

Abstract
Significance: Regulation of cell adhesions during tissue repair is fundamentally important for cell migration, proliferation, and protein production. All cells interact with extracellular matrix proteins with cell surface integrin receptors that convey signals from the environment into the nucleus, regulating gene expression and cell behavior. Integrins also interact with a variety of other proteins, such as growth factors, their receptors, and proteolytic enzymes. Re-epithelialization and granulation tissue formation are crucially dependent on the temporospatial function of multiple integrins. This review explains how integrins function in wound repair.
Recent Advances: Certain integrins can activate latent transforming growth factor beta-1 (TGF-β1) that modulates wound inflammation and granulation tissue formation. Dysregulation of TGF-β1 function is associated with scarring and fibrotic disorders. Therefore, these integrins represent targets for therapeutic intervention in fibrosis.
Critical Issues: Integrins have multifaceted functions and extensive crosstalk with other cell surface receptors and molecules. Moreover, in aberrant healing, integrins may assume different functions, further increasing the complexity of their functionality. Discovering and understanding the role that integrins play in wound healing provides an opportunity to identify the mechanisms for medical conditions, such as excessive scarring, chronic wounds, and even cancer.
Future Directions: Integrin functions in acute and chronic wounds should be further addressed in models better mimicking human wounds. Application of any products in acute or chronic wounds will potentially alter integrin functions that need to be carefully considered in the design.

Hannu Larjava, DDS, PhD, Dip. Perio.
Scope and Significance
Wounds are common in skin and oral mucosa, and they are caused by either trauma or surgery. Wound healing is a coordinated, progressive process which is used to restore the barrier function and integrity of the epithelium that involves blood coagulation, inflammation, re-epithelialization by migrating keratinocytes, granulation tissue formation, angiogenesis, and, eventually, tissue remodeling. The formation and regulation of cell adhesions via integrin receptors play a fundamental role during all phases of wound healing.1,2 In the present review, our main focus will be on integrin expression and functions during re-epithelialization and granulation tissue formation.
Translational Relevance
Oral mucosal wounds heal with minimal scarring compared with skin wounds. Integrins are likely to play a role in regulating this differential healing outcome, although direct evidence is still lacking. Understanding the pathways that are important for scarless healing will help in the development of therapies for the prevention of scar formation in skin.3 There are no commercial wound healing products in the market at the present time that are based on integrin functions. However, some therapies targeting αvβ6 integrin are being developed that show promise in reducing fibrosis.4 In addition, transferring oral cells to an extra-oral location to improve healing outcomes has been proposed.5
Clinical Relevance
Wound healing problems are very common, affecting millions of people worldwide every year. Some wounds cause morbidity, because they over-heal with extensive scarring and others, because they fail to heal, becoming chronic. At the molecular level, integrin-mediated adhesion is likely to participate in both conditions.
Overview
The process of wound healing has been recently reviewed and is, therefore, only briefly covered here (Fig. 1).6–8 Wound healing starts with blood clotting that initially seals the wound.9 Platelet activation during the primary hemostasis releases a number of important cytokines that provide chemotactic signals to inflammatory and resident cells. During this innate immune response, inflammatory cells (first neutrophil granulocytes, later macrophages, lymphocytes, and mast cells) which have been recruited to the wound site release more cytokines and chemokines that activate and recruit the resident epithelial cells and fibroblasts at the wound margins.10 Within 24 h after wounding, epithelial cells at the wound margins start to form cellular extensions into the clot and dissolve their hemidesmosomal adhesions to the basement membrane. Keratinocytes behind the leading edge start proliferating to seed more cells into the wound site. Keratinocytes migrate over the wound bed until they contact the front of the leading cells coming from the other side of the wound.11 The formation of granulation tissue starts simultaneously with re-epithelialization.8 Granulation tissue replaces the provisional wound matrix and provides a scaffold for connective tissue formation. Injury to the tissue also initiates angiogenesis (formation of new blood vessels from pre-existing blood vessels).12 Wound angiogenesis is tightly associated with granulation tissue formation and has many similarities to re-epithelialization. Similarly to the epithelial cells, the endothelial cells or their precursor cells in the pre-existing blood vessels become activated by cytokines and start to migrate to the wound provisional matrix. They create the granulation tissue along with the activated fibroblasts, mesenchymal progenitor cells (pericytes and other mesenchymal stem cells), and circulating fibroblast-like cells (fibrocytes) that also migrate to the provisional matrix. When a sufficient amount of collagen is produced into the granulation tissue, wound contraction is started by myofibroblasts that differentiate from local resident fibroblasts or other progenitor cells. This process pulls wound margins closer together, reducing the wound area and accelerating the wound closure. After wound contraction, granulation tissue remodeling takes place. During this process, myofibroblasts degrade, remodel, and re-organize the wound extracellular matrix (ECM). The maturation to connective tissue is slow and can continue for months if not years.
Figure 1.

Wound healing phases. The approximate timing of coagulation, inflammation, re-epithelialization, angiogenesis as well as granulation tissue formation and remodeling. Adapted from Häkkinen et al.8
Oral soft tissue healing proceeds with the same principles as in skin. However, in some parts of oral mucosa (gingiva, palatal mucosa), the result is a clinically scar-free healing with histological features of almost normal connective tissue; while healing in the skin often ends in a formation of a connective tissue scar with reduced tensile strength, disoriented collagen fibers, and other molecular alterations.8 Differences in integrin–ECM interactions between these two tissues likely influence the divergent healing outcomes.
Discussion of Findings and Relevant Literature
The playing field of wound healing: specialized ECM molecules in the provisional wound matrix and granulation tissue
The ECM is a highly dynamic structure that comprises hundreds of different proteins.13,14 The interactions between cells and their environment are bidirectional and dynamic—the cells can modulate the structure and composition of ECM, and the ECM, in turn, guides cell morphology and behavior, including proliferation, differentiation, survival, and migration in tissue homeostasis and during wound healing.1,2 Micro RNAs have been recently shown to provide one mechanism for the cells to control the composition of the ECM and the cell phenotype.15
In wounds, keratinocytes and fibroblasts encounter a multifaceted environment of ECM molecules, matrix-degrading enzymes, growth factors, and cytokines that are not present in their normal surroundings.8,11 The initial plasma-derived proteins, including fibrinogen, fibronectin, and vitronectin form the provisional wound matrix of the blood clot and act as a scaffold for further ECM accumulation.16–18 In addition, the developing granulation tissue contains ECM proteins that are released or synthesized by the wound cells at the injury site, such as osteopontin, thrombospondins, secreted protein acidic and rich in cysteine (SPARC), tenascins, collagens, and alternatively spliced forms of fibronectin.8,19–21 These proteins are only transitionally present in wounds, although their expression may continue long after the wound has clinically and histologically healed. The provisional basement membrane that supports keratinocyte migration during re-epithelialization contains extra domain A (EDA) fibronectin, tenascin-C, and laminin-332.11,22 Studies in knockout mice have provided considerable insight into the roles that ECM molecules play during various phases of wound healing in vivo (Table 1).23,24
Table 1.
Effect of deficiency or overexpression of certain extracellular matrix molecules on wound healing in mice
| ECM Molecule | Animal Model | Wound Healing Phenotypes |
|---|---|---|
| Fibrinogen | Knockout | Wound closure not affected but somewhat increased initial bleeding, defects in granulation tissue formation and reduced wound tensile strength, altered pattern of epithelial cell migration, and increased epithelial hyperplasia |
| Fibronectin | Knockout | Embryonic lethal phenotype |
| Plasma fibronectin | Hepatocyte-targeted knockout | Normal fibrinogenesis, hemostasis, and wound healing |
| Fibronectin EDA | Exclusion of EDA domain | Keratinocyte migration normal; blurred border between the new epidermis and granulation tissue; ulceration in the newly formed epidermis; influx of inflammatory cells to ulcerated area |
| Fibronectin EDA | Constitutive inclusion of EDA domain | Normal wound healing but a striking decrease in the levels of fibronectin in most of the organs |
| Laminin-332 | Knockout of α3 or γ2 subunit of laminin-332 | No wound healing data due to neonatal lethal phenotype; the animals exhibit epidermal/dermal blistering |
| Tenascin-C | Knockout | Skin wound re-epithelialization normal; defects in corneal wound re-epithelialization; reduced fibronectin expression in wounds |
| Vitronectin | Knockout | Re-epithelialization normal but slightly delayed dermal wound healing, decreased angiogenesis, and formation of larger blood vessels |
| Thrombospondin-1 | Overexpression (K14-promoter) | Healing of full-thickness skin wounds was greatly delayed with reduced granulation tissue formation and highly diminished wound angiogenesis |
| Thrombospondin-2 | Knockout | Accelerated wound healing with reduced inflammation and scarring but with irregularly organized and highly vascularized granulation tissue and thickened epithelium, possibly due to elevated levels of MMP-2 and MMP-9 |
| Osteopontin | Knockout | Significantly decreased level of debridement, greater disorganization of matrix, and altered collagen fibrillogenesis with small-diameter collagen fibrils |
| Osteonectin/SPARC | Knockout | Accelerated wound closure potentially due to decreased granulation tissue collagen content resulting in enhanced granulation tissue contractibility |
| EMILIN1 | Knockout | Accelerated wound closure due to fibroblast and keratinocyte hyperproliferation |
In this section, we briefly focus on ECM molecules that have an important role in the regulation of fibroblast and keratinocyte behavior and functions during wound healing, namely fibronectin, tenascin-C, collagens, and laminin-332.
Fibronectin
Fibronectins are ECM proteins that have a multidomain structure, in which the various domains serve different functions, such as binding to specific integrins and ECM molecules (Fig. 2A).18,25,26 Plasma fibronectin is produced by hepatocytes and circulates in blood. It becomes incorporated into the fibrin clot during blood clotting process either noncovalently or through covalent crosslinking by transglutaminase (activated factor XIII).
Figure 2.

