity in vitro.
MATERIALS AND METHODS
Construction of plasmids
Cloning of plasmid constructs used in transfection experiments was performed by standard techniques[12]. Mutagenesis reactions were performed through polymerase chain reaction (PCR)-mediated site-directed mutagenesis with the designed mutagenetic primers (Table 1). For the calculation of the nucleotide (ntd.) positions, the first thymine of the putative EcoRI site of HBV was defined as position 1 and then anti-clockwise was counted (GenBank accession number is AY040627, genotype C). Different kinds of mutations were introduced by amplification with Pwo-Taq DNA polymerase mix (expand high fidelity PCR system, Roche Molecular Biochemical, Germany).
Table 1.
Sequences of primers for mutagenesis reaction.
| Primer | Direction | Length | Position (nt) | 5’-sequence-3’ |
| A1762/G1764For | + | 34 | 1748-1781 | aggagattagttaaaggtctttgtactaggaggc |
| 1896/1899Rev | - | 20 | 1895-1876 | aaagccacccaaggcacagc |
| A1896For | + | 31 | 1876-1906 | gctgtgccttgggtggctttagggcatggac |
| A1899For | + | 34 | 1876-1909 | gctgtgccttgggtggctttgggacatggacatt |
| A1896A1899For | + | 34 | 1876-1909 | gctgtgccttgggtggctttaggacatggacatt |
| PS5For | + | 20 | 1260-1279 | gccgatccatactgcggaac |
| PS4Rev | - | 20 | 2386-2405 | gagaccttcgtctgcgaggc |
First, pUC18-HBV1.2-TA was constructed. Plasmid pUC18-HBV1.2-TA was derived from pALTER-HBV1.2 containing a 1.2×HBV full length (T1762/A1764 mutant type). The 1.2×HBV full length from pALTER-HBV1.2 and linearized pUC18 vector were obtained by restrictive endonuclease EcoRI and Hind III digestion. Both of them were purified by QIAquick gel extraction kit (Qiagen, Germany). Then the 1.2×HBV full length was orientedly subcloned into the linearized vector pUC18 to acquire pUC18-HBV1.2-TA (T1762/A1764).
Second, we constructed wild type recombinants. Plasmid pUC18-HBV1.2-WT was produced by the large primer PCR method. In brief, fragments which were prepared by expand high fidelity PCR system (Roche) were used as large primers. In the PCR system, pUC18-HBV1.2-TA was used as a template with repair primer (wild type) A1762/G1764For and primer PS4Rev. The PCR products were purified by QIAGEN gel extraction kit and then used as large primers. The large primers and primer PS5For were used to amplify template pUC18-HBV1.2-TA to obtain the wild type fragments of HBV which were ascertained by sequencing. The PCR products and pUC18-HBV1.2-TA were both digested by restrictive endonucleases EcoRI and Xmn I respectively. After ligation and subcloning, the fragments between EcoRI and Xmn I in pUC18-HBV1.2-TA were replaced by corresponding fragments derived from PCR products, then wild type constructs of pUC18-HBV1.2-WT were produced which had a wild type A1762/G1764 instead of a mutant type T1762A1764.
Third, we constructed 1896/1899 recombinants. One segment, designated as segment A was prepared by PCR using pUC18-HBV1.2 as template with the primers PS5For and 1896/1899Rev, this fragment was used as a common segment for all next round amplifications. Segment B was prepared with the primers A1896For and PS4Rev, while segment C was prepared with primers A1899For and PS4Rev and segment D with primers A1896A1899For and PS4Rev. Then A1896 mutated fragments were obtained by PCR using segments A and B as templates with primers PS4 and PS5. Restrictive endonucleases EcoRI and Xmn I were used to digest PCR products and pUC18-HBV1.2-WT, then fragments between EcoR I and Xmn I were exchanged between pUC18-HBV1.2-WT and PCR products by subcloning as described above. Plasmids containing the mutated site A1896 were obtained and named as pUC18-HBV1.2-A1896. Accordingly, plasmids pUC18-HBV1.2-A1899 and pUC18-HBV1.2-AA were obtained later. The detailed mutation patterns of all the produced plasmids are shown in Table 2 and Figure 1. Correctly introduced nucleotides were verified by sequencing.
Table 2.
Mutation patterns of the 4 recombinant constructs.
| Number | Recombinant constructs |
Mutation pattern |
|
| A1896 | A1899 | ||
| 1 | pUC18-HBV1.2-WT | - | - |
| 2 | pUC18-HBV1.2-A1896 | + | - |
| 3 | pUC18-HBV1.2-A1899 | - | + |
| 4 | pUC18-HBV1.2-AA | + | + |
Note: “+” indicates the construct containing the corresponding mutation, “-” indicates the constructs containing no corresponding mutation instead of wild type.
