

Abstract
AIM: To study the modulation of glutamate on post-ischemic intestinal and cerebral inflammatory responses in a ischemic and excitotoxic rat model.
METHODS: Adult male rats were subjected to bilateral carotid artery occlusion for 15 min and injection of monosodium glutamate intraperitoneally, to decapitate them at selected time points. Tumor necrosis factor alpha (TNF-α) level and nuclear factor kappa B (NF-κB) activity were determined by enzyme-linked immunosorbant assay (ELISA) and electrophoretic mobility shift assay (EMSA), respectively. Hemodynamic parameters were monitored continuously during the whole process of cerebral ischemia and reperfusion.
RESULTS: Monosodium glutamate (MSG) treated rats displayed statistically significant high levels of TNF-α in cerebral and intestinal tissues within the first 6 h of ischemia. The rats with cerebral ischemia showed a minor decrease of TNF-α production in cerebral and intestinal tissues. The rats with cerebral ischemia and treated with MSG displayed statistically significant low levels of TNF-α in cerebral and intestinal tissues. These results correlated significantly with NF-κB production calculated at the same intervals. During experiment, the mean blood pressure and heart rates in all groups were stable.
CONCLUSION: Glutamate is involved in the mechanism of intestinal and cerebral inflammation responses. The effects of glutamate on cerebral and intestinal inflammatory responses after ischemia are up-regulated at the transcriptional level, through the NF-κB signal transduction pathway.
Keywords: Cerebral ischemia, Glutamate, Intestine inflammatory responses, Cerebral inflammatory responses, NF-κB
INTRODUCTION
Cerebral ischemia, a major threatening pathologic process in many brain diseases, such as gastrointestinal ischemia and hemorrhage[1] is complicated by multiple organ dysfunction. In this process, proinflammatory cytokines, such as TNF-α, are elevated in the injured areas[2].
During an early stage of cerebral ischemia, glutamate, a ubiquitous excitatory amino acid in mammalian brains, is released from neurons and cumulated in intercellular space[3,4]. Glutamate and its receptors play key roles in pathology of ischemia injury[5], leading to the glutamate-calcium overload hypothesis[6,7]. According to this hypothesis, glutamate is implicated in the pathology in two ways: excessive accumulation of glutamate in the extracellular spaces during ischemia, and subsequent activation of glutamate receptors in postsynaptic cells[8,9].
Thousands of agents are intentionally added into the food we consume. Monosodium glutamate (MSG) is one of the widely-used food addictives in our daily diet[10]. MSG treatment provokes hormonal alterations and specific intestinal changes in smooth muscle reactivity to agonists[11]. Oral or intestinal stimulation with isotonic MSG solution could increase the discharge rate of gastric branches of the vagus nerve[12]. In addition, excitotoxic injury could also result in a marked stimulation of TNF-α mRNA expression in forebrain structure lesions[13]. Although intestinal inflammatory response is activated after the elevation of intracellular glutamate concentrations[14], the effect of glutamate on the gastrointestinal system after cerebral ischemia still remains unclear.
In the present study, we used a post-ischemic and excitotoxic animal model to investigate the effect of glutamate on inflammatory response of intestine and brain after cerebral ischemia. The involvement of glutamate in the mechanism of post-ischemic inflammatory response and was discussed.
MATERIALS AND METHODS
The rat TNF-α kit was purchased from DIACLONE (Besancon, France). [32P]ATP was purchased from Free Biotech (Beijing, China). A commercial kit for EMSA was purchased from PROMEGA (Madison, WI). All other reagents were of molecular biology grade and purchased from SIGMA. All reagents and plasticwares used throughout the experiments were pyrogen-free.
Animal and experimental protocol
Adult male Sprague-Dawley rats, purchased from the Animal Center of the Chinese Academy of Sciences (Shanghai, China), were divided into experimental groups in a random manner. All procedures were approved by the Institutional Animal Care Committee. The rats were housed in plexiglass cages at 23±1 °C, and had free access to food and water ad libitum for 7 d, and used for experiments when their body weights gained between 300-350 g. The rats were anesthetized with urethane (1000 mg/kg body weight, intraperitoneally). A polyethylene catheter was implanted in the tail vein for continuous infusion of solutions using a Razel Model WZ-50C syringe pump.