Structural and functional domains in fibronectin (FN), tenascin-C, and laminin-332, including major ECM and keratinocyte and fibroblast integrin binding sites. (A) FN consists of two similar subunits that are linked in an antiparallel orientation by two disulphide bridges at their C-termini. It is formed by repeating homologous type I, II, and III units, and it binds to a number of biologically important molecules, including heparin (and heparan sulfate proteoglycans), denatured collagen, fibrin, and tenascin-C (TN-C). FN has three sites of alternative splicing: type III repeats A and B as well as the CSIII segment. The binding site for α5β1, αvβ1, α8β1, and αvβ6 integrins is located in module III10. In addition, α5β1 integrin interacts with a second site, located in module III9. Integrins α4β1 and α9β1 bind to module IIIA. FN is present in blood plasma in a soluble, globular form, and its cell-binding sites are unexposed. The cell-binding sites become exposed when it is absorbed to fibrin and polymerizes into insoluble fibrils. (B) Three tenascin-C monomers are joined together via their N-terminal tenascin-C assembly (TA) domains to form a trimer. Two trimers are further linked together to form a hexamer. Each arm of mammalian tenascin-C consists of EGF-like repeats (EGFL) that are recognized by EGFR, FN type III-like repeats that contain binding sites for FN and heparin, and a C-terminal fibrinogen globe (FBG), also interacting with heparin. Nine additional type III repeats can be included or excluded by alternative RNA splicing (light green). Binding sites for integrins are located in module III3. (C) Laminin (LM) 332 is a T-shaped molecule consisting of three genetically distinct polypeptide chains, α3, β3, and γ2. The α3 chain contains five C-terminal globular domains (LG), the β3 chain an N-terminal globular domain (LN), and the γ2 chain an interrupting globular domain (L4) in the short arm. Proteolytic cleavage between LG3 and LG4 domains of the α3 chain results in a functional conversion of laminin-332 from a motility to an adhesion factor. LG3 domain contains the binding site for α3β1 and α6β4 integrins, whereas LG4 includes a heparin/heparan sulfate proteoglycan-binding site. The β3 and γ2 chains can also be cleaved. The γ2 L4 domain contains binding sites for nidogen and fibulin, which aid the incorporation of laminin-332 to the basement membrane. This arm also contains the binding site for α2β1 integrin. The β3 LN domain contains a binding site for type VII collagen and a laminin-332–laminin-311 interaction site, which facilitate laminin-332 integration to the ECM. Adapted from Larjava et al.25 ECM, extracellular matrix; EGF, epidermal growth factor; EGFR, EGF receptor. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Wound fibroblasts and keratinocytes produce cellular fibronectins that contain variable amounts of alternatively spliced extra domains A and B (EDA and EDB, respectively; Fig. 2A), which are not present in plasma fibronectin.18 Interestingly, fetal wounds in skin and airway mucosa that heal without scars show no induction of EDA fibronectin expression.27 Similarly, its expression is induced more transiently in porcine oral mucosal wounds that show minimal scarring than in scar forming skin wounds in the same animals.28 In addition, EDA fibronectin expression is associated with many fibrotic conditions, suggesting its important involvement in the regulation of fibroblast differentiation into myofibroblasts and matrix production.18
Tenascin-C
Tenascins belong to the family of matricellular proteins that regulate cell adhesion and cytokine activation. Other members of this group include thrombospondins and SPARCs. Collectively, these proteins appear to have their main function in reducing fibroblasts migration in the healing wounds, as evidenced by accelerated wound closure in the respective knockout mice (Table 1).21 Tenascin-C is a large, multi-domain hexameric glycoprotein that is capable of binding a multitude of matrix proteins and receptors (Fig. 2B).29 In normal oral mucosa (gingiva), it is strongly and constitutively expressed in the subepithelial connective tissue, whereas it is weakly expressed in or absent from healthy skin.28,30,31 During wound healing, its expression is strongly upregulated in both tissues. Tenascin-C has both adhesive and anti-adhesive functions depending on the cellular context. During wound healing, it can interact with EDA fibronectin and reduce fibroblast and keratinocyte adhesion to it, thus allowing faster cell migration.32 Tenascin-C can also promote fibroblast migration into fibrin-fibronectin matrix via its fibronectin type III repeats.33 Low tenascin-C expression may contribute to scarring. Oral mucosal and fetal skin wounds that heal with minimal scarring exhibit early and continuing expression of tenascin-C even after the tissue morphology is normalized, whereas skin wounds that form scars show transient expression, and tenascin-C is, thus, effectively downregulated before remodeling is complete.28,34 However, its aberrant expression may also be involved in fibrosis, as tenascin-C is highly expressed in scars, keloids, and lung fibrosis; whereas mice lacking its expression are protected from bleomycin-induced lung fibrosis.35,36 This protection is attributed to reduced accumulation myofibroblasts and impaired transforming growth factor (TGF)-β responsiveness of the tenascin-C null cells.
Collagens
During the active granulation tissue formation in the scarlessly healing human oral mucosal wounds, a number of collagen genes (collagen types I, III, IV, V, VI, and XV) are significantly upregulated as compared with the unwounded tissue.* Almost identical sets of genes continue to be elevated in human and pig hypertrophic scars in the skin,28,37 suggesting that the expression of these genes relates to both scarless and scar-forming wounds but that they may be ineffectively downregulated in scars. Early wounds contain mainly fibrillar types III and I collagen produced and then organized into fibrils by myofibroblasts that highly express these collagen types.28,38 Fibrillogenesis of type I collagen also requires type III and V collagens, fibronectin, and integrins.39,40 When the granulation tissue matures, active remodeling of collagen fibers will follow, leading to either a normal connective tissue orientation (basket weave) or a scar (parallel orientation).8
Laminin-332
Laminins are heterotrimers of α, β, and γ chains. They can be incorporated into the ECM by interactions with other ECM molecules.41 Laminin-332 (α3β3γ2, formerly known as laminin-5) is essential for the formation of hemidesmosomes and basement membranes, where it interacts with types VII (anchoring fibrils) and XVII collagen (BP180).41 Mutations to laminin-332 cause junctional epidermolysis bullosa, in which the affected patients suffer severe skin blistering due to the absence or poor development of hemidesmosomes.42
Laminin-332 polymerization and proteolytical processing affect its biological functions (Fig. 2C).22,41,43 For example, activated keratinocytes at the leading edges of wounds express high levels of unprocessed laminin-332 α3 chains; while in quiescent keratinocytes, these chains are cleaved, which changes laminin-332 function from migratory to adhesive and promotes hemidesmosome formation.44 Moreover, in vitro studies demonstrate that the leading migrating keratinocytes deposit the unprocessed laminin-332 at the rear of the cell, forming a trail of laminin-332 deposits.41 The laminin-332 β3 and γ2 chains can also be processed to promote epithelial cell migration (Fig. 2C).22,41
In addition to regulating epithelial cell functions, laminin-332 may also modulate granulation tissue formation, and mutations leading to deletion of the N-terminus of the α3 chain are associated with laryngo-onycho-cutaneous syndrome that leads to excessive granulation tissue formation in mucosal tissues.45
Integrins: cell attachment receptors and mediators of cellular signaling
Composition and extracellular ligand specificities of the integrin heterodimers
Integrins are the main mediators of cell attachment to ECM.46 All 24 integrin-type adhesion receptors are formed by 1 of the 18 α subunits and 1 of the 8 β subunits, which are bound together in a noncovalent manner (Fig. 3; Table 2).46–65 Integrins can be divided into four subfamilies based on their ligand specificity and/or phylogenetic comparison of the α subunits (Fig. 3).66 All multicellular animals have integrin heterodimers that recognize a specific three amino acid motifs, arginine-glycine-aspartic acid (RGD) in their extracellular ligands. The vertebrate heterodimers in this subfamily of the integrins are named α5β1, α8β1, αvβ1, αvβ3, αvβ5, αvβ6, αvβ8, and αIIbβ3, and their RGD-containing ECM and plasma protein ligands include fibronectin, vitronectin, fibrinogen, and thrombospondins (Table 2). The members of the laminin receptor subfamily, α3β1, α6β1, α7β1, and α6β4 integrins, can mediate cell adhesion to basement membranes in various tissues (Fig. 4A). Integrins α4β1, α4β7, and α9β1 form their own subfamily. They can recognize ECM ligands such as fibronectin, but in an RGD-independent manner. Integrins α4β1 and α4β7 can also bind to counter receptors in other cells, such as vascular cell adhesion molecule. Integrin α subunits with a special inserted (αI) domain or von Willebrand factor type A (αA) domain form the fourth subfamily. Integrin subunits α1, α2, α10, and α11 combine with the β1 subunit, and they bind numerous collagen types (Table 2). Integrins αDβ2, αLβ2, αMβ2, αXβ2, and αEβ7 are expressed in leukocytes, and their ligands include counter receptors such as intercellular adhesion molecules (ICAMs) and plasma proteins, including complement component C3b (Table 2; Fig. 4B).
Figure 3.

Integrin heterodimers. Schematic presentation of the 24 integrin receptors. Integrins α1β1, α2β1, α10β1, and α11β1: αI domain-containing collagen receptors; α5β1, α8β1, αvβ1, αvβ3, αvβ5, αvβ6, αvβ8, and αIIbβ3: RGD-binding integrins; α3β1, α6β1, α7β1, and α6β4: laminin receptors; α4β1, α9β1, and α4β7: α4/α9β1 integrin family; αDβ2, αLβ2, αMβ2, αXβ2, and αEβ7: leukocyte integrin subgroup. RGD, arginine-glycine-aspartic acid.
Table 2.