Figure 1.
Electrophoresis of mutagenesis PCR product. Lanes A-D: Different HBV fragments using different mutated primers, Lanes E-G: The second round PCR products.
Determination of the most optimum concentration of interferon-α in vitro
Huh7 cells were seeded at a density of 6×105/well in six-well plates (10 cm2 per well, Falcon) in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum and cultured at 37 °C in 5 mL/L CO2 1 d before transfection. The medium was changed 4 h before transfection. Five ug plasmid was transfected into nearly confluent Huh7 cells. Sixteen hours after transfection, Huh7 cells were shocked with 15% glycerol solution, and then incubated with a fresh culture medium for 24 h. To determine the most optimal concentration of interferon in vitro, different concentrations of interferon α-2b were added to the culture medium. The concentrations of IFN-α (Intron A, Schering-Plough Research Institute) used in the experiment were 62.5 IU/mL, 125 IU/mL, 250 IU/mL, 500 IU/mL, 1000 IU/mL, 2000 IU/mL, 4000 IU/mL, 8000 IU/mL. Three days after transfection, cell medium was collected to detect HBsAg, HBeAg and HBV DNA. By comparing the amount of HBsAg, HBeAg and HBV DNA secreted to the medium, we selected the concentration of interferon which could inhibit HBV most.
Transfection of HBV DNA by calcium phosphate precipitation
All plasmid DNAs were prepared and purified by QIAGEN EndoFree maxi kit (EndoFreeTM Plasmid Mega Kit, Qiagen, Gemany). The DNA was then quantified spectrophotometrically (a ratio of the optical density at 260 nm to that at 280 nm of 1.7 to 1.8).
Transfection was performed by the calcium phosphate precipitation method (Promega ProFection® mammalian transfection system-calcium phosphate) according to the supplied protocol. Human hepatoma cell line Huh7 was grown as a monolayer in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum at 37 °C in 5 mL/L CO2. Huh7 cells were plated at a density of 3.0×106 cells per Petri dish (Falcon) 1 d before transfection to reach 80% confluence at transfection. The medium was changed 4 h before transfection. Twenty ug corresponding HBV replication-competent plasmid was mixed with CaCl2 solution, then added dropwise to 2× HEPES-buffered saline (pH 7.05), and transfected into confluent Huh7 cells. Sixteen hours after transfection Huh7 cells were shocked with 15% glycerol solution, and then incubated with a fresh culture medium for 24 h. Medium with or without 1000 IU/mL IFN-α (interferon alfa-2b, intron A, Schering-Plough Research Institute) was then added and the culture medium was harvested 3 d after transfection.
Transfection efficiency was measured by cotransfection of 1.0 μg of reporter plasmid pSEAP2-Control-SV40 (Clontech) expressing secretable alkaline phosphatase under the control of the SV40 early promoter and the SV40 enhancer[13,14]. The SEAP coding sequence was followed by the SV40 late polyadenylation signal to ensure proper and efficient processing of the SEAP transcript in eukaryotic cells. The secreted SEAP enzyme was assayed directly from the culture medium. The reporter plasmid served as an internal control to monitor transfection efficiency and potential IFN-α-mediated cytotoxic effects or nonspecific inhibitory effects. Transfection efficiency in each cell culture plate was carefully controlled by determination of the SEAP enzymatic activity in the cell culture medium before addition of IFN-α, and was found to vary by less than 10% (data not shown).
The SEAP activity of the culture media was measured 24 h after transfection by performing a colorimetric assay according to the recommendation[13]. Briefly, twenty microliters of heat-treated (at 65 °C for 5 min) medium was adjusted to 1×SEAP assay buffer (1.0 mol/L diethanolamine pH9.8, 0.5 mmol/L MgCl2, 10 mmol/L L-homoarginine) in a final volume of 200 μL and prewarmed to 37 °C for 10 min in a 96-well flat-bottom culture dish. Twenty microlitres of prewarmed 120 mmol/L p-nitrophenylphosphate (pNPP) dissolved in 1×SEAP assay buffer was then added and mixed. A405 of the reaction mixture was read in a microplate reader at 5-min intervals. The change in absorbance was plotted and the maximum linear reaction rate was determined. The SEAP activity was expressed in A405 (data not shown). The amount of both HBsAg and HBV DNA was corrected by the SEAP activities.
Detection for HBsAg secreted to cell culture medium
The amount of HBV surface antigen (HBsAg) secreted into the cell culture medium was determined by enzyme-linked immunosorbent assay (ELISA, Hepanostika HBsAg Uni-Form II, Biomérieux bv, Netherlands) after removal of cell debris and stored at -20 °C until analysis. Total amount of HBsAg was calculated by comparison with a standard HBsAg-positive human serum and negative control sera provided by the manufacturer. If the A450 for HBsAg was more than 3.0, a 1:10 dilution of culture medium was made for further assay.
