

Abstract
A miniaturized fluorescent assay is described that monitors the conversion of NADPH to NADP+. The same assay format may also be used to measure NADH to NAD+ conversion. Examples of assay development and validation results are presented to illustrate the use of this protocol to screen an enzyme that consumes NADPH as a cofactor during conversion of substrate to a reduced product. Enzymatic assays are carried out low volume 384-well plates, in which the turnover of NADPH is monitored by the decrease in fluorescent emission at 460 nm between an initial fluorescence measurement and a second reading after 90 minutes. A follow-up assay is used to rule out false positive artifacts arising from compounds that fluoresce at 460 nm.
INTRODUCTION
We have optimized a miniaturized high-throughput screening assay that monitors the decrease in fluorescence at 460 nm upon conversion of NADPH to NADP+. To correct for the intrinsic fluorescence at 460 nm of some test compounds, two readings of each plate are obtained: one immediately after compound and reagent addition, and the second after 90 min incubation with the target enzyme. Nevertheless, a slight increase in compound fluorescence readings over 90 min is sufficient to give rise to a number of false positives (compounds that appeared to be inhibitors of the target enzyme but in fact are not). A counterscreen is used to identify these false positives and exclude them from further study.
Thus two assays are described here:
Primary Screening Assay
NADPH fluorescence assay: Enzyme activity is monitored by the decrease in NADPH fluorescence over 90 min.
Counterscreen Assay
Fluorescence change in the absence of enzyme and substrate: Fluorescence change due to test compounds is monitored in assay buffer alone.
The optimization of these assays and their use to confirm screening hits is exemplified by assay validation and HTS to discover inhibitors of two enzymes, RmlC and RmlD, essential for cell wall biosynthesis in Mycobacterium tuberculosis (Sivendran et al., 2010). The two enzymes are assayed together as a mixture: RmlC epimerizes the keto sugar nucleotide dTDP-4-keto-6-deoxy-D-xylo-hexulose, and RmlD then uses the cofactor NADPH to reduce the epimerized keto sugar nucleotide to dTDP-rhamnose (Figure 1).
Figure 1.
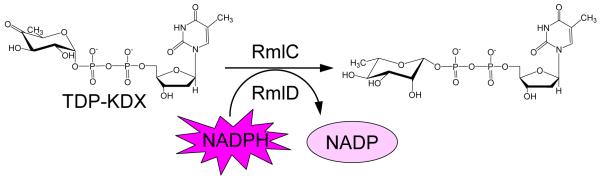
RmlC/D enzyme assay. RmlC epimerizes the keto sugar nucleotide dTDP-4-keto-6-deoxy-D-xylo-hexulose (TDP-KDX), and RmlD then uses the cofactor NADPH to reduce the epimerized keto sugar nucleotide to dTDP-rhamnose. Enzyme activity is determined by monitoring the decrease in NADPH fluorescence.
PROTOCOL
The protocol is divided under Procedure into the sections listed below. For simplicity and to provide a clear example, the protocol describes exactly the method used for the Rml enzymes assay. The Critical Parameters and Troubleshooting section describes how the Rml protocol may be modified and broadened to be generally applicable to a wider range of NADPH- or NADH-utilizing enzymes.
Steps 1-5. NADPH standard curve
Steps 6-11. Enzyme titration and timecourse
Steps 12-24. Km determinations
Steps 25-36. Positive control inhibitor dose-response and IC50 determination.
Steps 37-46. Quality control (QC) validation plate
Step 47. Primary Screening Assay (single concentration compound screening)
Steps 48-49. Data analysis and hit selection
Steps 50-53. Dose-response testing
Step 54. Curve fitting, IC50 determination, and hit confirmation
Steps 55-61. Counterscreen Assay (elimination of false positives)
Materials
Target enzyme(s). For the study used as an example here, the M. tb rhamnosyl biosynthetic enzymes dTDP-6-deoxy-D-xylo-4-hexulose 3,5-epimerase (RmlC) and dTDP-6-deoxy-L-lyxo-4-hexulose reductase (RmlD) were cloned and expressed in E. coli and purified by Michael McNeil and co-workers (Ma et al., 2001). These enzymes are not commercially available.
Substrates. TDP-6-deoxy-D-xylo-hexopyranosid-4-ulose (TDP-KDX) was synthesized enzymatically and stored frozen at a concentration of 1 mg/ml in 50 mM MOPS buffer, pH 7.4 at −80°C as previously described (Sivendran et al., 2010). This substrate is not commercially available. NADPH was purchased from Roche, dissolved in water to a concentration of 1 mM and stored frozen at −20°C.
Buffer components. MOPS and Triton X-100 were purchased from Sigma. MgCl2 and glycerol were purchased from Fisher.
Assay buffer. Final composition (1×): 50 mM MOPS, pH 7.4, containing 1 mM MgCl2, 0.01 % Triton X-100, 10 % glycerol. (Buffer without added glycerol may be stored as a 5× stock after filtration through a sterile 0.45 μm filter. Glycerol is added upon dilution of the buffer to the final 1× concentration.)
Positive control inhibitor. Thymidine diphosphate (TDP) was purchased from Sigma.
Assay plates. Black low-volume 384-well plate (Corning #3676).
Compound dilution plates. Polypropylene V-bottom 384-well plates (Greiner Bio-One #781280).
DMSO. Purchase analytical grade dry DMSO from Fisher or VWR.
Test compounds may be stored dissolved in DMSO (for example at 10 mM stock concentration) in 384-well polypropylene plates. Room temperature storage is recommended if the compounds are to be reused within a period of less than one week. For longer term storage, compounds stocks should be frozen at <−20°C.
Equipment Required
Plate reader capable of reading fluorescence in 384-well plates, for example, an EnVision multimode plate reader from Perkin Elmer.
Pipetting workstation equipped with 384-tip MDT pipetting head, for example, a JANUS from Perkin Elmer or equivalent (earlier model known as Evolution EP3).
For compound screening: Pipetting workstation equipped with pintool consisting of 384 pins with nominal transfer volume of 100 nl , for example, a JANUS MDT from Perkin Elmer or equivalent (earlier model known as Evolution EP3).
Reagent dispenser, for example, a Multidrop-384 reagent dispenser from Thermo Scientific.