Integrin expression and function in wound cells
| Integrin | Wound Cells Expressing | Ligands in Wounds | Cellular Functions in Wound Healing | Wound Phenotypes in Animal Models |
|---|---|---|---|---|
| α1β1 | ECs, FBLs, monocytes, macrophages, and myofibroblasts | COL I, III, IV, and V, LM-111 | Mediates VEGF-driven angiogenesis, negative feedback regulation of collagen synthesis in FBLs | The integrin α1 null mice exhibit increased collagen gene expression during early granulation tissue development and altered collagen fibril distribution |
| α2β1 | Platelets, KCs, ECs, and FBLs | COLs (types I and III with high affinity), LM-332, TN-C, MMP-1, and CCN1/Cyr6 | Mediates KC migration and VEGF-driven angiogenesis, may mediate platelet adhesion to COL, and contribute to collagen polymerization by FBLs | Increased wound angiogenesis in α2 integrin knockout mice |
| α3β1 | KCs, ECs, and FBLs | LM-332, other LMs | Regulates KC polarization during re-epithelialization, controls angiogenesis and TGF-β1–mediated responses | Integrin α3 null mice die shortly after birth with skin blisters and disrupted basement membranes; KC-targeted α3 integrin knockout mice exhibit slightly accelerated or not affected wound closure but impaired wound angiogenesis; retarded re-epithelialization α3 integrin null mouse skin grafts |
| α4β1 | Leukocytes, FBLs, and ECs | EDA-FN, OPN, several ADAMs, VCAM-1, and EMILIN1 | Interaction with EMILIN1 may control FBL proliferation and TGF-β1 processing | Genetic ablation of α4 integrin causes an embryonic lethal phenotype |
| α5β1 | Platelets, KCs, ECs, FBLs, and leukocytes | FN, CCN2/CTGF, and CCN3/NOV | Promotes KC migration, may be involved in platelet–blood vessel wall interactions | Genetic ablation of α5 integrin causes an embryonic lethal phenotype |
| α6β1 | Platelets, ECs, leukocytes, and FBLs | LMs, TSPs, CCN1/Cyr6, CCN2/CTGF, and CCN3/NOV | May be involved in platelet–blood vessel wall interactions and angiogenesis; interaction with CCN1/Cyr61 promotes myofibroblast senescence and controls fibrogenesis | Genetic ablation of α6 integrin causes a neonatal lethal phenotype; endothelial cell-targeted deletion of α6 integrin increases tumor angiogenesis but reduces postischemic vascular repair—these phenotypes may be partly caused by the loss of α6β4 in these cells as well; no wound healing data available |
| α7β1 | N/A (expressed by muscle cells, vascular smooth muscle cells, and in neurons) | TN-C, LM | ||
| α8β1 | Myofibroblasts | FN, VN, TN-C, and latent TGF-β1 | May contribute to fibrotic responses | No wound healing data available |
| α9β1 | KCs, FBLs, neutrophils, and ECs | EDA-FN, TN-C, OPN, several ADAMs, EMILIN1, and VEGFs | Regulates KC and FBL growth, neutrophil chemotaxis as well as EC migration and angiogenesis | Reduced keratinocyte proliferation and delayed wound closure in keratinocyte-targeted α9 integrin knockout mice; impaired formation of granulation tissue in mouse wounds treated with a blocking antibody against α9 integrin because of reduced adhesion and migration of FBLs |
| α10β1 | FBLs? | COLs | May mediate FBL adhesion to collagen | No wound healing data available |
| α11β1 | FBLs | COLs | Controls myofibroblast differentiation and may mediate FBL adhesion to collagen and contribute to collagen reorganization | No wound healing data available |
| αvβ1 | KCs, ECs | FN, VN, OPN, and latent TGF-β1 | Mediates KC adhesion during re-epithelialization | Difficult to address the specific function of this receptor due to the wide array of αv and β1 integrins |
| αvβ3 | ECs, platelets, FBLs, and macrophages | Fibrin(ogen),TN-C, OPN, latent TGF-β1, CCN1/Cyr6, CCN2/CTGF, and CCN3/NOV | Required for neoangiogenesis, modulates fibrin network structure and stability, mediates EC adhesion to CCN1/Cyr6 and CCN2/CTGF, EC survival, pericyte retention in blood vessels, and FBL proliferation | Mice lacking β3 integrin subunit show enhanced re-epithelialization but reduced KC proliferation due to increased TGF-β1 and enhanced dermal fibroblast infiltration into the wound. They also exhibit reduced macrophage numbers in wounds and increased angiogenesis in late wound healing. Peptide antagonists of αvβ3 integrin reduce granulation tissue formation and wound-induced angiogenesis in porcine wounds |
| αvβ5 | ECs, FBLs, and skin KCs | VN, OPN, latent TGF-β1, CCN1/Cyr6, CCN3/NOV, VEGF | May participate in FBL transformation to myofibroblasts, interaction with CCN1/Cyr61 mediates FBL migration | Wound healing is normal in β5 integrin knockout mice |
| αvβ6 | KCs | FN, VN, TN-C, latent TGF-β1 and -β3, and chromogranin A | Regulates inflammation and KC proliferation, contributes to basement membrane and granulation tissue remodeling | Re-epithelialization is normal in β6 integrin knockout mice if not challenged by age or diabetes; mice with keratinocyte-targeted β6 integrin overexpression are susceptible to chronic, nonhealing wounds but show faster re-epithelialization than wild-type mice after being challenged by a glucocorticoid treatment |
| αvβ8 | Dentritic cells, FBLs, and ECs | LM-111, COL IV, FN, and latent TGF-β1 | May regulate inflammation through TGF-β activation | No wound healing data available |
| α6β4 | KCs, ECs | LM-332, other LMs | Promotes KC adhesion, migration and proliferation, and EGFR signaling; regulates angiogenesis in ECs | Genetic ablation of β4 integrin causes a neonatal lethal phenotype with severe epidermal blistering; deletion of β4 integrin intracellular signaling domain caused decelerated wound re-epithelialization and reduced keratinocyte migration |
| αIIbβ3 | Platelets | Fibrin(ogen), FN, COL, VWF, CCN1/Cyr6, and CCN2/CTGF | Mediates platelet aggregation in clot formation, modulates fibrin network structure and stability | An antagonist of αIIbβ3 integrin has an antithrombotic effect in mice |
| α4β7 | Leukocytes, dentritic cells | OPN, VCAM, | Involved in leukocyte extravasation | Impaired immune response in β7 integrin knockout mice due to defective T-cell trafficking, no wound healing data available |
| αEβ7 | T-lymphocytes, dentritic cells | E-cadherin | Mediates leukocyte extravasation | Inflammatory skin lesions in αE integrin knockout mice, no wound healing data available |
| αLβ2 (LFA-1) | All leucocytes | Several ICAMs, JAM-1, and lumican | Mediates leukocyte extravasation across the endothelium | No wound healing data available |
| αMβ2 (Mac-1) | Monocytes, macrophages, NK, neutrophils, and T-cells | Fibrin(ogen), several ICAMs, heparin, plasminogen, FN, LMs, COL I, uPAR, CCN1/Cyr6, CCN2/CTGF, and others | Involved in leukocyte extravasation across the endothelium; promotes fibrinolysis and clearance of fibrin clots by monocytes and neutrophils in complex with uPAR and its ligand uPA | Attenuation of granulation tissue deposition and wound re-epithelialization in αM integrin knockout mice |
| αXβ2 | Monocytes, macrophages, dentritic cells, and NK | Fibrin(ogen), several ICAMs, COL I, heparin, and OPN | Involved in leukocyte extravasation | No wound healing data available |
| αDβ2 | Macrophages, eosinophils | ICAM-3, VCAM-1, and CCN1/Cyr6 | Involved in leukocyte extravasation | No wound healing data available |
The data is consolidated from the following articles: Häkkinen et al.,8 Koivisto et al.,11 Senger and Davis,12 Gardner et al.,47 Bouvard et al.,48 Grüner et al.,49 Reynolds et al.,50 AlDahlawi et al.,51 Grenache et al.,52 Sisco et al.,53 Blue et al.,54 Mitchell et al.,55 Nieswandt et al.,56 Germain et al.,57 Jacobsen et al.,58 Nakayama et al.,59 Chen and Lau,60 Oommen et al.,61 Worthington et al.,62 Bouvard et al.,63 Høye et al.,64 and Tan.65
ADAM, a disintegrin and metalloproteinase; CCN, Cyr61-CTGF-Nov; COL, collagen; CT, connective tissue; EDA, extra domain A; EC, endothelial cell; EGFR, epidermal growth factor receptor; FBL, fibroblast; FN, fibronectin; ICAM, intercellular adhesion molecule; KC, keratinocyte; LM, laminin; NK, natural killer cell; OPN, osteopontin; TGF, transforming growth factor; TN, tenascin; TSP, thrombospondin; uPAR, urokinase-type plasminogen activator receptor; VCAM, vascular cell adhesion molecule; VEGF, vascular endothelial growth factor; VN, vitronectin; VWF, von Willebrand factor.
Figure 4.
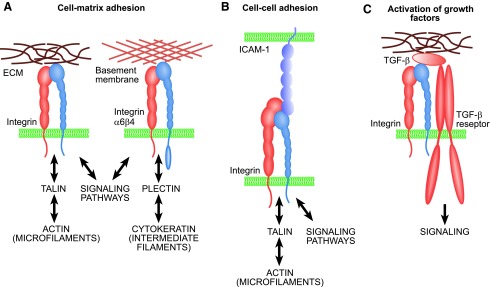
Basic functions of integrins. (A) Integrins mediate cell adhesion to ECM and form adhesion plaques and intracellular complexes with cytoskeletal and signaling proteins. Most integrins are connected to actin microfilaments, whereas hemidesmosomal α6β4 integrin is linked to cytokeratins via plectin protein. (B) Leukocyte integrins can mediate cell–cell adhesion. (C) Integrins also have numerous other functions, such as binding to growth factors. For example, integrins αvβ6 and αvβ8 are critical for in vivo activation of TGF-β1. TGF, transforming growth factor. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Integrin participation in embryonic development and distinct biological processes has been extensively studied using knockout and transgenic mouse models.48,67 In general, integrin heterodimers containing either β2 or β7 subunit participate in immunodefence, for example, by recognizing components of the complement system or by mediating leukocyte extravasation.65,68 The most important task of most other integrins is to anchor cells to tissues. However, these receptors also often participate in other processes, including innate immunity and cell regulation by growth factors. For example, the collagen receptor α2β1 integrin can also recognize members of the collectin family, namely C1q complement protein, mannose-binding lectin, and surfactant protein A.69 The same receptor can also bind to matrix metalloproteinase-1 (MMP-1; collagenase-1) to influence matrix remodeling.70 Furthermore, it can promote cell locomotion. Another example of the specialized functions of the integrins is the binding and in vivo activation of the latent TGF-β by the fibronectin receptor integrins αvβ6 and αvβ8 (Fig. 4C).62,71 Many other integrins can recognize various growth factors and act as assisting or enabling receptors for them.72 In addition to chemical signals, the integrin-based adhesion sites also mediate the effects of mechanical stress from the ECM.73
Integrins are bidirectional signaling molecules
Integrins mediate two-directional signaling across the plasma membrane—extracellular ligand binding to integrin promotes outside-in signaling, whereas binding of intracellular signalling molecules (e.g., talin) to the cytoplasmic tail of an integrin β subunit can initiate integrin inside-out signaling. Recent development has revealed the structural basis of the integrin-mediated activation of cellular signaling pathways.74,75 At the molecular level, the difference between an inactive and a fully activated receptor depends on a conformational change from a bent to an extended form with the integrin standing with the legs of the α and β subunits apart from each other.
In inside-out integrin activation, the relatively short intracellular parts of the integrin β subunits seem to play a critical role, as they contain binding sites for many cytoskeletal and signaling proteins.76,77 Talin, an actin-binding protein, can break the bonds between the α and β subunits and, consequently, induce an extension of the extracellular part, and it seems essential for integrin activation and for the formation of matrix adhesion sites or adhesion plaques (Figs. 4A and 5).77 The three members of the kindlin family can also bind to the intracellular tails of integrin β subunits and activate the receptors.78 Tyrosine phosphorylation is fundamentally linked with the regulation of integrin activity, and protein tyrosine kinases, such as focal adhesion kinase (FAK), p130Cas, and Src may initiate signaling cascades (Fig. 5).79 To date, the integrin adhesome network is reported to include at least 180 different scaffold and signaling proteins that can form more than 700 direct protein–protein interactions.80 Intracellular proteins that mediate integrin binding to actin microfilaments include vinculin, paxillin, tensin, filamin, and α-actinin; whereas signaling proteins such as RhoA family GTPases regulate reorganization of actin microfilaments in the adhesion plaques. In epithelial hemidesmosomes, α6β4 integrins are connected to intermediate filaments formed by cytokeratins (Fig. 4B).81 In these structures, plectin is a critical protein that is responsible for the β4 integrin–keratin interaction. Integrins also have links to microtubules. A pseudokinase/adaptor protein integrin-linked kinase (ILK) is essential for this interaction.82
Figure 5.