Real-time quantitative PCR to detect HBV DNA secreted to culture medium
Cell culture medium was collected 3 d after transfection and centrifuged at 14000 r/m rpm 10 min at 4 °C to remove cell debris. The supernatant was adjusted to 10 mmol/L MgCl2 and treated with 100 μg/mL DNase I for 60 min at 37 °C to remove the remaining recombinants. The reaction was stopped by adding EDTA to a final concentration of 25 mmol/L. HBV DNA in the cell medium was detected by fluorescent real-time quantitative PCR (commercially available assay kit, Roche Light cycler).
RESULTS
Replication-competent constructs containing HBV genome with mutations at precore region
By PCR-mediated site-directed mutagenesis method and molecular cloning, we obtained different HBV fragments containing varied mutations with different mutagenetic primers. The agarose gel electrophoresis of different HBV fragments is shown in Figure 1. Finally we acquired four kinds of replication-competent constructs containing HBV genome with mutations at the precore region. All the mutated sites were ascertained by sequencing. The sequences were analyzed by Chromas and Bioedit software. Therefore the desired mutations instead of the unrelated mutations were introduced. Part of the sequencing results at the mutation site are shown in Figure 2.
Figure 2.
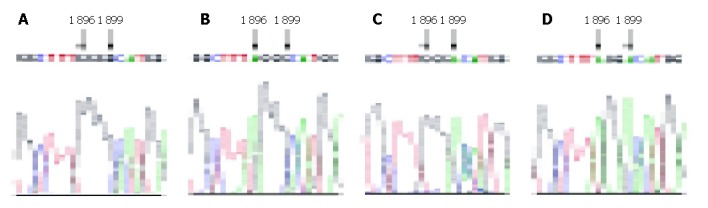
Local sequences of mutagenesis sites of 4 recombinants. Arrowhead indicates the mutated nucleosides. A: pUC18-HBV1.2-WT; B: pUC18-HBV1.2-A1896; C: pUC18-HBV1.2-A1899; D: pUC18-HBV1.2-AA.
HBV reconstructs were named as pHBV1.2 containing a 1.2-fold-overlength genome of HBV, genetype C, with a 5’ terminal redundancy encompassing enhancers I and II, the origin of replication (direct repeats DR1 and DR2), the X- and pregenomic/core promoter regions, the transcription initiation site of the pregenomic RNA, the unique polyadenylation site, and the entire X open reading frame as depicted in Figure 3. Such a recombinant was proven to initiate HBV replication efficiently and with high liver specificity in transfection experiments and in transgenic mice[15]. In this research, we obtained 4 recombinant constructs by orientation-cloning and PCR-mediated site-directed mutagenesis. As shown in Figure 3, the 4 constructs contained different combinations of mutations at HBV precore region.
Figure 3.

Sketch map of structure of plasmid pUC-HBV1.2 and the mutation sites of all constructed plasmids. Panel A: Upper line and box figure indicate the 1.2 copy HBV genome cloned into pUC18 EcoRI and Hind III sites. Different open reading frames are indicated with corresponding box. Lower letters indicate HBV BCP and precore region sequence, TA rich region and DR region are underlined. Scheduled nucleosides to be mutated are indicated with numeral; Panel B: Recombinant constructs and their corresponding mutation sites. The constructs are listed at left, long gray box indicate consensus sequences, mutation nucleosides and their sites are indicated.
Dose-dependent sensitivity to interferon in vitro
As described in Materials and Methods, the plasmid containing wild type HBV was transfected to Huh7 in six-well plates. Different concentrations of interferon-α (62.5 IU/mL, 125 IU/mL, 250 IU/mL, 500 IU/mL, 1000 IU/mL, 2000 IU/mL, 4000 IU/mL, 8000 IU/mL) were added and cell medium was collected 3 d after transfection. As shown in Figure 4, different concentrations of interferon-α had different effects on HBV antigen expression and viral replication. Along with increase of interferon-α concentration, HBsAg and HBeAg secretion decreased almost in the same extent in general. The results showed that interferon-α with 2000 IU/mL medium in vitro could inhibit HBV antigen expression to the highest extent. For the viral DNA secreted to the medium, the realtime PCR was used to detect HBV DNA. With different concentrations of interferon-α in cell medium, the amounts of secreted viral DNA were also different. HBV DNA decreased most in the medium with 1000 IU/mL interferon-α. According to previous reports and our experimental results, we selected the concentration 1000 IU/mL of interferon-α as the concentration of experiments in vitro.