The rats were randomly assigned into 4 groups (6 rats each group). Group A (the control group) accepted a pseudo-operation of cervical incision and was injected with the same volume of physiological saline intraperitoneally. Group B was administered 4 mg/kg body weight MSG after cervical incision. Group C and group D served as the ischemic groups. An ischemical model was established by 15-min of bilateral carotid artery occlusion and reperfusion afterwards[15,16]. Group D, however, accepted an additional MSG (4 mg/kg body weight) at the beginning of carotid artery occlusion.
In the experiments, the right femoral artery was intubated with a 25 G catheter prefilled with heparin solution and hemodynamical parameters were continuously observed by using a Datex monitor. Pre-ischemia was appointed at 0, 10, 20, 30, 60, 120 min after onset of the cerebral ischemia at 10, 20, 30, 60, 120 min, respectively.
The rats were decapitated at 6 h after ischemic insults. Blood plasma, cerebral cortex and small intestine were collected for TNF-α or NF-κB measurement. All samples were frozen immediately in liquid nitrogen, and stored at -80 °C.
Enzyme-linked immunosorbant assays (ELISA)
TNF-α concentrations in plasma, cerebral and intestinal tissues were quantified by ELISA according to the manufacturer’s instructions.
Nuclear protein extracts and electrophoretic mobility shift assay (EMSA)
Nuclear extracts from cerebral and intestinal tissues were prepared by hypotonic lysis followed by high salt extraction as described previously[17-19]. Intestinal tissue cells were incubated in 0.5 mL ice-cold buffer (10 mmol/L HEPES, pH 7.9, 10 mmol/L KCl, 2 mmol/L MgCl2, 0.1 mmol/L EDTA, 1 mmol/L dithiothreitol (DTT)), then in 0.5 mmol/L phenylmethysulfonyl fluoride (PMSF) for 15 min, and 50 μL NP-40 was added. After 30 s, the mixture was centrifuged at 5000 g for 10 min at 4 °C, the pellet was suspended in 50 μL ice-cold buffer (50 mmol/L HEPES, pH 7.9, 50 mmol/L KCl, 300 mmol/L NaCl, 0.1 mmol/L EDTA, 1 mmol/L DTT, 0.5 mmol/L PMSF, with 100 mL/L glycerol) and incubated on ice for 30 min and frequently mixed. After centrifugation at 12000 g for15 min at 4 °C, the supernatants were collected and stored at - 80 °C until use. Protein concentration was determined as described previously[20,21].
The NF-κB consensus oligonucleotide probe (5’-AGTTGAGGGGACTTTCCCAGGC-3’) was end-labeled with [γ-32P]-ATP with T4-polynucleotide kinase. Nuclear protein (intestine: 80 μg, cerebra: 60 μg) was pre-incubated for 10 min at room temperature (RT) in 9 μL of binding buffer, containing 10 mmol/L Tris-HCl (pH 7.5), 4% glycerol, 1 mmol/L MgCl, and 0.5 mmol/L EDTA, 0.5 mmol/L DTT, 50 mmol/L NaCl, and 0.05 mg/mL poly (di-dc). After the 32P-lebaled oligonucleotide probe was added, the incubation was continued for 20 min at RT. Reaction was stopped by adding 1 μL gel loading buffer, and the mixture was subjected to nondenaturing 4% polyacrylamide gel electrophoresis in 0.5×TBE buffer, pre-run at 300 V for 10 min. Electrophoresis was conducted at 390 V for 1 h. After electrophoresis, gels were transferred to Whatman DE-81 paper, dried and exposed to autoradiographic film (Fuji hyperfilm) with an intensifier screen at -70 °C[22].
Data presentation and statistical analysis
Data were presented as mean±SE, and compared by analysis of variance followed by one-way ANOVA and Tukey test (using SPSS 10.0 software equipped with a Pentium 4 computer). P<0.05 was accepted as statistically significant.
RESULTS
Hemodynamical parameters were stable during cerebral- ischemia/MSG insults
In the experiments, the mean artery pressure (MAP) fluctuated at 80-90 mmHg, and the heart rate was190-210 beats per minute (bpm). TNF-α was not detected in plasma, there was no significant difference in MAP or heart rate among all groups at any time point (Figures 1, 2).
Figure 1.
Mean artery pressure (MAP) changes during cerebral ischemia and MSG insults. There were no significant changes between inter-groups or intra-groups.