Cytoplasmic integrin signaling complexes. Binding of talin to the cytoplasmic tail of integrin β subunit causes a conformational change in the receptor that allows for binding of intracellular signaling molecules, such as tyrosine kinases FAK, Src, and p130Cas as well as other structural proteins, such as vinculin, which mediate integrin interaction with the actin cytoskeleton. These intracellular protein complexes allow for the translation of the integrin-ECM interaction to a change in cell shape and behavior (e.g., motility). FAK, focal adhesion kinase. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
In outside-in activation, ligand binding to an activated receptor leads to further conformational changes. Fibronectin and laminins attach to the integrin βI domain (also called βA domain) and trigger movement of the leg of the β subunit apart from that of the α subunit. In leukocyte and collagen receptor integrins, ligands bind to the αI domain (αA domain), inducing a conformational change first in this domain and, consequently, in the β subunit. In both cases, the separation of integrin legs promotes the binding of signaling proteins to integrin cytoplasmic tails to initiate cellular signaling.
In addition to the conformational changes, receptor clustering plays a major role in the regulation of integrin avidity and signaling. Many extracellular ligands are multivalent to promote receptor clustering, and they may also contain binding motifs with different affinity or integrin subtype specificity.
Integrins collaborate with other cell surface receptors and molecules to regulate cellular signaling
Many integrins associate functionally with growth factor receptors and regulate their number and stability on the cell surface by influencing their endocytosis and recycling.71,72 In addition, other cell surface receptors (e.g., urokinase-type plasminogen activator receptor [uPAR]), transmembrane adaptor proteins such as caveolin-1, or members of the transmembrane-4 superfamily (also called tetraspanins or tetraspans) and membrane lipids (gangliosides) modulate integrin function and cellular signaling.83–86 These binding partners may alter integrin conformation and affinity directly, modulate the avidity of integrins by regulating their clustering, assist in recruiting signaling molecules to cell adhesion sites, regulate receptor endocytosis, or facilitate cell migration through tissues. Integrins also cross talk with cadherins via reactive oxygen species (ROS)-mediated signaling to coordinate cell–matrix and cell–cell adhesions during tissue remodeling.87
Integrin-mediated cell migration
Cell movement involves focalized cell–matrix interactions. Integrins play a central role in cell migration, where cell adhesion contacts function as signaling centers, and the linkages between ECM and actin cytoskeleton allow adhesion sites to serve as traction sites for cell movement.88,89 Directional cell migration involves coordinated assembly of new adhesions at the front of the migrating cell into cellular protrusions and dissolution of adhesion contacts at their rear end. This process also involves integrin endocytosis and trafficking. Molecules that have been recently shown to regulate front–rear polarity in migrating cells include the small GTPases Rac1 and RhoG, ILK, Engulfment and Cell Motility-2, phosphatidylinositol-4,5-biphosphate, actinin-4, and syndecan-4.90–93 The maximum cell migration speed is facilitated by medium strength of cell adhesion—weak adhesion does not provide sufficient traction for cell movement, whereas too strong an adhesion will render the cells stationary.88 The substratum ligand level, the level of integrin expression in cells and integrin ligand-binding affinity contribute to the strength of cell adhesion. Therefore, the strength and turnover rates of cell attachments to the extracellular environment determine which cell shapes and forces are being generated during migration. For example, fibroblasts exhibit relatively slow invasive migration into the loose provisional wound matrix, whereas keratinocytes migrate relatively fast.88 Cells also have ways to modify the strength of their cellular adhesions to promote more efficient movement, for example, by focalized matrix degradation, by utilizing several low-to-moderate affinity integrins in concert or by expressing anti-adhesive ECM molecules underneath themselves.
Integrins regulate many aspects of wound healing, including cellular crosstalk
During wound healing, cells interact with the wound ECM molecules with their integrin receptors. Concomitantly, many integrins are functionally activated or their expression is either induced or upregulated in response to their contact with these ECM molecules. As evidenced earlier, many integrins can bind various ECM molecules and, conversely, many ECM molecules are recognized by several different integrin heterodimers. Due to their overlapping specificities and functional compensation, many integrin knockout animals display surprisingly mild wound healing phenotypes (Table 2).
There is extensive interaction between the different types of wound cells. ECM proteins and integrins play key roles in these interactions.94 For example, knocking out neutrophil and monocyte integrin αMβ2 delays skin wound re-epithelialization in mice; whereas elimination of αvβ3 integrin, which is expressed in wounds by platelets, endothelial cells, macrophages, and fibroblasts, accelerates it (Table 2).50,53 The formation of a new basement membrane zone is an example where interaction between keratinocytes and fibroblasts is crucially involved, as the basement membrane components are produced jointly by the two types of cells.94 Similarly, endothelial cells and pericytes collaborate in the assembly of vascular basement membranes during wound angiogenesis.95 Keratinocytes also cross talk with endothelial cells via integrins. For example, epidermal deletion of the α3 integrin subunit leads to impaired wound angiogenesis due to decreased expression of the pro-angiogenic mitogen-related protein-3 by the keratinocytes.55 Moreover, fibroblasts acquire the myofibroblast phenotype (see below) under the control of keratinocytes.94
Next, we will focus on integrin expression and functions during re-epithelialization and granulation tissue formation. Integrin functions in other wound cells (e.g., platelets or infiltrating macrophages) have been comprehensively reviewed elsewhere (Table 2).56,65,68 Much of the data presented are drawn from reports on human and animal skin wound healing and from experiments with cultured cells. Information about integrin functions during oral mucosal healing is also presented when available. It also needs to be kept in mind that, in addition to integrins, several nonintegrin adhesions regulate cell functions.1
Integrins control epithelial cell migration and proliferation during re-epithelialization
Mechanisms of re-epithelialization
After wounding, keratinocytes are exposed to a new pericellular environment that consists of proteins of the underlying connective tissue at the wound edge and the plasma-derived proteins present in the blood clot (Fig. 6A).11 Their contact with these pro-migratory ECM molecules, in collaboration with other factors that influence wound re-epithelialization, such as proteinases, which cleave matrix molecules and release matrix-bound growth factors, growth factors, and cytokines (such as the members of the epidermal growth factor [EGF] family, TGF-β1, and fibroblast growth factors [FGFs]) synthesized by other wound cells and by keratinocytes themselves, loss of cell–cell contacts as well as changes in the direction of the intraepithelial electrical field, and the concentrations of Mg2+ and Ca2+ in the wound fluid, promote keratinocyte transition to a migratory phenotype.11 The autocrine expression of heparin-binding EGF-like growth factor by the keratinocytes is, however, needed for the sustenance of the re-epithelialization process (Fig. 6A).96
Figure 6.

A schematic presentation of KC integrin expression and ECM molecule distribution during human oral mucosal healing. (A) The contact with the ECM molecules and pro-migratory growth factors (e.g., EGF) present in the wound clot activate the wound edge basal KCs. They dissolve their hemidesmosomal contacts with the BM and extend into the wound clot. As they migrate, wound KCs interact with the provisional BM they deposit underneath themselves. In this provisional matrix, they interact with EDA FN via α5β1, α9β1, and αvβ1 integrins, with TN-C via α9β1 integrin and with laminin-332 via α2β1, α3β1, and α6β4 integrins. KC migration is sustained by their autocrine expression of HB-EGF. (B) After wound edges have joined, BM is regenerated, and hemidesmosome re-assembly is initiated. At this point, αvβ6 integrin expression is induced in the wound KCs. It potentially interacts with and activates latent, ECM-bound TGF-β1 to regulate KC proliferation, inflammation, and granulation tissue remodeling. Migrating wound KCs express α2β1, α3β1, α5β1, α9β1, αvβ1, and α6β4 integrins; suprabasal early-wound KCs express β1 integrins; late-wound basal KCs express mainly α2β1, α3β1, α9β1, αvβ6, and α6β4 integrins, whereas the expression of α5β1 and αvβ1 integrins is downregulated; late wound suprabasal KCs express αvβ6 integrin. Mature BM consists mainly of laminin-332, other laminins, collagen types IV and VII, and tenascin-C; provisional BM consists mainly of laminin-332, EDA FN, and tenascin-C. Connective tissue (CT) contains type I collagen, other collagens, and FN. Wound clot (FC) contains fibrin, FN, and vitronectin. Granulation tissue (GT) contains EDA and EDB FNs, collagen types I and III, other collagens, tenascin-C, fibrin, vitronectin. KC, keratinocyte; BM, basement membrane; EDA/B, extra domain A/B; HB-EGF, heparin-binding EGF-like growth factor. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
The activated keratinocytes at the wound edge dissolve their hemidesmosomal contacts with the basement membrane, and their morphology becomes flattened and elongated with long lamellipodia that extend into the wound provisional matrix (Fig. 6A). Unlike epidermal keratinocytes that migrate under the blood clot, oral mucosal keratinocytes invade into and migrate through it (Fig. 7A, B).97 The leading keratinocytes need to proteolytically dissolve and remodel the fibrin barrier ahead of them via focalized activation of plasmin by the collaboration of integrins and uPAR on the keratinocyte cell surface to be able to migrate into the wound clot.98 In re-epithelialization, the keratinocytes move as a sheet and maintain some of their cell–cell connections, but the exact mechanism of their migration has not yet been conclusively established (Fig. 7A).11,99
Figure 7.

Active re-epithelialization of a 3-day-old human palatal mucosal wound. (A) Epithelial keratinocytes migrate into the wound provisional matrix (HE staining). Scale bar: 200 μm. (B) Higher magnification image illustrating fibrin (arrow) between the migrating cells and the CT collagen (Mallory's phosphotungstic acid hematoxylin staining). Scale bar: 50 μm. (C) High expression of α6β4 integrin at the leading keratinocytes (indirect immunofluorescence image). (D) High expression of α3β1 integrin at the leading keratinocytes (indirect immunofluorescence image). See Larjava et al. for experimental procedures.97 To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Keratinocytes adapt to the wound environment via changes in their integrin expression and localization
In healthy skin and oral mucosa, the basal epithelial cells express mainly α3β1, α2β1, α9β1, and α6β4 integrins, which anchor the cells to the underlying basement membrane and maintain the normal epithelial proliferation and differentiation patterns as well as the innate epithelial immune system, with additional weak expression of α5β1 and αvβ5 integrins.11,100 As an adaptation to the new extracellular environment in the wound as well as to the provisional basement membrane the keratinocytes assemble underneath themselves against the provisional wound matrix, there is a change in the expression levels and/or localization of the existing keratinocyte integrins and an induction of fibronectin-recognizing α5β1, αvβ1, and αvβ6 integrins.11 Expression of αvβ5 integrin in the migrating keratinocytes may depend on the severity of the trauma, as it is expressed in deep human and porcine excisional skin wounds, but not in smaller incisional human skin or oral mucosal wounds.101,102 For optimal motility on the wound composite matrix, keratinocytes seem to utilize several intermediate strength integrin–matrix interactions in cooperation. The strength of their cell adhesion may be regulated by focalized digestion of strong cell adhesion ligands, by assembly of matrices with reduced adhesiveness, by reduction of the strength of integrin affinity through cellular signaling, and by robust expression of low-affinity integrins.