Figure 4.
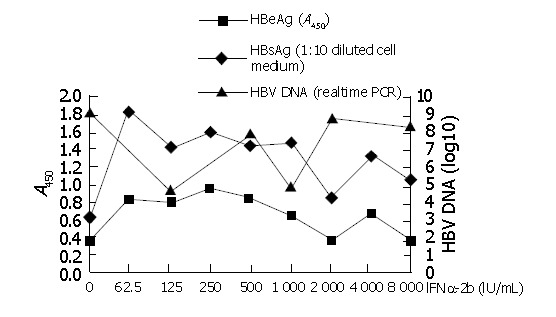
Detection of HBsAg, HBeAg, HBV DNA secreted to the cell medium with different concentrations of interferon.
Precore mutation decreased HBV replication
All the different recombinants were introduced into hepatoma cell Huh7 by calcium phosphate transient transfection method. Cell culture medium was collected 3 d after transfection and HBV DNA was detected by fluorescent real-time quantitative PCR. HBV DNA in the cell medium was given as viral copies per milliliter. As shown in Table 3 and Figure 4, all the recombinants could produce and secrete HBV DNA to the cell medium. When the amounts of HBV DNA in the medium were compared, replication efficiency was different in different recombinants. Recombinants containing precore mutations had lower replication efficiency than wild type. The recombinant containing the A1896 and A1899 double mutations had lower replication efficiency than the recombinant containing A1896 or A1899 single mutations.
Table 3.
HBsAg and HBV DNA secreted into cell culture medium with or without IFN (mean±SD).
| Recombinants |
HBsAg (A450) |
HBV DNA (realtime PCR, log10) |
||
| Without IFN | With IFN | Without IFN | With IFN | |
| Puc18-HBV1.2-WT | 2.63±0.1c | 1.96±0.07c | 5.02±0.06ac | 4.86±0.07c |
| Puc18-HBV1.2-A1896 | 1.43±0.05 | 1.46±0.07 | 4.43±0.09ac | 5.53±0.07c |
| Puc18-HBV1.2-A1899 | 1.97±0.15 | 1.96±0.12 | 4.80±0.05 | 4.90±0.06 |
| Puc18-HBV1.2-AA | 2.6±0.05 | 2.5±0.06 | 4.06±0.07ac | 4.46±0.05c |
n = 3,
P<0.05 (WT in comparison with MT without IFN),
P<0.05 (without IFN in comparison with with IFN). WT: wild type, MT: mutated type.
Precore mutation exerted no effect on expression of HBsAg
Cell culture medium was collected 3 d after transfection with recombinant constructs. The amount of hepatitis B virus surface antigen (HBsAg) secreted into the cell culture medium was determined by ELISA. All the recombinants could express and secrete HBsAg into cell medium. When the relative amount of HBsAg was compared (Table 3), a slight difference was found among all the recombinants containing different precore mutation combinations. But there was no significant difference by statistic analysis (P>0.05). The result indicated that precore mutation might have no effect on expression of HBsAg.
Recombinants containing precore mutation were resistant to interferon
Sixteen hours after transfection, Huh7 cells were shocked with 15% glycerol solution, and then incubated with fresh culture medium for 24 h. Fresh complete medium with and without 1000 IU/mL IFN-α was added and the culture medium was harvested 3 d after transfection. HBV DNA in the cell medium was detected by fluorescent real-time quantitative PCR. By comparing HBV DNA in the cell medium with or without addition of IFN, we could know the sensitivity to IFN of different recombinants. The level of HBV DNA was decreased in pUC18-HBV1.2-WT recombinants after IFN was added while others with precore mutations were not. HBV DNA in the medium from the recombinants containing HBV precore mutations such as A1896 and/or A1899 did not decrease in amount after IFN was added. Thus recombinants containing precore mutations were resistant to interferon, precore mutations might be one indicator of HBV resistance to IFN.
Effect of IFN on secretion of HBsAg
As shown in Table 3, HBsAg secreted to the medium was slightly decreased in recombinants with HBV wild type after IFN was added. While after interferon was added, HBsAg in the medium of recombinants with HBV precore mutations did not decrease.
DISCUSSION
Chronic HBV infection remains a major public health problem worldwide as well as a therapeutic challenge. Despite a high rate of viral clearance in immunocompetent adults and the availability of efficient vaccines, a large proportion of the world’s population (400 million) are chronically infected with HBV. This is due to the fact that vertical transmission of HBV in neonates leads to a chronic infection in 90% of cases. Chronic HBV infection may result in a wide spectrum of liver diseases including acute self-limiting hepatitis or chronic infection. There are still 250 000 deaths each year resulted from hepatitis B. While HBV is generally regarded as noncytopathic, HBV-induced liver diseases are mediated by cytotoxic T-lymphocyte (CTL) lysis of infected hepatocytes[16,17]. Nevertheless, activity of HBV-induced liver diseases may also relate to viral factors. The coexistence of repeated cycles of HBV replication and immune lysis of infected hepatocytes are associated with fibrosis, cirrhosis, and hepatocellular carcinoma.