Software required
Microsoft Excel, OpenHTS (CeuticalSoft), or ActivityBase (IDBS) (or equivalent): for percent inhibition calculations, and evaluation of data sets and selection of hits.
GraphPad Prism (or equivalent): for graphing data and curve fitting for IC50 calculation.
Procedure
NADPH standard curve
1. Dissolve NADPH in water to a concentration of 1 mM.
2. NADPH dilution. Serially dilute NADPH in one column of 384-well polypropylene plate by 1.5-fold dilutions in assay buffer (1×). Add 105 μl of 80 μM NADPH in assay buffer to the first well and 35 μl of assay buffer to 9 of the remaining wells in the column. Serially transfer 70 μl from the first well to the second, then from the second to the third, and so on until 70 μl is added to and then discarded from the 9th well to yield 9 concentrations of NADPH ranging from 3-80 μM and one well containing buffer alone.
3. Assay start. Transfer 10 μl per well of serially diluted NADPH (Step 2 above) to 3 columns of 384-well assay plate to give triplicate wells at each concentration of NADPH.
4. Read fluorescence (excitation 340 nm, emission 460 nm).
5. Standard curve. Plot fluorescence against NADPH concentration. Fit to a straight line by linear regression in GraphPad Prism or similar curve fitting and graphing program.
Enzyme titration and timecourse
6. Enzyme dilution. Make dilutions of enzymes (2× final concentration in assay) in assay buffer (1×). In the case of RmlC and RmlD, a useful starting point was provided by an earlier 96-well absorbance assay, in which optimal concentrations were determined to be 1.75 × 10−4 μg/μl RmlC and 4.35 × 10−4 μg/μl RmlD (Ma et al., 2001). To confirm that these concentrations are also appropriate for the 384-well fluorescent assay, prepare solutions to give final enzyme concentrations of 0.5, 1.0, 1.5, 2.0, and 3.0 times the above values in the 10-μl fluorescent assay.
7. Substrate and cofactor stock. Mix substrate and NADPH cofactor (2× final concentration in assay) in assay buffer (1×). Based on prior assay results (Ma et al., 2001), use final assay concentrations of 400 μM TDP-KDX and 50 μM NADPH. Thus a solution containing 800 μM TDP-KDX and 100 μM NADPH should be prepared in 1× assay buffer. Add 5 μl per well of this TDP-KDX/NADPH solution to a block of 15 wells (5 down by 3 across) in a 384-well assay plate using a multichannel pipette. Include an adjacent block of 15 blank wells containing NADPH cofactor without substrate.
8. Assay start. Using a multichannel pipette, transfer 5 μl of enzyme dilutions to substrate and cofactor mix and blanks to give triplicates of each enzyme concentration in both substrate and cofactor mix and blank wells (final assay volume 10 μl).
9. Timecourse. Read fluorescence (excitation 340 nm, emission 460 nm) immediately and at 3 minute intervals thereafter for 180 min. Incubate plate between reads at 25°C. (Provided the assay plate temperature does not exceed 25°C it may be left uncovered for the duration of the assay; some evaporation from the assay wells will occur, but this should not be enough to adversely affect the results.)
Data analysis
10. For each concentration of enzyme(s), plot fluorescence at each timepoint (0-180 min). Also plot fluorescence of blanks to ensure that change over timecourse is minimal. For screening, select the concentration of enzyme(s) giving the largest total change in fluorescence that also displays a linear change in fluorescence over 90 min.
11. From the NADPH standard curve (Step 5) convert change in fluorescence to amount of NADPH converted. This allows determination of the total amount of NADPH converted over the 3 hr timecourse.
Km determinations
The goal of the Km determination studies is to select appropriate concentrations of substrate and NADPH cofactor for compound screening. To maximize the sensitivity of the assay to enzyme inhibitors, substrate and cofactor should be used close to their respective Km values. Substrate Km is determined by varying substrate concentration at a fixed concentration of cofactor, and likewise cofactor Km is determined by varying cofactor concentration at a fixed concentration of substrate. (For further explanation and discussion of this methodology see Km determinations under Critical Parameters and Troubleshooting. Useful literature references: Assay Guidance Manual, Enzyme General 2010, How to Measure Km (listed under Internet resources) and Fersht (1985).)
Determine Substrate Km
12. Serially dilute TDP-KDX in one column of a 384-well polypropylene compound dilution plate by 1.5-fold dilutions in assay buffer (1×) containing 30 μM NADPH cofactor to yield 10 concentrations of substrate ranging from 0-2000 μM in the presence of a fixed concentration of NADPH cofactor.
13. Add 5 μl of each TDP-KDX dilution per well to 2 columns of a 384-well assay plate using a multichannel pipette, giving duplicate wells of each TDP-KDX dilution.
14. Assay start. Add 5 μl per well of a mixture of 3.5 × 10−4 μg/μl RmlC and 8.7 × 10−4 μg/μl RmlD in assay buffer (1×). Thus, each 10 μl assay has 15 μM NADPH, 1.75 × 10−4 μg/μl RmlC, 4.35 × 10−4 μg/μl RmlD, and 0-1000 μM TDP-KDX substrate.
15. Read fluorescence (excitation 340 nm, emission 460 nm) immediately and after 90 min at 25°C.
16. Proceed to Step 21 for data analysis.
Determine NADPH cofactor Km
17. Serially dilute NADPH in one column of a 384-well polypropylene compound dilution plate by 1.5-fold dilutions in assay buffer (1×) containing 400 μM TDP-KDX to give 12 concentrations of cofactor ranging from 0-200 μM in the presence of a fixed concentration of TDP-KDX substrate.
18. Add 5 μl per well of each NADPH dilution to 2 columns of a 384-well assay plate using a multichannel pipette, giving duplicate wells of each NADPH dilution.
19. Assay start. Add 5 μl per well of a mixture of 3.5 × 10−4 μg/μl RmlC and 8.7 × 10−4 μg/μl RmlD in assay buffer (1×). Thus, each 10 μl assay has 200 μM substrate, 1.75 × 10−4 μg/μl RmlC, 4.35 × 10−4 μg/μl RmlD, and 0-100 μM NADPH.