Integrins guide keratinocyte migration directionality during re-epithelialization
Integrins are involved in the guidance of the migration directionality of wound edge keratinocytes. Integrin α2β1 binding to fibrillar dermal collagen at the wound edge and in the granulation tissue with high affinity leads to induction of MMP-1 expression and focalized denaturation of the collagen matrix.103 Keratinocytes do not express any known receptors for denatured collagen, but it can bind other pro-migratory ECM molecules, such as fibronectin, and this process may help regulate the directionality of keratinocyte migration in wounds.103 Integrin α6β4 regulates the migratory behavior of keratinocytes by determining laminin-332 organization underneath the epithelial front, whereas α3β1 integrin may help in maintaining keratinocyte migration directionality by binding to the laminin-332 that is newly deposited on the wound bed at the rear of the cell, thus regulating their polarization, adhesion, and migration velocity during re-epithelialization.104,105
Pro-migratory signaling by α6β4 integrin is needed for keratinocyte migration
Chemotactic factors (e.g., EGF) promote the mobilization of α6β4 integrin from hemidesmosomes to the keratinocyte protrusions by increasing the phosphorylation of the β4 integrin cytoplasmic domain (Fig. 7C).106,107 Signaling through this receptor is required for normal re-epithelialization (Table 2). Recently, α6β4 integrin was shown to complex with C4.4A (a structural homologue of uPAR), MMP-14, and disintegrin-metalloproteinase TACE to promote keratinocyte migration on laminin-332, possibly through focalized laminin-332 degradation.108 In addition, it may regulate the expression of α2β1 and α3β1 integrins in keratinocytes.109
Integrins of the β1 family are essential for wound re-epithelialization
The expression of β1 integrins is strongly upregulated after wounding in the wound-edge keratinocytes and in several suprabasal keratinocyte layers, and they are critical for proper re-epithelialization (Fig. 6A).110 Recently, it was shown that adaptor protein 4.1R regulates the expression of β1 integrins in wound keratinocytes, and its loss results in defective re-epithelialization.111 Wounding causes an increase in the expression of α2β1, α3β1, and α9β1 integrins as well as their relocation onto the basal cell membrane of the basal keratinocytes and an induction of αvβ1 and α5β1 integrin expression (Fig. 6A).25 The importance of these individual β1 integrins for re-epithelialization is not yet completely understood. For example, α3β1 integrin, which is upregulated in migrating epithelial front in wounds (Fig. 6D), has been reported to either promote or inhibit keratinocyte migration and re-epithelialization depending on the experimental model both in vitro and in vivo (Table 2). β1 integrins can also modify each other's ligand binding. For example, α3β1 integrin is a trans-dominant inhibitor of α2β1 and α5β1 integrins, and it can moderate their binding affinity toward collagen and fibronectin, respectively, during re-epithelialization.112
Fibronectin receptors α5β1 and αvβ1 are induced in the migrating wound keratinocytes (Fig. 6A). Unlike α5β1 integrin, which is required for keratinocyte migration and fibronectin matrix assembly, αvβ1 integrin is a low-affinity fibronectin receptor that only weakly supports keratinocyte migration.113 It may, thus, facilitate keratinocyte migration on the underlying EDA fibronectin during re-epithelialization by supporting cell attachment without decelerating the migration speed. In addition, deposition of tenascin-C underneath the migrating epithelium reduces the affinity of α5β1 integrin toward fibronectin to facilitate keratinocyte migration.32 Migrating keratinocytes also express low levels of αvβ6 integrin in the re-epithelializing oral mucosal and skin wounds, where it co-localizes with tenascin-C and EDA fibronectin, but its main role comes later in the wound healing.
Integrins regulate keratinocyte proliferation in wounds
While migrating wound keratinocytes do not divide, the basal keratinocytes adjacent to the wound edge start proliferating 48–72 h after the injury to supply more migratory cells to the wound, and these cells may be amended by a putative keratinocyte stem cell population that is localized in the basal keratinocyte layer against the connective tissue papilla area in oral mucosal epithelium and in interfollicular epidermis in skin.114–116 In addition, epidermal stem cells residing in hair follicle bulges and sebaceous glands contribute to skin re-epithelialization.116 Apart from mediating keratinocyte migration, integrins also influence their proliferation during re-epithelialization. Integrin α9β1 seems essential for keratinocyte proliferation at the wound edge, as mice with α9 integrin-deficient keratinocytes exhibit retarded wound re-epithelialization due to reduced proliferation of keratinocytes (Table 2). In addition to tenascin-C and EDA fibronectin, α9β1 integrin was recently shown to interact with another ECM component, elastic microfibril interface–located protein 1 (EMILIN1), to regulate keratinocyte proliferation.24 The interaction of α5β1 integrin with fibronectin may also contribute to keratinocyte proliferation in addition to promoting their adhesion and motility on this matrix.117
Epithelial αvβ6 integrin regulates inflammation and granulation tissue remodeling
When the migrating epithelial fronts originating from the wound edges have joined and cover the wound surface, β1 integrin expression is downregulated, and α6β4 integrin binding to laminin-332 is restored.25,43 New hemidesmosomes start forming along the wound epithelium, and they function as nucleation sites for the basement membrane restorations (Fig. 6B).44 During this later phase of wound healing, αvβ6 integrin expression is significantly upregulated in the basal and several suprabasal keratinocyte layers (Fig. 6B).102 It may mediate TGF-β1 activation and regulate ECM deposition in the granulation tissue (Fig. 8),62 as its expression coincides with the peak expression and activity of this cytokine.118 In addition, αvβ6 integrin may have other functions, such as regulation of keratinocyte proliferation and inflammation as well as remodeling of the basement membrane zone and removal of the fibrin provisional matrix during late wound healing.11,119
Figure 8.

Schematic presentation illustrating how integrins could activate latent TGF-β1 during wound healing. In wounds, macrophages are the main source of TGF-β1, although other cells produce it as well. TGF-β1 is produced in a latent form, in which active TGF-β1 (red circle) is confined inside latency-associated peptide (LAP) that renders it inactive. LAP contains the RGD recognition signal for integrins. LAP further binds latent TGF-β1-binding protein (LTBP) to create a “large latent complex” that subsequently binds to ECM, specifically to FN. Target cells with LAP RGD-recognizing integrins (keratinocytes with αvβ6 integrin and fibroblasts with αvβ5 integrin) attach to the large complex, create a tractional force with their cytoskeleton (red actin filaments), and pull, leading to a conformational change in the complex and release of the active TGF-β1 that can now bind to its receptor complex (TGFβR). Active TGF-β1 signals via smad proteins and stimulate keratinocyte migration and ECM production but inhibit their proliferation. In pericytes and fibroblasts, active TGF-β1 stimulates cell differentiation to myofibroblasts and their ECM production. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Fibroblast integrins mediate cell migration as well as granulation tissue formation and remodeling
Connective tissue is repaired by fibroblasts or progenitor cells (pericytes and other mesenchymal stem cells) that are activated by serum and other factors (e.g., TGF-β1, platelet-derived growth factor [PDGF], and FGF-2) that are released from platelets and by other wound cells, such as macrophages and keratinocytes.8 Additional activating signals include the disruption of cell–cell connections, changes in oxygen tension, exposure to new ECM molecules, and changes in mechanical tension in the tissue itself.8,120–122 In addition, the circulating fibroblast-like cells (fibrocytes) may serve as important precursor cells to pericytes in wound healing.123 Connective tissue cell activation extends far from the wound edge, as evidenced by localization of the induction of fibroblast activating protein (FAP or seprase) expression at cells distant from the wound edge.124
Activated fibroblasts and pericytes migrate from the adjacent subepithelial connective tissue and blood vessels, respectively, to the wound bed where they differentiate into myofibroblasts, synthesize the granulation tissue ECM, and remodel it to either normal or scar tissue.8 Cytokines, such as TGF-β1, PDGF, and Cyr61-CTGF-Nov (CCN)2/connective tissue growth factor (CTGF), that are secreted by platelets and macrophages are important for pericyte and connective tissue fibroblast differentiation into myofibroblasts (Fig. 9). In addition, their differentiation depends on the presence of EDA fibronectin and mechanical tension experienced by the cells from the ECM through integrin-mediated signaling (Fig. 9).125
Figure 9.

A simplified illustration depicting how pericytes differentiate to matrix-producing myofibroblasts. Chemotactic PDGF, TGF-β1, and CCN2/CTGF signaling by platelets and macrophages causes the pericytes to detach from the vessel wall and produce EDA FN and collagen. Full differentiation of pericytes into myofibroblasts, evidenced by αSMA expression, depends on three essential elements, namely TGF-β1, EDA FN, and tension that can be created in the matrix produced by the pericytes. Myofibroblasts interact with EDA FN via α5β1, αvβ3, and αvβ5 integrins and with collagen via α2β1 and α11β1 integrins. Latent TGF-β1 complex is bound into the FN matrix and possibly activated via αvβ5 integrin and by other mechanisms. Whether the wound heals scarless, with scars, or becomes fibrotic depends on both the presence of myofibroblasts and macrophage-derived TGF-β1. αSMA, alpha–smooth muscle actin; CCN, Cyr61-CTGF-Nov; CTGF, connective tissue growth factor. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Integrins are required for fibroblast infiltration into the wound clot
Normal fibroblasts and granulation tissue fibroblasts (mixture of fibroblasts and myofibroblasts) express a large repertoire of integrins, including α1β1, α2β1, α3β1, α5β1, α11β1, αvβ1, αvβ3, and αvβ5, that the cells can use for binding to a variety of ECM molecules such as collagens, fibronectins, other blood clot components, CCN2/CTGF, and others (Table 2).8 However, many individual integrins are dispensable for normal connective tissue healing at least in the mouse (Table 2), although they appear to have specific functions in cultured fibroblasts. This may be due to compensatory mechanisms by other integrins with overlapping functions. Due to significant differences between murine and human wound healing, the consequences of integrin deficiencies in human beings may be more significant.