Current strategies for treating hepatitis B are focused on clearance of active HBV infection through suppression of viral replication. Interferon-α (IFN-α) and nucleoside analogs (lamivudine and adefovir) have been approved for their clinical use by FDA. The sustained virological response rate to IFN-α, however, ranges between 20% and 40%[3,4]. The actual rate of sustained response in non selected patients is supposed to be lower. The resistance to antiviral treatment by some viruses is due to positive selection of variants harboring mutations that confer resistance[18]. Variations at the precore region have been reported to be associated with the response to IFN therapy[19]. The mutation G to A switch at position 1896 of the pre-core region of the HBV genome that leads to a translational stop codon in the leader sequence of HBeAg protein could result in the inhibition of protein synthesis[20]. Such variants selected during seroconversion from HBeAg to anti-HBe are responsible for HBeAg-negative chronic hepatitis B, which represents up to 50-95% of the chronic hepatitis B cases followed up in liver units in Europe and has an increasing prevalence from north to south[21]. Recently a total of 694 consecutive chronic HBV-infected patients seen in 17 liver centers in the US during a one-year period were enrolled[22]. Precore variants are found in 27% of chronic hepatitis B (CHB) patients and more common in hepatitis B e antigen (HBeAg)-negative patients than in HBeAg-positive patients (38% vs 9%, P<0.001). The precore stop codon variants are detected in a median of 60% (range 0-100%) of HBeAg-negative patients, 92% in the Mediterranean, 50% in Asia Pacific and 24% in the USA and Northern Europe[23]. Anti-hepatitis e antigen-positive chronic hepatitis B is a progressive liver disease associated with precore mutants and poor response to interferon. It is still unknown why this variant is related to IFN responsiveness, and whether it is IFN-sensitive or resistant, or whether it is directly or causally related to responsiveness. Previous studies have not fully answered these questions and the results are contradictory[10].
Some studies have shown that mutant-type diseases (anti-HBe-positive/HBeAg-negative) are less responsive to IFN given for 6-12 mo while wild-type responds relatively well to IFN treatment. Naoumov et al[24] sequenced precore/core region in 46 serum samples obtained before, during, and after interferon treatment of 12 patients. No significant changes occurred in the precore/core regions in responders after seroconversion to anti-hepatitis B e antigen, but multiple variations persisted in nonresponders during treatment and new mutations occurred with the relapse of hepatitis. Other studies had contradictory results. Shindo et al[25] investigated 93 patients with chronic hepatitis B treated with IFN alpha. The results suggest that the precore wild type and mutant have similar sensitivity to IFN. Schepis et al[26] found core gene variability did not seem to be involved either in the outcome of infection or in the responses to IFN treatment in children with chronic HBV infection. Zampino et al[27] showed that G1896A precore stop codon mutation was not detected before, during and after interferon treatment in young Caucasian cancer survivors who acquired HBV infection during chemotherapy for malignancies. Some researchers[10] have found that precore mutant HBV could influence the response to interferon when it reaches significant serum levels (> 20% of total viremia).
For the study of HBV infection, no permissive cell line or small animals are available. Stable cell lines with integrated HBV genomes, e.g., HepG2.2.15 cells[28], are commonly used for assessing the action of drugs on HBV replication. HBV-transgenic mice have been proved to be very useful for immunological studies[29]. However, stable cell lines as well as transgenic mice have the disadvantage that, unlike in natural infection, HBV replicates from an integrated genome which cannot be eliminated. In contrast to almost all previous studies with cell lines containing chromosomally integrated HBV DNA or transfection of cells with subgenomic HBV fragments[5,30-32], we developed recombinant constructs in which 1.2-fold-overlength HBV containing precore mutations were inserted. All the different recombinants were introduced into hepatoma cell line Huh7 by calcium phosphate transient transfection method. The constructs efficiently initiated virus replication and antigen expression in transiently transfected hepatoma cell line Huh7, which could be demonstrated by measuring HBsAg and HBV DNA secreted to the cell culture medium. All the recombinant constructs were replication-competent. By comparing the amount of HBV DNA into the medium, we found that the replication efficiency was different in different recombinants. Whether HBV harboring single A1896 and A1899 precore mutations affects replication efficiency or not is still uncertain.