20. Read fluorescence (excitation 340 nm, emission 460 nm) immediately and after 90 min at 25°C.
Data analysis (for both Km determinations)
21. For each well, subtract fluorescence after 90 min from fluorescence at start of assay to determine change in fluorescence.
22. From the NADPH standard curve (Step 5), convert change in fluorescence to amount of NADPH converted over 90 min. This may be expressed as a rate (e.g., μM NADPH/min), allowing determination of Vmax (maximal velocity of the enzymatic conversion at saturating concentration of substrate.
23. Plot rate of NADPH conversion against concentration (of substrate or cofactor, respectively).
24. Fit rate of NADPH conversion vs. concentration plot to sigmoidal curve using GraphPad Prism or similar curve fitting and graphing program. The curve fit will approach a maximum rate at high concentrations of the x-value (substrate or cofactor concentration). This maximum rate represents Vmax, the maximal velocity of the enzymatic conversion at saturating concentration of the varied reagent (substrate or cofactor). The Km value determined from the curve fit is defined as the concentration of varied reagent (substrate or cofactor) that gives a rate equal to half the Vmax.
Positive control inhibitor dose-response and IC50 determination
The aim of this procedure is to determine the IC50 value (inhibitory concentration giving half maximal response) for TDP, a known inhibitor of the RmlC/D enzymes (Sivendran et al., 2010). The measured IC50 is then compared with previously determined values to ensure that the enzymes show the expected sensitivity to inhibition.
TDP dose-response
25. Dissolve TDP in water to a concentration of 75 mM. Store 100 μl portions in 500 μl Eppendorf tubes frozen at −20°C.
26. Serially dilute TDP in one column of a 384-well polypropylene compound dilution plate by 2-fold dilutions in water from a top concentration of 75 mM to yield 16 concentrations of positive control inhibitor ranging from 2.3 μM to 75 mM (Note that this will be 2.5× the final concentration in assay).
27. Add 4 μl of water per well to columns 1-2 and 23-24 of a 384-well assay plate using a multichannel pipette. (Columns 1 and 23 will be used as blanks, and columns 2 and 24 as no-inhibitor controls.) (See Figure 2: location of blanks is labeled as such, and no-inhibitor controls are labeled Negative Controls.)
28. Add 4 μl per well of serially diluted TDP (from Step 26 above) to columns 3, 4, and 5 of the assay plate using a multichannel pipette, giving triplicate columns of TDP dilutions.
29. Add 5 μl per well of assay buffer (1.67×) containing 5.26 × 10−4 μg/μl RmlC, 13.06 × 10−4 μg/μl RmlD, and 50 μM NADPH to columns 1-5 and 23-24 using a multichannel pipette or reagent dispenser (columns 6-22 are empty for this experiment). Final reagent concentrations: 2.63 × 10−4 μg/μl Rml C, 6.53 × 10−4 μg/μl Rml D, 25 μM NADPH in 1x assay buffer.
30. Add 1 μl per well of assay buffer (1.67×) into columns 1 and 23 (blank) using a reagent dispenser.
31. Assay start. Add 1 μl of 2 mM TDP-KDX substrate (diluted in 1.67x assay buffer) per well to columns 2-5 and 24 using a reagent dispenser. Final concentration of TDP-KDX: 200 μM.
32. Read fluorescence (excitation 340 nm, emission 460 nm) immediately and after 90 min at 25°C.
Figure 2.

Assay plate format. The layout of blanks and negative controls shown here is also used for the QC Plate and the dose-response plates.
IC50 determination
33. For each well, subtract fluorescence after 90 min from fluorescence at start of assay to determine change in fluorescence.
- 34. Calculate percent activity using the fluorescence change at each TDP concentration and the mean fluorescence change of the plate blanks (columns 1 and 23), and the mean fluorescence change of the plate controls (columns 2 and 24) [see plate map in Figure 2]:
(Equation 1) 35. Plot percent activity against concentration of inhibitor (use log scale for inhibitor concentration [x-axis] and linear scale for percent activity [y-axis]).
36. Fit percent activity vs. log of concentration to a sigmoidal dose-response curve. Use the four parameter logistic equation entitled “log(inhibitor) vs. response -- Variable slope” in GraphPad Prism or an equivalent curve fitting equation in another graphing program. Report IC50, Hill slope (defined as slope at mid-point of curve), and top and bottom values. For further explanation and discussion of IC50 determination see Assay Guidance Manual, Enzyme General 2010, IC50 determination (listed under Internet resources). For a very extensive discussion of non-linear regression, with numerous examples including IC50 curves, see the GraphPad Prism Regression Guide (listed under Internet resources).
Quality control (QC) validation plate
The IC50 value determined for the positive control inhibitor TDP (Steps 33-36) is used to set the concentration for a QC plate to monitor the consistency of the assay between plates and between experiments performed on different days.
QC plate set up
37. Dissolve TDP in DMSO to a concentration of 40.3 mM.
38. Add 20 μl per well of TDP in DMSO to columns 3-22 of a polypropylene V-bottom 384-well plate (TDP storage plate). Add 20 μl per well of DMSO alone to columns 1-2 and 23-24 for blanks and negative controls [see plate map in Figure 2]. Plate may be used immediately or sealed with an adhesive plate seal and stored at room temperature for up to 2 weeks before use.
Testing of QC plate
39. Dispense 4 μl of water per well to a 384-well assay plate using reagent dispenser.
40. Pintool transfer TDP in DMSO from TDP storage plate (120 nl per well). Final concentrations of TDP and DMSO: 500 μM and 1.2%, respectively.
41. Add 5 μl per well of assay buffer (1.67×) containing 5.26 × 10−4 μg/μl RmlC, 13.06 × 10−4 μg/μl RmlD, and 50uM NADPH. Final reagent concentrations: 2.63 × 10−4 μg/μl Rml C, 6.53 × 10−4 μg/μl Rml D, 25 μM NADPH.
42. Add 1 μl per well of assay buffer (1.67×) into columns 1 and 23 (blank) using reagent dispenser.
43. Assay start. Add 1 μl of 2 mM TDP-KDX substrate in 1.67× assay buffer to each well in columns 2-22 and 24 using reagent dispenser. Final concentration of TDP-KDX: 200 μM.