Fibroblasts express many β1 integrins. Of these, it can be speculated that α5β1 integrin plays the most critical role in vivo during connective tissue cell invasion into the wound clot and their subsequent migration in the fibrin–fibronectin-containing 3D wound environment.1 It was recently shown that dermatopontin, a dermal ECM protein, colocalizes with fibrin and fibronectin in the wound clot, where it may enhance fibroblast adhesion to the provisional matrix via α5β1 integrin, accelerating fibroblast cell adhesion to the provisional matrix in the initial stage of wound healing.126 Syndecan-4 triggers the endocytosis and trafficking of the fibroblast α5β1 integrin, which leads to immigration of the cells into the wound bed.127 Interestingly, granulation tissue fibroblasts seem to have a reduced ability to adhere to fibronectin via α5β1 integrin that may help them in being more mobile in the early fibronectin-rich granulation tissue matrix.128 The presence of tenascin-C can also promote the α5β1 integrin-dependent fibroblast migration into the granulation tissue.33
Integrin αvβ3 may be inhibitory to fibroblast infiltration into the wound clot, because it can suppress TGF-β1–mediated signaling. Notably, mice lacking β3 integrins show faster epidermal wound healing with enhanced dermal fibroblast infiltration into wounds (Table 2).50
Both β1 and αv integrins are involved in myofibroblast differentiation and in granulation tissue synthesis and remodeling
About 1 week after wounding, the connective tissue cells within the wound granulation tissue start expressing alpha–smooth muscle actin (αSMA) in the cytoskeletal stress fibers, becoming myofibroblasts. The integrins of the β1 family appear important in myofibroblast differentiation and function. Mouse fibroblasts deficient in β1 integrins show reduced expression of αSMA, CCN2/CTGF, and type I collagen as well as lowered ability to activate latent TGF-β, and they fail to differentiate into myofibroblasts.129 This leads to delayed cutaneous wound closure and diminished granulation tissue formation in the β1 integrin null animals. In vitro, TGF-β1–induced αSMA expression and collagen matrix contraction are blocked by antibodies against α5 integrin in both human oral mucosal and dermal fibroblasts.130 TGF-β1 from macrophages can upregulate both EDA fibronectin and α5β1 integrin expression in fibroblasts, leading to activation of FAK, an essential signaling protein for myofibroblast differentiation.131 Integrin signaling proteins kindlin-2 and ILK are also required for the TGF-β1–induced myofibroblast differentiation and function.132,133 Indeed, defective granulation tissue formation is seen in mice with fibroblast-specific ILK ablation.134 Inhibition of α9β1 integrin by a specific antibody also inhibits granulation tissue formation during skin wound healing.59
Fibroblasts interact with fibrillar collagens via α1β1, α2β1, and α11β1 integrins (Table 2). Mice lacking individual collagen receptors show only subtle changes in granulation tissue ECM remodeling, however (Table 2). It is likely that in the absence of α1β1 or α2β1 integrin, the other collagen receptors can compensate in most functions, such as regulation of MMP expression and collagen fibrillogenesis. Interestingly, it was recently shown that α11β1 is induced by mechanical strain and by members of the TGF-β family, and it promotes myofibroblast differentiation.135,136 However, there is no wound healing data available that show the effects of α11β1 deficiency or blockage in granulation tissue formation or remodeling.
In addition, αv integrins may be involved in myofibroblast differentiation. Blocking of αvβ5 or αvβ3 integrin suppresses TGF-β1-induced myofibroblast differentiation of both oral and dermal fibroblasts in cell culture.137 Furthermore, increased expression of αvβ5 integrin induces αSMA expression in fibroblasts and promotes their responsiveness to TGF-β1 via RGD motif-dependent recruitment of latent TGF-β complex into the cell surface and its activation (Fig. 8).138 Integrin αvβ5 also appears to regulate α2β1 integrin function in myofibroblasts, promoting their persistent myofibroblast phenotype.139
Tension within granulation tissue increases as it is getting compacted by myofibroblasts. Cells under minimal tension in early granulation tissue express α2β1 integrin that regulates fine collagen fibril organization into thick collagen fibers.140 Thicker fibers create a rigid matrix, generating more tension, which leads to cell switching to αvβ3 integrin and noncollagen ECM interactions.140 However, fibroblast differentiation into myofibroblasts and wound contraction are not affected by αvβ3 integrin deficiency at least in the mouse skin wound healing model (Table 2),50 suggesting that compensatory mechanisms exist in vivo.
Usually, myofibroblasts are driven into senescence and converted from ECM-producing cells into ECM-degrading cells in the remodeling phase of wound repair.141,142 This process is mediated by collaborative interaction of α6β1 integrin and cell surface heparan sulphate proteoglycans with matricellular protein CCN1/Cyr61 and by subsequent production of ROS.142 Myofibroblasts persist in fibrotic conditions and pathological scars, where they continue to produce matrix.141 However, the number of myofibroblasts in the oral mucosal wounds, which heal with minimal scarring, is higher than in skin wounds,143 suggesting that the presence of myofibroblasts per se does not predict scarring or fibrosis.
Integrins are essential for angiogenesis
Wound angiogenesis is an integral part of granulation tissue formation. It begins with endothelial cell activation by hypoxia-induced production of vascular endothelial growth factor (VEGF), FGF-2/bFGF, and other pro-angiogenic growth factors by various types of wound cells, such as macrophages.12 Consequently, the vascular basement membrane is degraded, and vascular sprouting is initiated into the provisional wound matrix; this process involves proliferation and migration of endothelial cells (as well as endothelial progenitor cells) and vessel-associated pericytes.12 Integrins play a central role in wound neo-vascularization. In addition to supporting endothelial cell and pericyte migration, they act as co-receptors for growth factor receptors, such as the VEGF and angiopoietin receptors, and support vascular basement membrane assembly.12 The exact roles of individual integrins in wound angiogenesis are still unclear. Deposition of fibronectin is critical for angiogenic development. However, endothelial cells can interact with it by using several different fibronectin receptors in a compensatory manner and in collaboration with pericyte integrins.12,144 Information about the role of integrins in adult vascular remodeling comes mostly from studies on tumor angiogenesis, which may be mechanistically different from wound vascularization. These studies point to α6β4 integrin-mediated signaling playing a role in the initial endothelial cell activation, whereas at least αvβ3 and αvβ5 (when attached to matrix-bound ECM molecules), α9β1 (in interaction with VEGFs) as well as α1β1 and α2β1 integrins can participate in vascular sprouting.12,61,145
Integrin expression and functions may be altered in chronic wounds and hypertrophic scars
Disruption of the normal wound healing sequence, especially prolonged inflammation, may result in over-healing (hypertrophic scar) or a failure to heal (chronic wound). Impaired wound healing may result from inadequate or excessive integrin signaling, which is accompanied by defective ECM that fails to support re-epithelialization and normal fibroblast function as well as from insufficient angiogenesis which results in poor tissue oxygenation. Therefore, activation or inhibition of certain integrins may provide an effective way to influence wound healing outcomes.
Many ECM molecules (e.g., fibronectin and collagen) are abnormally glycated in chronic wounds that are associated with diabetes. This reduces the binding of different ECM molecules to each other and integrin-mediated epithelial adhesion to ECM, resulting in defective re-epithelialization.2 Notably, chronic wounds exhibit drastically decreased epithelial expression of α5β1 integrin, which results in reduced integration of fibronectin into the provisional basement membrane, increased fibronectin degradation, and a failure in keratinocyte migration and re-epithelialization.146 In addition, increased keratinocyte integrin expression may contribute to the formation of chronic wounds. The expression of αvβ6 integrin is strongly upregulated in the epidermis of human chronic wounds, and its constitutive overexpression in mouse epidermis is associated with over activation of TGF-β1 and increased susceptibility for chronic fibrotic ulcers in these animals.147 However, the responsiveness of myofibroblasts to the stimulatory action of TGF-β1 is diminished in chronic wounds; this is possibly due to a failure in myofibroblast integrin activation and cross-talk, which is caused by the degraded wound ECM, especially EDA fibronectin.141,146 Integrin αvβ6 signaling may also promote the production of MMP-9 by the keratinocytes, which is shown to be upregulated in chronic wounds where it contributes to the excessive ECM degradation.146,148
Members of the CCN family of matricellular proteins are ligands for many integrins (Table 2),60 and they critically regulate myofibroblast fate as well as ECM production and remodeling in wounds.142 In chronic human wounds, the expression of CCN1/Cyr61 is increased, and CCN2/CTGF is significantly reduced compared with normally healing wounds.149 The excessive expression of CCN1/Cyr61 may result in premature α6β1-integrin-mediated myofibroblast senescence that is associated with early down-regulation of ECM (e.g., collagen) production, up-regulation of matrix-degrading proteases, and ongoing breakdown of the granulation tissue before the re-epithelialization is completed, contributing to the wound chronicity.142,146 In hypertrophic scars, myofibroblasts persist and they continue to produce excessive matrix possibly because of a reversed CCN1/Cyr61 and CCN2/CTGF balance.147 Elevated levels of CCN2/CTGF are one of the hallmarks of fibrosis, and it potentiates TGF-β1 actions in myofibroblasts to produce a sustained fibrotic response.60
Take-Home Messages.
• Integrins are essential cell surface receptors that bind proteins in the extracellular and wound provisional matrix.
• Keratinocytes and fibroblasts have multiple integrins with overlapping matrix-binding capabilities, and one can compensate for the lack of another.
• Expression of many integrins is induced during wound healing, and their localization in tissues may be altered.
• Integrins regulate wound re-epithelialization and granulation tissue formation at several levels. The most important integrins for epithelial migration are α5β1 and α6β4 integrins, while α5β1 integrin appears critical for granulation tissue formation. In addition, other integrins participate, especially when healing is compromised or when it leads to fibrotic outcomes.
• Certain integrins can activate TGF-β1 and potentially regulate fibrosis. These integrins are potential targets for anti-fibrosis therapies.
• Most of the information regarding wound healing is derived from mouse or cell culture studies that poorly reflect human mucosal or skin healing. Therefore, future research should dissect integrin functions in animal models that better mimic human mucosal and skin wound healing.
Abbreviations and Acronyms
- αSMA
alpha–smooth muscle actin
- ADAM
a disintegrin and metalloproteinase
- BM
basement membrane
- CCN
Cyr61-CTGF-Nov
- COL
collagen
- CT
connective tissue
- CTGF
connective tissue growth factor
- EC
endothelial cell
- ECM
extracellular matrix
- EDA/B
extra domain A/B
- EGF
epidermal growth factor
- EGFR
EGF receptor
- EMILIN
elastic microfibril interface–located protein
- FAK
focal adhesion kinase
- FAP
fibroblast activating protein
- FBL
fibroblast
- FC
wound (fibrin) clot
- FGF
fibroblast growth factor
- FN
fibronectin
- GT
granulation tissue
- HB-EGF
heparin-binding EGF-like growth factor
- ICAM
intercellular adhesion molecule
- ILK
integrin-linked kinase
- KC
keratinocyte
- LAP
latency-associated peptide
- LM
laminin
- LTBP
latent TGF-β1–binding protein
- MMP
matrix metalloproteinase
- NK
natural killer cell
- OPN
osteopontin
- PDGF
platelet-derived growth factor
- RGD
arginine-glycine-aspartic acid
- ROS
reactive oxygen species
- SPARC
secreted protein acidic and rich in cysteine
- TGF
transforming growth factor
- TN
tenascin
- TSP
thrombospondin
- uPAR
urokinase-type plasminogen activator receptor
- VCAM
vascular cell adhesion molecule
- VEGF
vascular endothelial growth factor
- VN
vitronectin
- VWF
von Willebrand factor
Acknowledgments and Funding Sources
The original research performed by the authors was supported by the Canadian Institutes of Health Research.