Interferons exert their cellular activities by binding to specific membrane receptors on the cell surface. Once bound to the cell membrane, interferons initiate a complex sequence of intracellular events including induction of certain enzymes, suppression of cell proliferation, immunomodulating activities such as enhancement of the phagocytic activity of macrophages and augmentation of the specific cytotoxicity of lymphocytes for target cells, and inhibition of virus replication in virus-infected cells. It has been reported that type I interferons have immunomodulatory properties as well as direct antiviral activity[33]. Previous results obtained in in vivo studies could not show whether precore mutation of HBV is directly related to between IFN responsiveness. We used an in vitro system by hepatoma cell lines (as described before) to investigate the relationship between precore mutation and IFN responsiveness. By comparing HBV DNA in the cell medium, we found that the production of HBV DNA was decreased in HBV wild type after adding IFN but not in HBV precore mutations. It demonstrates that IFN could directly suppress viral replication in Huh7 cells transiently transfected with wild type HBV DNA in vitro. These in vitro experiment results indicate that the mechanism of IFN’s antiviral effect is not only by immune regulation but also by direct or indirect suppression of viral replication. IFN can not suppress viral replication in Huh7 cells transiently transfected with mutant type HBV DNA in vitro while it suppresses wild type. Meanwhile our results imply that the antiviral effect of IFN in vitro is dose-dependent. Based on the above results, we conclude that interferon has direct anti-viral effects, HBV harboring precore mutations influences the sensitivity of IFN. Therefore precore mutations might at least partly affect the response of IFN in vivo.
As discussed before, IFN might have direct antiviral effects on HBV. HBsAg secreted to the medium was slightly decreased in recombinants with HBV wild type after adding IFN while HBsAg did not decrease in recombinants with precore mutations. The results imply that the antiviral mechanism of IFN is not only by affecting viral replication but also by affecting viral antigen expression of HBV wild type. Precore mutations might hamper this mechanism. These results are similar to previous studies. Recent studies in chimpanzees have shown that in the initial phase of acute HBV-infection a non-cytolytic downregulation of HBV-replication takes place before cytotoxic T-cell mediated destruction of HBV-antigen[34]. Various in vitro studies have examined the inhibitory effects of IFN-α/β on HBV replication. Rang et al[35] showed that IFN-α might reduce the stability of HBV RNA in transiently transfected Huh7-cells and Caselmann et al[6] observed a decrease of pregenomic HBV RNA in HepG2 cells stably transfected with the HBV-genome when they were treated with IFN-β. Hamasaki et al[36] demonstrated a reduction of HBs-antigen mRNA-levels by IFN-α in hepatoma cells with integrated HBV-DNA. Thus type I IFNs inhibit HBV replication and protein production by a variety of mechanisms.
Our experiments indicate that HBV with precore stop mutation is resistant to IFN treatment in vitro. The result has been ascertained by some studies in vivo. The anti-viral mechanisms of IFN are very complicated. Further studies are needed to explore the mechanisms of HBV precore mutation resistance to IFN therapy. The pathogenicity and the treatment of HBV precore mutation remain to be investigated.
Footnotes
Supported by the Major State Basic Research Development Program of China (973 Program), No. G1999054106
Edited by Wang XL and Zhu LH
References
- 1.de Franchis R, Hadengue A, Lau G, Lavanchy D, Lok A, McIntyre N, Mele A, Paumgartner G, Pietrangelo A, Rodés J, et al. EASL International Consensus Conference on Hepatitis B. 13-14 September, 2002 Geneva, Switzerland. Consensus statement (long version) J Hepatol. 2003;39 Suppl 1:S3–25. [PubMed] [Google Scholar]
- 2.Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. 2000;64:51–68. doi: 10.1128/mmbr.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niederau C, Heintges T, Lange S, Goldmann G, Niederau CM, Mohr L, Häussinger D. Long-term follow-up of HBeAg-positive patients treated with interferon alfa for chronic hepatitis B. N Engl J Med. 1996;334:1422–1427. doi: 10.1056/NEJM199605303342202. [DOI] [PubMed] [Google Scholar]