44. Read fluorescence (excitation 340 nm, emission 460 nm) immediately and after 90 min at 25°C.
Calculation of percent inhibition
45. For each well, subtract fluorescence after 90 min from fluorescence at start of assay to determine change in fluorescence.
- 46. Calculate percent inhibition using the fluorescence change in each TDP test well (columns 3-22) and the mean fluorescence change of the plate blanks (columns 1 and 23), and the mean fluorescence change of the plate negative controls (columns 2 and 24) [see plate map in Figure 2]:
(Equation 2)
Primary Screening Assay (single concentration compound screening)
47. Follow Steps 39-44 above, except that pintool transfer (Step 40) into each assay plate is from a compound storage plate containing test compounds in DMSO in columns 3-22 and DMSO alone in columns 1-2 and 23-24. The final test compound concentration should not exceed 25 μM. (e.g., transfer of 120 nl of 10 mM compound in DMSO gives 12 μM compound in 1.2% DMSO in the final assay volume.) Reserve wells in columns 1-2 and 23-24 for negative controls and blanks treated with DMSO but no compound (as shown in Figure 2). Each screening run should include testing of at least one QC plate (Steps 37-46) per 20 test compound plates.
Data analysis and hit selection
48. For data analysis, follow Steps 45-46 above.
49. For hit selection, select hits based on percent inhibition exceeding a defined threshold. For the hits to be statistically significant, and not just due to random scatter in the data, the threshold should be below the mean of the plate controls by a minimum of 3× standard deviation of the negative controls. The standard deviation of the negative controls in enzyme inhibition assays typically ranges from 5-10%; thus the minimal hit threshold may be set at 20-30% inhibition. To select for only higher potency hits, the threshold may be set at 50% inhibition.
Dose-response testing
The assay protocol is identical to single compound screening (Step 47), except that compounds are tested at multiple concentrations obtained by serial dilution. A suitable layout is 16 two-fold dilutions of each compound, giving an assay concentration range of 50 μM to 1.5 nM. In this case, compounds may be serially diluted vertically down each plate such that columns 3-22 each contain the dilutions of a different compound. The protocol for serial dilution in DMSO is as described in Steps 50-53 below. The resulting dose-response plates may be used immediately or sealed with an adhesive plate seal and stored at room temperature for up to 1 week before use.
50. Add 20 μl of each compound stock (4.2 mM in DMSO) arranged 20 per plate in wells A3 to A22 of 384-well V-bottom polypropylene plates.
51. Add 20 μl of DMSO to wells A1, A2, A23, and A24.
52. Add 10 μl per well of DMSO to the entire plate except Row A using a reagent dispenser.
53. Two-fold serially dilute compounds by transfer of 10 μl row-by-row from Row A to Row P using a single row of disposable tips, then discard 10 μl from Row P. The resulting dose-response plates contain 16 two-fold dilutions of each compound, ranging from 4.2 mM to 125 nM, arranged one compound per column in columns 3–22. Proceed to Step 47. Pintool transfer from dose-response plates into assay plates as described in Step 47 gives a final compound dilution range of 50 μM to 1.5 nM.
Curve fitting, IC50 determination, and hit confirmation
54. Following dose-response testing (Steps 50-53), fit the data and determine IC50 values as described in Steps 33-36.
Compounds giving an IC50 below the top concentration tested (50 μM) are deemed to be confirmed hits.
Counterscreen Assay (elimination of false positives)
Confirmed hits from the Primary Screening Assay (Steps 47-54) must be tested in the Counterscreen Assay to eliminate false positives due to compound fluorescence that increases between the 0 min and 90 min reads during the assay.
Dose-response plate testing
55. Dispense 4 μl of water per well to a 384-well assay plate using reagent dispenser.
56. Pintool transfer compound in DMSO (120 nl per well) from dose-response plate (Step 53) into assay plate to give a final compound dilution range of 50 μM to 1.5 nM.
57. Assay start. Add 6 μl per well of assay buffer (1.67×) using a reagent dispenser.
58. Read fluorescence (excitation 340 nm, emission 460 nm) immediately and after 90 min at 25°C.
EC50 determination
59. For each well, subtract fluorescence at start of assay from fluorescence after 90 min to determine fluorescence increase (if any).
60. For each compound, plot fluorescence increase against compound concentration (use log scale for compound concentration [x-axis] and linear scale for fluorescence increase [y-axis]).
61. Fit fluorescence increase vs. log of concentration to a sigmoidal dose-response curve. Use a four parameter logistic equation in GraphPad Prism or a similar curve fitting and graphing program. Report EC50, Hill slope (defined as slope at mid-point of curve), and top and bottom values. The compound concentration at the mid-point of the curve is reported as EC50 (effective concentration giving half maximal response), rather than IC50 as reported in Step 36, as the compound gives rise to an increase in assay signal rather than the decrease observed in Step 36. For a very extensive discussion of non-linear regression, with numerous examples including EC50 curves, see the GraphPad Prism Regression Guide (listed under Internet resources).
Elimination of false positive actives
Compounds giving a measurable increase in fluorescence over 90 min and an EC50 < 3 × IC50 (determined in Step 54) are deemed to be false positive artifacts and should be eliminated from further study. A compound EC50 value less than 3-fold greater than its IC50 determined in Step 54 strongly suggests that the compound “activity” in the primary screening assay was due to its intrinsic fluorescence and not to inhibition of either of the enzymes RmlC and RmlD. Conversely, compounds that gives no measurable EC50 in the counterscreen assay, or an EC50 much higher than the corresponding IC50 obtained in the primary screening assay, are unlikely the be false positives due to intrinsic fluorescence. These compounds should therefore be selected for further study as inhibitors of RmlC and RmlD. This is explained in more detail under Critical Parameters and Troubleshooting, Counterscreen Assay.
COMMENTARY
Background Information
Monitoring of the conversion of NADH to NAD+ (or NADPH to NADP+) has been used for many years to assay the activity of enzymes catalyzing oxidation-reduction reactions mediated by NADH/NAD as cofactor. An assay for lactate dehydrogenase (LDH) that monitors the decrease in absorbance at 340 nm upon conversion of NADH to NAD was published in 1963 (Bergmeyer and Bernt, 1963). Two characteristics of absorbance-based methods such as this are not well-suited to high-throughput microplate-based compound screening:
A relatively small change in absorbance limits the sensitivity of the assay and requires that it is monitored as a timecourse
Absorbance measurements in microplates require a flat well bottom and a liquid depth of several millimeters.