Author Disclosure and Ghostwriting
No competing financial interests exist. The content of this article was expressly written by the authors listed. No ghostwriters were used to write this article.
About the Authors
Dr. Leeni Koivisto, PhD, is a Research Associate in the Laboratory of Periodontal Biology, Department of Oral Biological and Medical Sciences in the Faculty of Dentistry at the University of British Columbia (Vancouver, Canada). Dr. Jyrki Heino, MD, PhD, is a Professor of Biochemistry and Head of the Department of Biochemistry and Food Chemistry at the University of Turku (Turku, Finland) as well as the Scientific Director of BioCity Turku, the common biocenter of the University of Turku and the Åbo Akademi University (Turku, Finland). Dr. Lari Häkkinen, DDS, PhD, is an Associate Professor in the Department of Oral Biological and Medical Sciences in the Faculty of Dentistry at the University of British Columbia. Dr. Hannu Larjava, DDS, PhD, Dip. Perio., is a Professor and the Chair of the Division of Periodontics in the Faculty of Dentistry at the University of British Columbia.
Larjava H and Häkkinen L, unpublished gene expression profiling of pig wounds performed in 2006, University of British Columbia.
References
- 1.Geiger B. and Yamada KM: Molecular architecture and function of matrix adhesions. Cold Spring Harb Perspect Biol 2011; 3:a005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schultz GS, Davidson JM, Kirsner RS, Bornstein P, and Herman IM: Dynamic reciprocity in the wound microenvironment. Wound Repair Regen 2011; 19:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larjava H, Wiebe C, Gallant-Behm C, Hart DA, Heino J, and Häkkinen L: Exploring scarless healing of oral soft tissues. J Can Dent Assoc 2011; 77:b18. [PubMed] [Google Scholar]
- 4.Nishimura SL: Integrin-mediated transforming growth factor-β activation, a potential therapeutic target in fibrogenic disorders. Am J Pathol 2009; 175:1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stephens P. and Genever P: Non-epithelial oral mucosal progenitor cell populations. Oral Dis 2007; 13:1. [DOI] [PubMed] [Google Scholar]
- 6.Shaw TJ. and Martin P: Wound repair at a glance. J Cell Sci 2009; 122:3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sonnemann KJ. and Bement WM: Wound repair: toward understanding and integration of single-cell and multicellular wound responses. Annu Rev Cell Dev Biol 2011; 27:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Häkkinen L, larjava H, and Koivisto L: Granulation tissue formation and remodeling. Endod Top 2012; 24:94 [Google Scholar]
- 9.Oakley C. and Larjava H: Hemostasis, coagulation and complications. Endod Top 2012; 24:4 [Google Scholar]
- 10.Turabelize A. and DiPietro LA: Inflammation and wound healing. Endod Top 2012; 24:26 [Google Scholar]
- 11.Koivisto L, Häkkinen L, and Larjava H: Reepithelialization of wounds. Endod Top 2012; 24:59 [Google Scholar]
- 12.Senger DR. and Davis GE: Angiogenesis. Cold Spring Harb Perspect Biol 2011; 3:a005090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu P, Takai K, Weaver VM, and Werb Z: Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol 2011; 3:a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hynes RO. and Naba A: Overview of the matrisome—an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol 2012; 4:a004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rutnam ZJ, Wight TN, and Yang BB: miRNAs regulate expression and function of extracellular matrix molecules. Matrix Biol 2013; 32:74–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weisel JW: Fibrinogen and fibrin. Adv Protein Chem 2005; 70:247. [DOI] [PubMed] [Google Scholar]
- 17.Preissner KT. and Reuning U: Vitronectin in vascular context: facets of a multitalented matricellular protein. Semin Thromb Hemost 2011; 37:408. [DOI] [PubMed] [Google Scholar]
- 18.To WS. and Midwood KS: Plasma and cellular fibronectin: distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair 2011; 4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buback F, Renkl AC, Schulz G, and Weiss JM: Osteopontin and the skin: multiple emerging roles in cutaneous biology and pathology. Exp Dermatol 2009; 18:750. [DOI] [PubMed] [Google Scholar]
- 20.Shoulders MD. and Raines RT: Collagen structure and stability. Annu Rev Biochem 2009; 78:929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mosher DF. and Adams JC: Adhesion-modulating/matricellular ECM protein families: a structural, functional and evolutionary appraisal. Matrix Biol 2012; 31:155. [DOI] [PubMed] [Google Scholar]
- 22.Rousselle P. and Beck K: Laminin 332 processing impacts cellular behavior. Cell Adh Migr 2012; 7:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maclauchlan S, Skokos EA, Agah A, Zeng J, Tian W, Davidson JM, Bornstein P, and Kyriakides TR: Enhanced angiogenesis and reduced contraction in thrombospondin-2-null wounds is associated with increased levels of matrix metalloproteinases-2 and -9, and soluble VEGF. J Histochem Cytochem 2009; 57:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Danussi C, Petrucco A, Wassermann B, Pivetta E, Modica TM, Del Bel Belluz L, Colombatti A, and Spessotto P: EMILIN1-α4/α9 integrin interaction inhibits dermal fibroblast and keratinocyte proliferation. J Cell Biol 2011; 195:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larjava H, Koivisto L, Häkkinen L, and Heino J: Epithelial integrins with special reference to oral epithelia. J Dent Res 2011; 90:1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwarzbauer JE. and DeSimone DW: Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harb Perspect Biol 2011; 3:a005041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li-Korotky HS, Hebda PA, Lo CY, and Dohar JE: Age-dependent differential expression of fibronectin variants in skin and airway mucosal wounds. Arch Otolaryngol Head Neck Surg 2007; 133:919. [DOI] [PubMed] [Google Scholar]
- 28.Wong JW, Gallant-Behm C, Wiebe C, Mak K, Hart DA, Larjava H, and Häkkinen L: Wound healing in oral mucosa results in reduced scar formation as compared with skin: evidence from the red Duroc pig model and humans. Wound Repair Regen 2009; 17:717. [DOI] [PubMed] [Google Scholar]
- 29.Midwood KS. and Orend G: The role of tenascin-C in tissue injury and tumorigenesis. J Cell Commun Signal 2009; 3:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Häkkinen L, Hildebrand HC, Berndt A, Kosmehl H, and Larjava H: Immunolocalization of tenascin-C, α9 integrin subunit, and αvβ6 integrin during wound healing in human oral mucosa. J Histochem Cytochem 2000; 48:985. [DOI] [PubMed] [Google Scholar]
- 31.Csiszar A, Wiebe C, Larjava H, and Häkkinen L: Distinctive molecular composition of human gingival interdental papilla. J Periodontol 2007; 78:304. [DOI] [PubMed] [Google Scholar]
- 32.Ingham KC, Brew SA, and Erickson HP: Localization of a cryptic binding site for tenascin on fibronectin. J Biol Chem 2004; 279:28132. [DOI] [PubMed] [Google Scholar]
- 33.Trebaul A, Chan EK, and Midwood KS: Regulation of fibroblast migration by tenascin-C. Biochem Soc Trans 2007; 35:695. [DOI] [PubMed] [Google Scholar]
- 34.Whitby DJ, Longaker MT, Harrison MR, Adzick NS, and Ferguson MW: Rapid epithelialisation of fetal wounds is associated with the early deposition of tenascin. J Cell Sci 1991; 99:583. [DOI] [PubMed] [Google Scholar]
- 35.Sible JC, Rettig WJ, Eriksson E, Smith SP, and Oliver N: Gene expression of tenascin is altered in normal scars and keloids. Wound Repair Regen 1995; 3:37. [DOI] [PubMed] [Google Scholar]
- 36.Carey WA, Taylor GD, Dean WB, and Bristow JD: Tenascin-C deficiency attenuates TGF-β-mediated fibrosis following murine lung injury. Am J Physiol Lung Cell Mol Physiol 2010; 299:L785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu KQ, Carrougher GJ, Couture OP, Tuggle CK, Gibran NS, and Engrav LH: Expression of collagen genes in the cones of skin in the Duroc/Yorkshire porcine model of fibroproliferative scarring. J Burn Care Res 2008; 29:815 Erratum in: 2011; 32: 569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Häkkinen L, Westermarck J, Kähäri VM, and Larjava H: Human granulation-tissue fibroblasts show enhanced proteoglycan gene expression and altered response to TGF-β1. J Dent Res 1996; 75:1767. [DOI] [PubMed] [Google Scholar]
- 39.Liu X, Wu H, Byrne M, Krane S, and Jaenisch R: Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci U S A 1997; 94:1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kadler KE, Hill A, and Canty-Laird EG: Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol 2008; 20:495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tzu J. and Marinkovich MP: Bridging structure with function: structural, regulatory, and developmental role of laminins. Int J Biochem Cell Biol 2008; 40:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schneider H, Mühle C, and Pacho F: Biological function of laminin-5 and pathogenic impact of its deficiency. Eur J Cell Biol 2007; 86:701. [DOI] [PubMed] [Google Scholar]
- 43.Kariya Y, Sato H, Katou N, Kariya Y, and Miyazaki K: Polymerized laminin-332 matrix supports rapid and tight adhesion of keratinocytes, suppressing cell migration. PLoS One 2012; 7:e35546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Litjens SH, de Pereda JM, and Sonnenberg A: Current insights into the formation and breakdown of hemidesmosomes. Trends Cell Biol 2006; 16:376. [DOI] [PubMed] [Google Scholar]
- 45.McLean WH, Irvine AD, Hamill KJ, Whittock NV, Coleman-Campbell CM, Mellerio JE, Ashton GS, Dopping-Hepenstal PJ, Eady RA, Jamil T, Phillips R, Shabbir SG, Haroon TS, Khurshid K, Moore JE, Page B, Darling J, Atherton DJ, Van Steensel MA, Munro CS, Smith FJ, and McGrath JA: An unusual N-terminal deletion of the laminin α3a isoform leads to the chronic granulation tissue disorder laryngo-onycho-cutaneous syndrome. Hum Mol Genet 2003; 12:2395. [DOI] [PubMed] [Google Scholar]
- 46.Barczyk M, Carracedo S, and Gullberg D: Integrins. Cell Tissue Res 2010; 339:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gardner H, Broberg A, Pozzi A, Laato M, and Heino J: Absence of integrin α1β1 in the mouse causes loss of feedback regulation of collagen synthesis in normal and wounded dermis. J Cell Sci 1999; 112:263. [DOI] [PubMed] [Google Scholar]