Figure 2.
Heart rate changes during cerebral ischemia and MSG insults. There were no significant changes between inter-groups or intra-groups.
MSG provoked TNF-α production both in intestine and in brain
Compared with control animals, the level of TNF-α was elevated 3-fold both in MSG-treated rat intestine (6.08±0.52 vs 2.03±0.63 pg/g pro, P<0.05 , n = 6) and in cerebra (4.94±0.51 vs 1.85±0.32 pg/g pro, P<0.05 , n = 6) (Figure 3).
Figure 3.
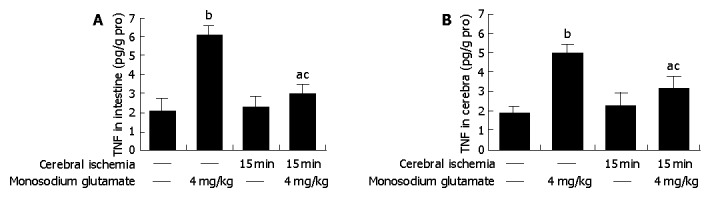
TNF-α production in intestine (A) and cerebra (B). TNF-α concentrations elevated in MSG-treated and ischemia/MSG-treated rats. Compared with that of MSG-treated rats, TNF-α levels deceased one-fold in ischemia/MSG-treated rats. aP<0.05 vs control, bP<0.01 vs control, cP<0.05 vs MSG group.
15-min cerebral ischemia increased TNF-α production in intestine and brain
TNF-α concentrations were elevated both in cerebra and intestine of the ischemic group, but did not differ significantly from the data of control (Figure 3).
Ischemia/MSG treatments increased TNF-α production but attenuated MSG-induced TNF-α elevation in intestine and brain
Cerebral-ischemic rats treated with MSG also had a high level of TNF-α (intestine: 3.02±0.49 vs 2.03±0.63 pg/g pro, cerebra: 3.09±0.69 vs 1.85±0.32 pg/g pro, P<0.05 , n = 6). Compared with MSG-treatment, the values were deceased both in intestine and cerebra (intestine: 3.02±0.49 vs 6.08±0.52 pg/g pro, cerebra: 3.09±0.69 vs 4.94±0.51 pg/g pro, P<0.05 , n = 6) (Figure 3).
NF-κB activation in intestine
Both MSG-treatment and cerebral ischemia insults induced a significantly higher intensity of NF-κB in the intestine than in controls (P<0.05). Compared with MSG-treatment, NF-κB activity decreased with ischemia-MSG-treatment (Figure 4A).
Figure 4.
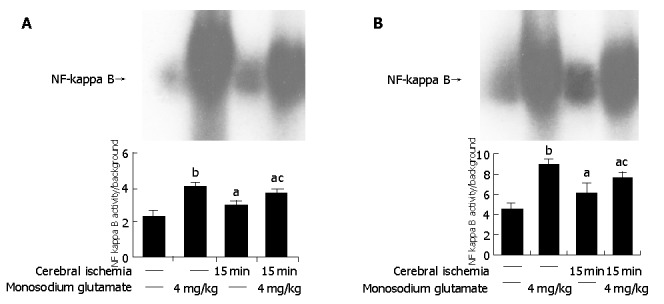
NF-κB activation in intestinal tissues (A) and in cerebral tissue (B). Lane1: controls, lane 2: MSG-treated rats, lane 3:cerebral-ischemic rats, lane 4: NF-κB intensity of ischemia/MSG-treated rats aP<0.05 vs control, bP<0.01 vs control, cP<0.05 vs MSG-treatment.
NF-κB activation in brain
The similar results were found in the brain. MSG-treatment and cerebral ischemia induced a significantly higher intensity of NF-κB in the cerebra than in control (P<0.05). Compared with MSG-treatment, NF-κB activity decreased significantly in ischemia-MSG-treatment compared with control (P<0.05) (Figure 4B).
DISCUSSION
In the present study, we demonstrated that 15-min cerebral ischemia plus exogenous glutamate or single exogenous glutamate could provoke significant inflammation responses in the central nervous system and gastrointestinal system characterized by an excess production of TNF-α and activation of NF-κB. However, the inflammatory response either in brain or in intestine was not prominent during cerebral ischemia injury.