The first characteristic limits throughput, because each plate has to be monitored for at least 15 mins. By contrast, an assay with a single endpoint read allows for the processing of multiple plates in this time. The second characteristic requires an assay volume of at least 50 μl in 384-well plates. Absorbance assays in 1536-well plates are not practical. This limitation leads to increased reagent consumption and cost compared with lower volume formats of 384- and 1536-well assays, which may be run in volumes of 10 μl or less.
Several approaches to NADH monitoring have been taken to avoid the limitations of absorbance assays. NADH fluoresces at 460 nm (excitation at 340 nm), and decrease in fluorescence may be used to monitor NADH to NAD conversion (Moran and Schnellmann, 1996). To increase detection sensitivity, NADH may be quantified by coupling enzymatic conversion to NAD with the generation of resorufin, a highly fluorescent molecule with an emission maximum of 590 nm (excitation 530 nm). One assay uses NADH oxidase to generate hydrogen peroxide (H2O2) from NADH, followed by horseradish peroxidase to convert Amplex Red to resorufin in the presence of H2O2 (Batchelor and Zhou, 2002). Another method uses the enzyme diaphorase to convert resazurin to resorufin in the presence of NADPH (Batchelor and Zhou, 2004).
Critical Parameters and Troubleshooting
The protocol above describes exactly the method used for the Rml enzymes assay (Sivendran et al., 2010), and the Anticipated Results section shows results obtained using this assay. The section here describes how the Rml protocol may be modified and broadened as needed to be generally applicable to a wider range of NADPH- or NADH-utilizing enzymes.
Enzyme titration and timecourse
The purpose of the enzyme titration and timecourse (Steps 6-11) is to determine an enzyme concentration that produces a linear decrease in NADPH fluorescence over 90 min while maximizing the total change in fluorescence over this time. Longer reaction times result in evaporation from low volume 384-well assay plates, leading to more scatter in the data. Shorter times will require more enzyme in order to produce a detectable amount of product. Deviation from linearity beyond a certain point in the timecourse generally indicates depletion of the substrate. It is important to adjust enzyme concentration and time to achieve linearity; a non-linear timecourse reduces the sensitivity of the assay to enzyme inhibition.
Factors to consider in applying this protocol to other enzymes
Enzyme dilution. Serial dilution of the target enzyme(s) in a single column in a polypropylene 384-well V-bottom plate provides enzyme dilutions in the appropriate format for direct transfer to an assay plate. The concentrations and dilution range used depends on the extent of enzyme activity information already available. In the absence of prior data suggesting a suitable enzyme concentration, 7 semi-log dilutions ranging from a 1/10 dilution of the enzyme stock to 1/10,000 will cover a broad range of enzyme activities. Typically, the target enzyme has already been assayed for activity during purification and characterization. Although established assays may not be suitable for HTS, they may help to guide the choice of enzyme concentrations to test. In the case of RmlC and RmlD, the activities had already been determined in an NADPH absorbance assay (see Introduction), so the enzyme concentrations were varied over a small range (8.75 × 10−5–5.25 × 10−4 μg/μl RmlC and 2.175 × 10−4 –1.31 × 10−3 μg/μl RmlD).
Substrate and cofactor stock. In the absence of results from a prior enzyme activity assay, several combinations of substrate and NAD(P)H cofactor may need to be tested ranging in concentration from 1-500 μM. An enzyme assay will rarely require concentrations outside this range. Typically, the enzyme will have already been tested for activity, so the conditions of this earlier assay will guide the choice of suitable substrate and cofactor concentrations. In the case of the 384-well Rml enzyme titration and timecourse experiments, 400 μM TDP-KDX and 50 μM NADPH were used based on previous results obtained in a 96-well absorbance assay. Subsequently, these values were adjusted for the HTS assay based on Km determinations carried out in the 384-well assay (Steps 12-24; and see below under Km determinations for more information about using Km values to select reaction conditions).
Timecourse (Step 9 in protocol). It is not useful to extend the timecourse of a 10 μl assay beyond 180 min at 25°C, because evaporation causes significant variability in the data. If the enzyme requires incubation at 37°C, acceptable data cannot be obtained beyond 1 hr.
Km determinations
The goal of the Km determination studies is to select appropriate concentrations of substrate and NAD(P)H cofactor for compound screening. To maximize the sensitivity of the assay to enzyme inhibitors regardless of their mechanism of inhibition, substrate and cofactor should be used close to their respective Km values. In the case of the Rml assay used to exemplify this protocol, TDP-KDX substrate was used at its Km of 200 μM, but NADPH was used at twice its Km of 12.5 μM. Use of NADPH at a concentration two times higher than its Km gave a more robust and linear change in fluorescence over the 90 min timecourse of the assay. The relationship of inhibitor IC50 to substrate concentration and how this relates to the mechanism of inhibition is explained in the Assay Guidance Manual, Section 13, Performing MOA Studies (listed under Internet Resources). Detailed discussion of this topic may also be found in Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems (Segel, 1975).
Factors to consider in applying this protocol to other enzymes
Ultimately, the Km of the substrate should be determined in the presence of cofactor fixed at its Km, and vice versa. This may require several iterations as the Km values for each are refined. In the absence of prior results, Km determinations may need to be carried out initially with the fixed component at a high concentration, close to saturation. The analysis becomes simpler if it appears that the Km of one component is not affected by the concentration of the other.
Data analysis. Typically, Km data show simple saturation binding kinetics, and may be fit accordingly. In the case of the Rml enzymes, the Km curves were sigmoidal (Figure 4). This may be indicative of enzyme cooperativity or allosteric modulation by substrate or cofactor, but can also arise for other reasons (Segel, 1975). For the purposes of assay development, the Km values obtained from the sigmoidal curves may be used without further analysis to determine the cause of the sigmoidal behavior.
Figure 4.