- 4.Zuckerman AJ, Lavanchy D. Treatment options for chronic hepatitis. Antivirals look promising. BMJ. 1999;319:799–800. doi: 10.1136/bmj.319.7213.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayashi Y, Koike K. Interferon inhibits hepatitis B virus replication in a stable expression system of transfected viral DNA. J Virol. 1989;63:2936–2940. doi: 10.1128/jvi.63.7.2936-2940.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caselmann WH, Meyer M, Scholz S, Hofschneider PH, Koshy R. Type I interferons inhibit hepatitis B virus replication and induce hepatocellular gene expression in cultured liver cells. J Infect Dis. 1992;166:966–971. doi: 10.1093/infdis/166.5.966. [DOI] [PubMed] [Google Scholar]
- 7.Davis MG, Jansen RW. Inhibition of hepatitis B virus in tissue culture by alpha interferon. Antimicrob Agents Chemother. 1994;38:2921–2924. doi: 10.1128/aac.38.12.2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marrone A, Zampino R, Luongo G, Utili R, Karayiannis P, Ruggiero G. Low HBeAg serum levels correlate with the presence of the double A1762T/G1764A core promoter mutation and a positive response to interferon in patients with chronic hepatitis B virus infection. Intervirology. 2003;46:222–226. doi: 10.1159/000072431. [DOI] [PubMed] [Google Scholar]
- 9.Raimondo G, Schneider R, Stemler M, Smedile V, Rodino G, Will H. A new hepatitis B virus variant in a chronic carrier with multiple episodes of viral reactivation and acute hepatitis. Virology. 1990;179:64–68. doi: 10.1016/0042-6822(90)90274-u. [DOI] [PubMed] [Google Scholar]
- 10.Brunetto MR, Giarin M, Saracco G, Oliveri F, Calvo P, Capra G, Randone A, Abate ML, Manzini P, Capalbo M. Hepatitis B virus unable to secrete e antigen and response to interferon in chronic hepatitis B. Gastroenterology. 1993;105:845–850. doi: 10.1016/0016-5085(93)90903-p. [DOI] [PubMed] [Google Scholar]
- 11.Brunetto MR, Oliveri F, Rocca G, Criscuolo D, Chiaberge E, Capalbo M, David E, Verme G, Bonino F. Natural course and response to interferon of chronic hepatitis B accompanied by antibody to hepatitis B e antigen. Hepatology. 1989;10:198–202. doi: 10.1002/hep.1840100213. [DOI] [PubMed] [Google Scholar]
- 12.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A Laboratory Manual, 2nded. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 13.Berger J, Hauber J, Hauber R, Geiger R, Cullen BR. Secreted placental alkaline phosphatase: a powerful new quantitative indicator of gene expression in eukaryotic cells. Gene. 1988;66:1–10. doi: 10.1016/0378-1119(88)90219-3. [DOI] [PubMed] [Google Scholar]
- 14.Cullen BR, Malim MH. Secreted placental alkaline phosphatase as a eukaryotic reporter gene. Methods Enzymol. 1992;216:362–368. doi: 10.1016/0076-6879(92)16033-g. [DOI] [PubMed] [Google Scholar]
- 15.Guidotti LG, Matzke B, Schaller H, Chisari FV. High-level hepatitis B virus replication in transgenic mice. J Virol. 1995;69:6158–6169. doi: 10.1128/jvi.69.10.6158-6169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chisari FV, Ferrari C, Mondelli MU. Hepatitis B virus structure and biology. Microb Pathog. 1989;6:311–325. doi: 10.1016/0882-4010(89)90073-9. [DOI] [PubMed] [Google Scholar]
- 17.Suri D, Schilling R, Lopes AR, Mullerova I, Colucci G, Williams R, Naoumov NV. Non-cytolytic inhibition of hepatitis B virus replication in human hepatocytes. J Hepatol. 2001;35:790–797. doi: 10.1016/s0168-8278(01)00215-x. [DOI] [PubMed] [Google Scholar]
- 18.Carman W, Thomas H, Domingo E. Viral genetic variation: hepatitis B virus as a clinical example. Lancet. 1993;341:349–353. doi: 10.1016/0140-6736(93)90146-8. [DOI] [PubMed] [Google Scholar]
- 19.Fattovich G, McIntyre G, Thursz M, Colman K, Giuliano G, Alberti A, Thomas HC, Carman WF. Hepatitis B virus precore/core variation and interferon therapy. Hepatology. 1995;22:1355–1362. [PubMed] [Google Scholar]
- 20.Carman WF, Jacyna MR, Hadziyannis S, Karayiannis P, McGarvey MJ, Makris A, Thomas HC. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet. 1989;2:588–591. doi: 10.1016/s0140-6736(89)90713-7. [DOI] [PubMed] [Google Scholar]