It has been reported that TNF-α is the most important proinflammatory cytokine, which is released early after an inflammatory stimulus[23], contributing to both morbidity and mortality in conditions of ischemia-reperfusion injury. Among the cytokines produced in intestinal mucosae during inflammation, TNF-α is particularly important because of its biological effects on intestine and other systems[24].
In cytoplasms, NF-κB is associated with its inhibitory subunits, inhibitory kappa B (IκB), which prevents it from translocating into nuclei[25]. Many stimuli including endotoxin, induce the phosphorylation and degradation of IκB, freeing NF-κB from NF-κB/IκBs complexes and enabling it to translocat into nuclei[25,26], where it regulates gene transcription. Many effector genes, including those encoding cytokines and adhesion molecules, are in turn regulated by NF-κB. Some anti-inflammatory agents (e.g., salicylates, dexamethasone) can inhibit NF-κB, suggesting that it is an important molecular target for the modulation of inflammatory diseases[27,28].
Cytokines and inflammation in the central nervous system (CNS) have been primarily studied in the context of autoimmune and infectious diseases. In this study, we found that the production of TNF-α in the central nervous system and gastrointestinal system was increased and NF-κB activity was increased after MSG treatment, indicating that the inflammatory responses in the central nervous and gastrointestinal systems can be enhanced by glutamate after cerebral ischemia. Glutamate could induce gene transcription in numerous physiological and pathological conditions. Among the glutamate-responsive transcription factors, NF-kappaB has been mainly implicated in neuronal survival and death. Glutamate could induce a rapid reduction of I kappaB alpha levels and nuclear translocation of the NF-kappaB subunit p65. The glutamate-induced reduction of I kappaB alpha levels is blocked by the N-methyl-D-aspartate inhibitor MK801[29]. Inflammatory mediators are involved in the pathogenesis of focal ischemic brain damage and regulated at transcriptional level[30].
It seems that cerebral ischemia can attenuate the intestinal inflammatory response during an exogenous glutamate challenge. Cerebral ischemia might protect intestinal tissues against exogenous glutamate insults. The intestine may be more resistant to glutamate-induced toxic effects after cerebral ischemic injury. Perhaps this effect is associated with the distinctive role of glutamate and its receptors in the brain-gut axis[31]. Excitatory amino acid (EAA) transmission is in the center of the brain-gut axis, the dorsal vagal complex. However, further investigations need to be carried out.
However, we do not advocate administrating glutamate, because we found that exogenous glutamate exacerbated inflammation of the central nervous system after cerebral ischemia injury. After all, glutamate is the most important toxic EAA in the pathogenesis of cerebral ischemia-reperfusion injury, and systemic glutamate can be absorbed from portal vein system and may penetrate the blood-brain-barrier (BBB) during or after cerebral ischemia, since the BBB is destroyed after cerebral ischemia-reperfusion injury[23].
In conclusion, exogenous glutamate can provoke inflammatory responses in brain and intestine. The effect of glutamate on inflammatory response after ischemia is regulated at transcriptional level. Ischemic treatment can attenuate glutamate-induced TNF-α production both in brain and in intestine.
ACKNOWLEDGEMENTS
The authors thank Drs. Gen-Bao Feng, Zhi-Qiang Zhou, Wei-Yan Li, Man-Lin Duan, Si-Hai Zhu, Hong-Yan Liu and Jing Chen for their technical assistance and friendly help.