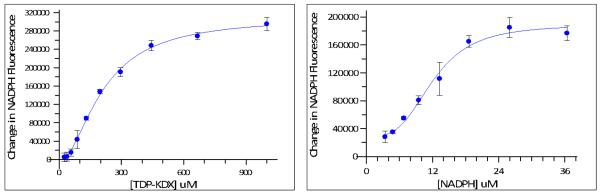
Km determinations for TDP-KDX (left panel) and NADPH (right panel). The protocol was based on that described under Steps 12-24, Km determinations. Data points represent the mean of triplicate determinations; error bars depict standard deviation. Left-Hand Panel. The concentration of TDP-KDX was varied from 0 to 1000 μM in the presence of a fixed concentration of NADPH (25 μM). The fluorescence change in the presence of a mixture of 2.63 × 10−4 μg/μl Rml C, 6.53 × 10−4 μg/μl Rml D in 1× assay buffer was monitored over 90 min at 25°C. Data were fit to a sigmoidal plot using GraphPad Prism, and the Km of TDP-KDX was calculated to be 200 μM. Right-Hand Panel. The concentration of NADPH was varied from 0 to 36 μM in the presence of a fixed concentration of TDP-KDX (200 μM). The fluorescence change in the presence of a mixture of 2.63 × 10−4 μg/μl Rml C, 6.53 × 10−4 μg/μl Rml D in 1× assay buffer was monitored over 90 min at 25°C. Data were fit to a sigmoidal plot using GraphPad Prism, and the Km of NADPH was calculated to be 12.5 μM.
Positive control inhibitor dose-response and IC50 determination
The aim of this procedure is to determine IC50 value for TDP, a known inhibitor of the RmlC/D enzymes (Sivendran et al., 2010). The measured IC50 is then compared with previously determined values to ensure that the enzymes show the expected sensitivity to inhibition. The IC50 value is used to set the concentration for a QC plate to monitor the consistency of the assay over time.
Factors to consider in applying this protocol to other enzymes
A known inhibitor of an enzyme target to be subjected to HTS is a valuable positive control to use as a benchmark to track the sensitivity of the enzyme towards inhibition during assay development and over the course of the screening campaign. A suitable inhibitor does not need to be potent provided it is soluble at a concentration that gives 30-70% inhibition. It should ideally be a reversible, substrate-competitive inhibitor that does not covalently modify or denature the protein.
Quality control (QC) validation plate
The aim of this procedure is to deliver TDP inhibitor by pintool to an entire 384-well assay plate (320 wells, columns 3-22 at a concentration slightly over its IC50 value. The calculated percent inhibition (expected to be approximately 65%) is used to monitor well-to-well variability and consistency of the assay over time.
Factors to consider in applying this protocol to other enzymes
Figure 6 reveals three outliers (wells 48, 78, and 215) that appear to deviate significantly from the remainder of the TDP-inhibited data. When outliers such as these are observed, a repeat of the experiment will reveal whether there is a problem associated with a specific pin in the pintool or a specific channel in the reagent dispenser. If the outliers are not replicated at the same well locations between experiments, then they may be attributed to random data scatter. If further refinement of the liquid handling steps fails to reduce the occurrence of such outliers, screening may nonetheless proceed. False positive outliers will be eliminated during dose-response testing (steps 50-53) and other follow-up assays. The occasional false negative (a true inhibitor missed during HTS) may also be deemed acceptable, provided the incidence is very low.
Figure 6.
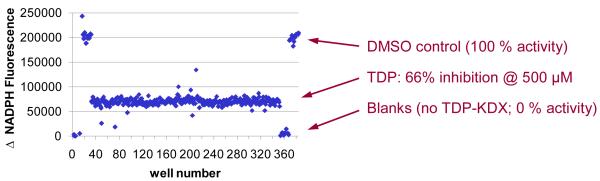
Representative QC plate data. Each point represents the change in NADPH fluorescence over 90 min at 25°C in a single well in a 384-well assay plate. The protocol is described under Steps 37-45, Quality control (QC) validation plate. TDP was added to test wells (columns 3-22) at a final concentration of 500 μM.
Dose-response testing
Factors to consider in applying this protocol to other enzymes
It is important that compounds are serially diluted in DMSO. Transfer of each dilution into the assay plate by pintool then gives a constant percent DMSO in each well, avoiding the possibility of well-to-well differences caused by the effect of DMSO alone on the enzyme activity. Serial dilution of compounds from a DMSO stock into buffer also may result in compound precipitation.
Counterscreen Assay (elimination of false positives)
The purpose of the counterscreen assay is to test hits from the Primary Screening Assay (Steps 47-54) to eliminate false positives due to compound fluorescence that increases between the 0 min and 90 min reads in the assay. One cause of the increase in compound fluorescence over time might be slow evaporation, which would tend to concentrate the fluorescent compound in the center of the round bottom plate well.
An explanation for how the change in compound fluorescence gives rise to a dose response curve that appears similar to that of a bona fide enzyme inhibitor is presented in Figures 7 and 8. The percent increase in compound fluorescence is independent of compound concentration; thus the absolute fluorescence change increases with increasing concentration. The concentration of NADPH is fixed, so the fluorescence decrease due to enzymatic conversion of NADPH does not change from well to well. Therefore, as the concentration of a fluorescent compound is increased, the magnitude of its fluorescence “drift” relative to the decrease in NADPH fluorescence becomes larger, resulting in a dose-dependent increase in “inhibition” (Figure 7). In the case of highly fluorescent compounds the increase in fluorescence at high concentrations is so large that the overall fluorescence in the assay increases over the 90 min assay timecourse, giving negative percent activity values (Figure 8). In primary HTS, negative percent activity corresponds to percent inhibition >100% (see Equation 2). Examples of highly fluorescent compounds (e.g., coumarins) giving percent inhibition values up to 1500% may be found in PubChem assay IDs 1532 and 1533 (see Internet Resources).
Figure 7.