- 21.Zoulim F, Tong S, Trépo C. Study of HBV replication capacity in relation to sequence variation in the precore and core promoter regions. Methods Mol Med. 2004;95:235–246. doi: 10.1385/1-59259-669-X:235. [DOI] [PubMed] [Google Scholar]
- 22.Chu CJ, Keeffe EB, Han SH, Perrillo RP, Min AD, Soldevila-Pico C, Carey W, Brown RS, Luketic VA, Terrault N, et al. Prevalence of HBV precore/core promoter variants in the United States. Hepatology. 2003;38:619–628. doi: 10.1053/jhep.2003.50352. [DOI] [PubMed] [Google Scholar]
- 23.Funk ML, Rosenberg DM, Lok AS. World-wide epidemiology of HBeAg-negative chronic hepatitis B and associated precore and core promoter variants. J Viral Hepat. 2002;9:52–61. doi: 10.1046/j.1365-2893.2002.00304.x. [DOI] [PubMed] [Google Scholar]
- 24.Naoumov NV, Thomas MG, Mason AL, Chokshi S, Bodicky CJ, Farzaneh F, Williams R, Perrillo RP. Genomic variations in the hepatitis B core gene: a possible factor influencing response to interferon alfa treatment. Gastroenterology. 1995;108:505–514. doi: 10.1016/0016-5085(95)90080-2. [DOI] [PubMed] [Google Scholar]
- 25.Shindo M, Hamada K, Koya S, Sokawa Y, Okuno T. The clinical significance of core promoter and precore mutations during the natural course and interferon therapy in patients with chronic hepatitis B. Am J Gastroenterol. 1999;94:2237–2245. doi: 10.1111/j.1572-0241.1999.01299.x. [DOI] [PubMed] [Google Scholar]
- 26.Schepis F, Verucchi G, Pollicino T, Attard L, Brancatelli S, Longo G, Raimondo G. Outcome of liver disease and response to interferon treatment are not influenced by hepatitis B virus core gene variability in children with chronic type B hepatitis. J Hepatol. 1997;26:765–770. doi: 10.1016/s0168-8278(97)80240-1. [DOI] [PubMed] [Google Scholar]
- 27.Zampino R, Marrone A, Cirillo G, del Giudice EM, Utili R, Karayiannis P, Liang TJ, Ruggiero G. Sequential analysis of hepatitis B virus core promoter and precore regions in cancer survivor patients with chronic hepatitis B before, during and after interferon treatment. J Viral Hepat. 2002;9:183–188. doi: 10.1046/j.1365-2893.2002.00347.x. [DOI] [PubMed] [Google Scholar]
- 28.Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci USA. 1987;84:1005–1009. doi: 10.1073/pnas.84.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guidotti LG, Borrow P, Hobbs MV, Matzke B, Gresser I, Oldstone MB, Chisari FV. Viral cross talk: intracellular inactivation of the hepatitis B virus during an unrelated viral infection of the liver. Proc Natl Acad Sci USA. 1996;93:4589–4594. doi: 10.1073/pnas.93.10.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tur-Kaspa R, Teicher L, Laub O, Itin A, Dagan D, Bloom BR, Shafritz DA. Alpha interferon suppresses hepatitis B virus enhancer activity and reduces viral gene transcription. J Virol. 1990;64:1821–1824. doi: 10.1128/jvi.64.4.1821-1824.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berthillon P, Crance JM, Leveque F, Jouan A, Petit MA, Deloince R, Trepo C. Inhibition of the expression of hepatitis A and B viruses (HAV and HBV) proteins by interferon in a human hepatocarcinoma cell line (PLC/PRF/5) J Hepatol. 1996;25:15–19. doi: 10.1016/s0168-8278(96)80322-9. [DOI] [PubMed] [Google Scholar]
- 32.Romero R, Lavine JE. Cytokine inhibition of the hepatitis B virus core promoter. Hepatology. 1996;23:17–23. doi: 10.1002/hep.510230103. [DOI] [PubMed] [Google Scholar]
- 33.Pfeffer LM, Dinarello CA, Herberman RB, Williams BR, Borden EC, Bordens R, Walter MR, Nagabhushan TL, Trotta PP, Pestka S. Biological properties of recombinant alpha-interferons: 40th anniversary of the discovery of interferons. Cancer Res. 1998;58:2489–2499. [PubMed] [Google Scholar]
- 34.Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825–829. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- 35.Rang A, Günther S, Will H. Effect of interferon alpha on hepatitis B virus replication and gene expression in transiently transfected human hepatoma cells. J Hepatol. 1999;31:791–799. doi: 10.1016/s0168-8278(99)80279-7. [DOI] [PubMed] [Google Scholar]
- 36.Hamasaki K, Nakata K, Nakao K, Mitsuoka S, Tsutsumi T, Kato Y, Shima M, Ishii N, Tamaoki T, Nagataki S. Interaction of interferon-alpha with interleukin-1 beta or tumor necrosis factor-alpha on hepatitis B virus enhancer activity. Biochem Biophys Res Commun. 1992;183:904–909. doi: 10.1016/0006-291x(92)90569-7. [DOI] [PubMed] [Google Scholar]