Footnotes
Assistant Editor Guo SY Edited by Wang XL and Ren SY
References
- 1.Chen DC, Jing BW. Acute stress-induced neurogenic gastrointestinal mucosa lesions. Chin J Practical Internal Med. 1997;17:641–642. [Google Scholar]
- 2.Li JS, Zhao JM, Guo SD, Zhang WH, Zhao J, Gao AS, Li JG. Changes and significance of prostaglandin and tumorous necrotic factor in aged rats with gastrointestinal injury after brain ischemia ia reperfusion. Chin J Gerontol. 2001;21:354–356. [Google Scholar]
- 3.Yusa T. Increased extracellular ascorbate release reflects glutamate re-uptake during the early stage of reperfusion after forebrain ischemia in rats. Brain Res. 2001;897:104–113. doi: 10.1016/s0006-8993(01)02099-6. [DOI] [PubMed] [Google Scholar]
- 4.Tang XC, Rao MR, Hu G, Wang H. Alterations of amino acid levels from striatum, hippocampus, and cerebral cortex induced by global cerebral ischemia in gerbil. Acta Pharmacol Sin. 2000;21:819–823. [PubMed] [Google Scholar]
- 5.Nakane H, Yao H, Ibayashi S, Kitazono T, Ooboshi H, Uchimura H, Fujishima M. Protein kinase C modulates ischemia-induced amino acids release in the striatum of hypertensive rats. Brain Res. 1998;782:290–296. doi: 10.1016/s0006-8993(97)01331-0. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto H, Mitani A, Cui Y, Takechi S, Irita J, Suga T, Arai T, Kataoka K. Neuroprotective effect of mild hypothermia cannot be explained in terms of a reduction of glutamate release during ischemia. Neuroscience. 1999;91:501–509. doi: 10.1016/s0306-4522(98)00648-4. [DOI] [PubMed] [Google Scholar]
- 7.Guo J, Meng F, Fu X, Song B, Yan X, Zhang G. N-methyl-D-aspartate receptor and L-type voltage-gated Ca2+ channel activation mediate proline-rich tyrosine kinase 2 phosphorylation during cerebral ischemia in rats. Neurosci Lett. 2004;355:177–180. doi: 10.1016/j.neulet.2003.10.076. [DOI] [PubMed] [Google Scholar]
- 8.Monnerie H, Shashidhara S, Le Roux PD. Effect of excess extracellular glutamate on dendrite growth from cerebral cortical neurons at 3 days in vitro: Involvement of NMDA receptors. J Neurosci Res. 2003;74:688–700. doi: 10.1002/jnr.10797. [DOI] [PubMed] [Google Scholar]
- 9.Pellegrini-Giampietro DE. The distinct role of mGlu1 receptors in post-ischemic neuronal death. Trends Pharmacol Sci. 2003;24:461–470. doi: 10.1016/S0165-6147(03)00231-1. [DOI] [PubMed] [Google Scholar]
- 10.Walker R, Lupien JR. The safety evaluation of monosodium glutamate. J Nutr. 2000;130:1049S–1052S. doi: 10.1093/jn/130.4.1049S. [DOI] [PubMed] [Google Scholar]
- 11.Sukhanov SN, de Andrade IS, Dolnikoff MS, Ferreira AT. Neonatal monosodium glutamate treatment alters rat intestinal muscle reactivity to some agonists. Eur J Pharmacol. 1999;386:247–252. doi: 10.1016/s0014-2999(99)00751-7. [DOI] [PubMed] [Google Scholar]
- 12.Niijima A. Effects of oral and intestinal stimulation with umami substance on gastric vagus activity. Physiol Behav. 1991;49:1025–1028. doi: 10.1016/0031-9384(91)90218-d. [DOI] [PubMed] [Google Scholar]
- 13.Szaflarski J, Burtrum D, Silverstein FS. Cerebral hypoxia-ischemia stimulates cytokine gene expression in perinatal rats. Stroke. 1995;26:1093–1100. doi: 10.1161/01.str.26.6.1093. [DOI] [PubMed] [Google Scholar]
- 14.Koçdor H, Koçdor MA, Astarcioğlu H, Fadiloğlu M. Serum tumor necrosis factor-alpha, glutamate and lactate changes in two different stages of mechanical intestinal obstruction. Turk J Gastroenterol. 2003;14:115–120. [PubMed] [Google Scholar]
- 15.Homi HM, Mixco JM, Sheng H, Grocott HP, Pearlstein RD, Warner DS. Severe hypotension is not essential for isoflurane neuroprotection against forebrain ischemia in mice. Anesthesiology. 2003;99:1145–1151. doi: 10.1097/00000542-200311000-00022. [DOI] [PubMed] [Google Scholar]
- 16.Pei L, Li Y, Zhang GY, Cui ZC, Zhu ZM. Mechanisms of regulation of tyrosine phosphorylation of NMDA receptor subunit 2B after cerebral ischemia/reperfusion. Acta Pharmacol Sin. 2000;21:695–700. [PubMed] [Google Scholar]