The effect on the NADPH assay of change in compound fluorescence over time increases with compound concentration. The percent increase in compound fluorescence is independent of compound concentration; thus the absolute fluorescence change increases with increasing concentration. The concentration of NADPH is fixed, so the fluorescence decrease due to enzymatic conversion of NADPH does not change from well to well. Therefore, as the concentration of a fluorescent compound is increased, the magnitude of its fluorescence “drift” relative to the decrease in NADPH fluorescence becomes larger, resulting in a dose-dependent increase in “inhibition”. Both Panels. The purple shaded area represents compound fluorescence, which increases the total fluorescence above the baseline fluorescence in the absence of added compound as shown. The green line represents the decrease in NADPH fluorescence over time. The fluorescence measured in the assay (shown as an orange line) is the sum of the fluorescence due to NADPH and the compound fluorescence. As an illustrative example, compound fluorescence is shown increasing by 50% over the course of the 90 min assay. Left-hand Panel. At low compound concentration, a 50% increase in compound fluorescence partially offsets the decrease in NADPH fluorescence, resulting in apparent 15% inhibition of NADPH conversion. Right-hand Panel. At high compound concentration, a 50% increase in compound fluorescence almost entirely offsets the decrease in NADPH fluorescence, resulting in apparent 85% inhibition of NADPH conversion.
Figure 8.

Upper Left-hand Panel. A moderately fluorescent compound gives a dose response curve similar to that of a bona fide enzyme inhibitor. Lower Left-hand Panel. A highly fluorescent compound gives rise to an increase in fluorescence larger than the decrease in NADPH fluorescence, giving apparent negative percent activity at a high concentration of compound. Right-hand Panel. Decrease in NADPH fluorescence over time is shown by a green line. In the presence of a high concentration of the moderately fluorescent compound giving the dose-response curve shown in the Upper Left-hand Panel, increase in compound fluorescence over the course of the assay offsets the decrease in NADPH fluorescence to the extent that the amount of NADPH conversion over 90 min is reduced to 15% of the conversion in the absence of added compound (see purple line labeled 15% activity). (See Figure 7 for further explanation of this effect.) In the presence of the highly fluorescent compound giving the dose-response curve shown in the Lower Left-hand Panel, increase in compound fluorescence over the course of the assay is greater in magnitude than the decrease in NADPH fluorescence, giving an overall increase in fluorescence over the course of the assay. Calculation of percent activity from a fluorescence increase using Equation 1 gives a negative value, in this case apparent −70% activity at a high compound concentration (see red line labeled −70% activity). Negative percent activity values (and the corresponding percent inhibition values >100 calculated using Equation 2) are not meaningful in the context of bona fide enzyme inhibition. Therefore, the appearance of a dose-response curve such as the one shown in the Lower Right-hand Panel immediately suggests that the compound “activity” is an artifact of its intrinsic fluorescence.
Factors to consider in applying this protocol to other enzymes
A significant number of compounds in screening libraries are fluorescent under the detection conditions used here (excitation 340 nm, emission 460). Therefore, it is important to follow HTS based on NAD(P)H fluorescence with the Counterscreen Assay.
Anticipated Results
Enzyme titration and timecourse
Figure 3 shows an enzyme timecourse in which the selected dilution of Rml enzymes mixture gives a linear decrease in NADPH fluorescence over 180 min. The amount of NADPH converted to NADP+ may be calculated from the fluorescence change using the NADPH standard curve (Steps 1-5).
Figure 3.
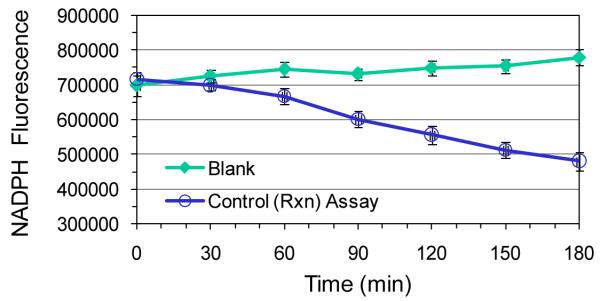
Enzyme activity timecourse. Example of dilution of Rml enzymes mixture that gives linear timecourse over 180 min. The protocol was based on that described under Steps 6-10, Enzyme titration and timecourse. Data points represent the mean of triplicate determinations; error bars depict standard deviation. Control Assay: The fluorescence of a mixture of 2.63 × 10−4 μg/μl Rml C, 6.53 × 10−4 μg/μl Rml D, 25 μM NADPH, and 200 μM TDP-KDX in 1× assay buffer was monitored over 180 min at 25°C. Blank: The Control mixture without TDP-KDX was used to determine the change in NADPH fluorescence in the absence of enzymatic activity. (The Rml enzymes do not convert NADPH to NADP in the absence of TDP-KDX.)
Km determinations
Figure 4 shows Km determinations for TDP-KDX and NADPH using the Rml enzymes. Note that the sigmoidal curves, as seen in this Figure, are fairly unusual (see also point (b) under Critical Parameters and Troubleshooting, Km determinations).
Positive control inhibitor dose-response and IC50 determination
Figure 5 shows a dose-response curve for TDP inhibition of RmlC/D. Non-linear regression analysis (Steps 33-36) gave the curve fit shown on the figure and an IC50 of 388 μM.
Figure 5.

Dose-response curve for TDP inhibition of enzymes RmlC/D. Data points represent a single well at each concentration. The protocol is described under Steps 25-36, Positive control inhibitor dose-response and IC50 determination. TDP was tested at concentrations ranging from 1 μM to 30 mM. From these data, the IC50 for TDP inhibition was determined to be 388 μM.
Quality control (QC) validation plate
Figure 6 shows representative QC plate data. TDP at a concentration of 500 μM gave 66% inhibition, essentially identical to the potency predicted based on the IC50 value of 388 μM determined from the dose-response curve shown in Figure 5.
Summary of RmlC/D Screening Results
This section provides a brief summary of the outcome of RmlC/D screening, dose-response testing, and counterscreening. A total of 201,368 unique compounds (265,000 including duplicates) from the Molecular Libraries Small Molecule Repository (MLSMR) were screened at a concentration of 5.5 μM as mixtures of 4 per well in 384-well plates. A total of 2328 mixtures that showed greater than 30% inhibition were selected as hits. (The standard deviation of the mean of the negative controls in each plate was <10% of the mean, so the 30% inhibition threshold for hit selection was at least 3 standard deviations below the mean of the negative controls.) Subject to availability, all 4 individual compounds in each mixture selected as a hit were reordered from the MLSMR and retested one compound per well in duplicate at a concentration of 5.5 μM. Out of 5266 compounds retested individually, 470 gave >30% inhibition. The very low retest rate is to be expected given the likelihood that the activity of each mixture stemmed from only one compound in that mixture. Only 388 out of the 470 compounds active at 5.5 μM were available for dose-response testing; of these, 372 gave an IC50 <50 μM. The counterscreen assay revealed 358 of these compounds to be false positives due to intrinsic compound fluorescence. The remaining 14 were selected as confirmed hits for further study.