- 17.Shimada T, Watanabe N, Ohtsuka Y, Endoh M, Kojima K, Hiraishi H, Terano A. Polaprezinc down-regulates proinflammatory cytokine-induced nuclear factor-kappaB activiation and interleukin-8 expression in gastric epithelial cells. J Pharmacol Exp Ther. 1999;291:345–352. [PubMed] [Google Scholar]
- 18.Sun J, Wang XD, Liu H, Xu JG. Ketamine suppresses intestinal NF-kappa B activation and proinflammatory cytokine in endotoxic rats. World J Gastroenterol. 2004;10:1028–1031. doi: 10.3748/wjg.v10.i7.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tian J, Lin X, Guan R, Xu JG. The effects of hydroxyethyl starch on lung capillary permeability in endotoxic rats and possible mechanisms. Anesth Analg. 2004;98:768–774, table of contents. doi: 10.1213/01.ane.0000099720.25581.86. [DOI] [PubMed] [Google Scholar]
- 20.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 21.Yuksel M, Okajima K, Uchiba M, Okabe H. Gabexate mesilate, a synthetic protease inhibitor, inhibits lipopolysaccharide-induced tumor necrosis factor-alpha production by inhibiting activation of both nuclear factor-kappaB and activator protein-1 in human monocytes. J Pharmacol Exp Ther. 2003;305:298–305. doi: 10.1124/jpet.102.041988. [DOI] [PubMed] [Google Scholar]
- 22.Vancurova I, Miskolci V, Davidson D. NF-kappa B activation in tumor necrosis factor alpha-stimulated neutrophils is mediated by protein kinase Cdelta. Correlation to nuclear Ikappa Balpha. J Biol Chem. 2001;276:19746–19752. doi: 10.1074/jbc.M100234200. [DOI] [PubMed] [Google Scholar]
- 23.Yang GY, Gong C, Qin Z, Liu XH, Lorris Betz A. Tumor necrosis factor alpha expression produces increased blood-brain barrier permeability following temporary focal cerebral ischemia in mice. Brain Res Mol Brain Res. 1999;69:135–143. doi: 10.1016/s0169-328x(99)00007-8. [DOI] [PubMed] [Google Scholar]
- 24.Chen LW, Egan L, Li ZW, Greten FR, Kagnoff MF, Karin M. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med. 2003;9:575–581. doi: 10.1038/nm849. [DOI] [PubMed] [Google Scholar]
- 25.Butcher BA, Kim L, Johnson PF, Denkers EY. Toxoplasma gondii tachyzoites inhibit proinflammatory cytokine induction in infected macrophages by preventing nuclear translocation of the transcription factor NF-kappa B. J Immunol. 2001;167:2193–2201. doi: 10.4049/jimmunol.167.4.2193. [DOI] [PubMed] [Google Scholar]
- 26.Helmer KS, Chang L, Cui Y, Mercer DW. Induction of NF-kappaB, IkappaB-alpha, and iNOS in rat gastric mucosa during endotoxemia. J Surg Res. 2002;104:46–52. doi: 10.1006/jsre.2002.6404. [DOI] [PubMed] [Google Scholar]
- 27.Xia L, Pan J, Yao L, McEver RP. A proteasome inhibitor, an antioxidant, or a salicylate, but not a glucocorticoid, blocks constitutive and cytokine-inducible expression of P-selectin in human endothelial cells. Blood. 1998;91:1625–1632. [PubMed] [Google Scholar]
- 28.Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V, et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277:16639–16647. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 29.Schölzke MN, Potrovita I, Subramaniam S, Prinz S, Schwaninger M. Glutamate activates NF-kappaB through calpain in neurons. Eur J Neurosci. 2003;18:3305–3310. doi: 10.1111/j.1460-9568.2003.03079.x. [DOI] [PubMed] [Google Scholar]
- 30.Ginis I, Jaiswal R, Klimanis D, Liu J, Greenspon J, Hallenbeck JM. TNF-alpha-induced tolerance to ischemic injury involves differential control of NF-kappaB transactivation: the role of NF-kappaB association with p300 adaptor. J Cereb Blood Flow Metab. 2002;22:142–152. doi: 10.1097/00004647-200202000-00002. [DOI] [PubMed] [Google Scholar]
- 31.Hornby PJ. Receptors and transmission in the brain-gut axis. II. Excitatory amino acid receptors in the brain-gut axis. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1055–G1060. doi: 10.1152/ajpgi.2001.280.6.G1055. [DOI] [PubMed] [Google Scholar]