For more details see the following Assay IDs in PubChem (see Internet Resources):
Primary Screening Assay (single concentration compound screening): Assay IDs 1532 and 1533.
Data analysis and hit selection: Assay IDs 1532 and 1533.
Dose-response testing: Assay ID 1695.
Curve fitting, IC50 determination, and hit confirmation: Assay ID 1695.
Counterscreen Assay (elimination of false positives): Assay ID 1696.
Time Considerations
NADPH standard curve
Total assay time is 5 min. Allow 1-2 hr to plan experiment and set up assay. Allow 1-2 hr after assay to analyze data.
Enzyme titration and timecourse
Total assay time is 90 min. Allow 4-5 hr to plan experiment and set up assay. Allow 4-5 hr after assay to analyze data.
Km determinations
Total assay time is 90 min. Allow 4-5 hr to plan experiment and set up assay. Allow 4-5 hr after assay to analyze data.
Positive control inhibitor dose-response and IC50 determination
Total assay time is 90 min. Allow 2-3 hr to plan experiment and set up assay. Allow 2-3 hr after assay to analyze data.
Quality control (QC) validation plate
Allow 2-3 hr to make up positive control compound storage plate. This may be reused for multiple assays. Total assay time is 90 min. Allow 1-2 hr before and after for set up and data collection, respectively.
Primary Screening Assay (single concentration compound screening)
Total assay time is 90 min. Allow 1-2 hr before and after for set up and data collection, respectively. Up to 30 plates may be nested for batchwise testing. For a 30-plate screening run, allow 3-4 hr before starting for assay set up, and 1-2 hr following the assay for clean up and data collection.
Data analysis and hit selection
Allow 1 day per week during screening.
Dose-response testing
Allow 1 day to select hits and serial dilute. Testing time similar to Positive control inhibitor dose-response and IC50 determination above.
Curve fitting, IC50 determination, and hit confirmation
Allow 1 day for data analysis per 100-200 compounds.
Counterscreen Assay (elimination of false positives)
Dose-response plates made up in G above may be reused. Allow 1 day for assay and data analysis per 100 compounds.
Acknowledgements
Assay development and validation and high-throughput screening was supported by the NIH Molecular Libraries Screening Center Network (Grant U54HG003915-02).
Literature Cited
- Batchelor RH, Zhou M. A resorufin-based fluorescent assay for quantifying NADH. Anal Biochem. 2002;305:118–9. doi: 10.1006/abio.2001.5596. [DOI] [PubMed] [Google Scholar]
- Batchelor RH, Zhou M. Use of cellular glucose-6-phosphate dehydrogenase for cell quantitation: applications in cytotoxicity and apoptosis assays. Anal Biochem. 2004;329:35–42. doi: 10.1016/j.ab.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Bergmeyer HU, Bernt E. Lactate dehydrogenase. In: Bergmeyer HU, editor. Methods of Enzymatic Analysis. Academic Press; London: 1963. pp. 574–579. [Google Scholar]
- Fersht A. Enzyme Structure and Mechanism. 2nd edition Freeman; New York: 1985. [Google Scholar]
- Ma Y, Stern RJ, Scherman MS, Vissa VD, Yan W, Jones VC, Zhang F, Franzblau SG, Lewis WH, McNeil MR. Drug targeting Mycobacterium tuberculosis cell wall synthesis: genetics of dTDP-rhamnose synthetic enzymes and development of a microtiter plate-based screen for inhibitors of conversion of dTDP-glucose to dTDP-rhamnose. Antimicrob Agents Chemother. 2001;45:1407–16. doi: 10.1128/AAC.45.5.1407-1416.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran JH, Schnellmann RG. A rapid beta-NADH-linked fluorescence assay for lactate dehydrogenase in cellular death. J Pharmacol Toxicol Methods. 1996;36:41–4. doi: 10.1016/1056-8719(96)00071-8. [DOI] [PubMed] [Google Scholar]
- Segel I. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. Wiley; New York: 1975. [Google Scholar]
- Sivendran S, Jones V, Sun D, Wang Y, Grzegorzewicz AE, Scherman MS, Napper AD, McCammon JA, Lee RE, Diamond SL, McNeil M. Identification of triazinoindol-benzimidazolones as nanomolar inhibitors of the Mycobacterium tuberculosis enzyme TDP-6-deoxy-d-xylo-4-hexopyranosid-4-ulose 3,5-epimerase (RmlC) Bioorg Med Chem. 2010;18:896–908. doi: 10.1016/j.bmc.2009.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Internet Resources
- Results and analysis from screening 265,000 compounds against RmlC/D enzymes. http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1532&loc=ea_ras http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1533&loc=ea_ras.
- Dose response testing following RmlC/D HTS and hit confirmation by curve fitting and IC50 determination. http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1695&loc=ea_ras.
- Dose response testing to eliminate fluorescent compounds as false positives following RmlC/D HTS. http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1696&loc=ea_ras.
- Section of the Assay Guidance Manual that describes how to measure Km. http://spotlite.nih.gov/assay/index.php/Enzyme_General_2010#How_to_Measure_Km.
- Section of the Assay Guidance Manual that describes how to determine IC50 values. http://spotlite.nih.gov/assay/index.php/Enzyme_General_2010#IC50_Determination.
- Section of the Assay Guidance Manual that describes the relationship of enzyme inhibitor IC50 to substrate concentration and how this relates to the mechanism of inhibition. http://spotlite.nih.gov/assay/index.php/Section13:Performing_MOA_Studies.
- GraphPad Prism Regression Guide, which provides a highly detailed discussion and many examples of non-linear regression. http://www.graphpad.com/downloads/docs/Prism5Regression.pdf.