- 48.Bouvard D, Brakebusch C, Gustafsson E, Aszódi A, Bengtsson T, Berna A, and Fässler R: Functional consequences of integrin gene mutations in mice. Circ Res 2001; 89:211. [DOI] [PubMed] [Google Scholar]
- 49.Grüner S, Prostredna M, Schulte V, Krieg T, Eckes B, Brakebusch C, and Nieswandt B: Multiple integrin-ligand interactions synergize in shear-resistant platelet adhesion at sites of arterial injury in vivo. Blood 2003; 102:4021. [DOI] [PubMed] [Google Scholar]
- 50.Reynolds LE, Conti FJ, Lucas M, Grose R, Robinson S, Stone M, Saunders G, Dickson C, Hynes RO, Lacy-Hulbert A, and Hodivala-Dilke K: Accelerated re-epithelialization in β3-integrin-deficient- mice is associated with enhanced TGF-β1 signaling. Nat Med 2005; 11:167. [DOI] [PubMed] [Google Scholar]
- 51.AlDahlawi S, Eslami A, Häkkinen L, and Larjava HS: The αvβ6 integrin plays a role in compromised epidermal wound healing. Wound Repair Regen 2006; 14:289. [DOI] [PubMed] [Google Scholar]
- 52.Grenache DG, Zhang Z, Wells LE, Santoro SA, Davidson JM, and Zutter MM: Wound healing in the α2β1 integrin-deficient mouse: altered keratinocyte biology and dysregulated matrix metalloproteinase expression. J Invest Dermatol 2007; 127:455. [DOI] [PubMed] [Google Scholar]
- 53.Sisco M, Chao JD, Kim I, Mogford JE, Mayadas TN, and Mustoe TA: Delayed wound healing in Mac-1-deficient mice is associated with normal monocyte recruitment. Wound Repair Regen 2007; 15:566. [DOI] [PubMed] [Google Scholar]
- 54.Blue R, Kowalska MA, Hirsch J, Murcia M, Janczak CA, Harrington A, Jirouskova M, Li J, Fuentes R, Thornton MA, Filizola M, Poncz M, and Coller BS: Structural and therapeutic insights from the species specificity and in vivo antithrombotic activity of a novel αIIb-specific αIIbβ3 antagonist. Blood 2009; 114:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mitchell K, Szekeres C, Milano V, Svenson KB, Nilsen-Hamilton M, Kreidberg JA, and DiPersio CM: α3β1 integrin in epidermis promotes wound angiogenesis and keratinocyte-to-endothelial-cell crosstalk through the induction of MRP3. J Cell Sci 2009; 122:1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nieswandt B, Varga-Szabo D, and Elvers M: Integrins in platelet activation. J Thromb Haemost 2009; 7:206. [DOI] [PubMed] [Google Scholar]
- 57.Germain M, De Arcangelis A, Robinson SD, Baker M, Tavora B, D'Amico G, Silva R, Kostourou V, Reynolds LE, Watson A, Jones JL, Georges-Labouesse E, and Hodivala-Dilke K: Genetic ablation of the α6-integrin subunit in Tie1Cre mice enhances tumour angiogenesis. J Pathol 2010; 220:370. [DOI] [PubMed] [Google Scholar]
- 58.Jacobsen JN, Steffensen B, Häkkinen L, Krogfelt KA, and Larjava HS: Skin wound healing in diabetic β6 integrin-deficient mice. APMIS 2010; 118:753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakayama Y, Kon S, Kurotaki D, Morimoto J, Matsui Y, and Uede T: Blockade of interaction of α9 integrin with its ligands hinders the formation of granulation in cutaneous wound healing. Lab Invest 2010; 90:881. [DOI] [PubMed] [Google Scholar]
- 60.Chen C-C. and Lau LF: Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol 2009; 41:771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oommen S, Gupta SK, and Vlahakis NE: Vascular endothelial growth factor A (VEGF-A) induces endothelial and cancer cell migration through direct binding to integrin α9β1: identification of a specific α9β1 binding site. J Biol Chem 2011; 286:1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Worthington JJ, Klementowicz JE, and Travis MA: TGFβ: a sleeping giant awoken by integrins. Trends Biochem Sci 2011; 36:47. [DOI] [PubMed] [Google Scholar]
- 63.Bouvard C, De Arcangelis A, Dizier B, Galy-Fauroux I, Fischer AM, Georges-Labouesse E, and Helley D: Tie2-dependent knockout of α6 integrin subunit in mice reduces post-ischaemic angiogenesis. Cardiovasc Res 2012; 95:39. [DOI] [PubMed] [Google Scholar]
- 64.Høye AM, Couchman JR, Wewer UM, Fukami K, and Yoneda A: The newcomer in the integrin family: integrin α9 in biology and cancer. Adv Biol Regul 2012; 52:326. [DOI] [PubMed] [Google Scholar]
- 65.Tan SM: The leucocyte β2 (CD18) integrins: the structure, functional regulation and signalling properties. Biosci Rep 2012; 32:241. [DOI] [PubMed] [Google Scholar]
- 66.Johnson MS, Lu N, Denessiouk K, Heino J, and Gullberg D: Integrins during evolution: evolutionary trees and model organisms. Biochim Biophys Acta 2009; 1788:779. [DOI] [PubMed] [Google Scholar]
- 67.Heino J, Huhtala M, Käpylä J, and Johnson MS: Evolution of collagen-based adhesion systems. Int J Biochem Cell Biol 2009; 41:341. [DOI] [PubMed] [Google Scholar]
- 68.Evans R, Patzak I, Svensson L, De Filippo K, Jones K, McDowall A, and Hogg N: Integrins in immunity. J Cell Sci 2009; 122:215. [DOI] [PubMed] [Google Scholar]
- 69.Zutter MM. and Edelson BT: The α2β1 integrin: a novel collectin/C1q receptor. Immunobiology 2007; 212:343. [DOI] [PubMed] [Google Scholar]
- 70.Dumin JA, Dickeson SK, Stricker TP, Bhattacharyya-Pakrasi M, Roby JD, Santoro SA, and Parks WC: Pro-collagenase-1 (matrix metalloproteinase-1) binds the α2β1 integrin upon release from keratinocytes migrating on type I collagen. J Biol Chem 2001; 276:29368. [DOI] [PubMed] [Google Scholar]
- 71.Munger JS. and Sheppard D: Cross talk among TGF-β signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol 2011; 3:a005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ivaska J. and Heino J: Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu Rev Cell Dev Biol 2011; 27:291. [DOI] [PubMed] [Google Scholar]
- 73.Roca-Cusachs P, Iskratsch T, and Sheetz MP: Finding the weakest link: exploring integrin-mediated mechanical molecular pathways. J Cell Sci 2012; 125:3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim C, Ye F, and Ginsberg MH: Regulation of integrin activation. Annu Rev Cell Dev Biol 2011; 27:321. [DOI] [PubMed] [Google Scholar]
- 75.Fu G, Wang W, and Luo BH: Overview: structural biology of integrins. Methods Mol Biol 2012; 757:81. [DOI] [PubMed] [Google Scholar]
- 76.Legate KR. and Fässler R: Mechanisms that regulate adaptor binding to β-integrin cytoplasmic tails. J Cell Sci 2009; 122:187. [DOI] [PubMed] [Google Scholar]
- 77.Anthis NJ. and Campbell ID: The tail of integrin activation. Trends Biochem Sci 2011; 36:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Larjava H, Plow EF, and Wu C: Kindlins: essential regulators of integrin signalling and cell-matrix adhesion. EMBO Rep 2008; 9:1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spassov DS, Wong CH, and Moasser MM: Trask phosphorylation defines the reverse mode of a phosphotyrosine signaling switch that underlies cell anchorage state. Cell Cycle 2011; 10:1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zaidel-Bar R. and Geiger B: The switchable integrin adhesome. J Cell Sci 2010; 123:1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.de Pereda JM, Ortega E, Alonso-García N, Gómez-Hernández M, and Sonnenberg A: Advances and perspectives of the architecture of hemidesmosomes: lessons from structural biology. Cell Adh Migr 2009; 3:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wickström SA, Lange A, Montanez E, and Fässler R: The ILK/PINCH/parvin complex: the kinase is dead, long live the pseudokinase! EMBO J 2010; 29:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Salanueva IJ, Cerezo A, Guadamillas MC, and del Pozo MA: Integrin regulation of caveolin function. J Cell Mol Med 2007; 11:969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Regina Todeschini A. and Hakomori SI: Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim Biophys Acta 2008; 1780:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eden G, Archinti M, Furlan F, Murphy R, and Degryse B: The urokinase receptor interactome. Curr Pharm Des 2011; 17:1874. [DOI] [PubMed] [Google Scholar]
- 86.Bassani S. and Cingolani LA: Tetraspanins: Interactions and interplay with integrins. Int J Biochem Cell Biol 2012; 44:703. [DOI] [PubMed] [Google Scholar]
- 87.Goitre L, Pergolizzi B, Ferro E, Trabalzini L, and Retta SF: Molecular crosstalk between integrins and cadherins: do reactive oxygen species set the talk? J Signal Transduct 2012; 2012:807682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Friedl P. and Wolf K: Plasticity of cell migration: a multiscale tuning model. J Cell Biol 2010; 188:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huttenlocher A. and Horwitz AR: Integrins in cell migration. Cold Spring Harb Perspect Biol 2011; 3:a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bass MD, Williamson RC, Nunan RD, Humphries JD, Byron A, Morgan MR, Martin P, and Humphries MJ: A syndecan-4 hair trigger initiates wound healing through caveolin- and RhoG-regulated integrin endocytosis. Dev Cell 2011; 21:681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thapa N. and Anderson RA: PIP2 signaling, an integrator of cell polarity and vesicle trafficking in directionally migrating cells. Cell Adh Migr 2012; 6:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hamill KJ, Hopkinson SB, Skalli O, and Jones JC: Actinin-4 in keratinocytes regulates motility via an effect on lamellipodia stability and matrix adhesions. FASEB J 2013; 27:546–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ho E. and Dagnino L: Epidermal growth factor induction of front-rear polarity and migration in keratinocytes is mediated by integrin-linked kinase and ELMO2. Mol Biol Cell 2012; 23:492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Werner S, Krieg T, and Smola H: Keratinocyte-fibroblast interactions in wound healing. J Invest Dermatol 2007; 127:998. [DOI] [PubMed] [Google Scholar]
- 95.Stratman AN. and Davis GE: Endothelial cell-pericyte interactions stimulate basement membrane matrix assembly: influence on vascular tube remodeling, maturation, and stabilization. Microsc Microanal 2012; 18:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Poumay Y. and de Rouvroit CL: HB-EGF, the growth factor that accelerates keratinocyte migration, but slows proliferation. J Invest Dermatol 2012; 132:2129. [DOI] [PubMed] [Google Scholar]
- 97.Larjava H, Salo T, Haapasalmi K, Kramer RH, and Heino J: Expression of integrins and basement membrane components by wound keratinocytes. J Clin Invest 1993; 92:1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.D'Alessio S. and Blasi F: The urokinase receptor as an entertainer of signal transduction. Front Biosci 2009; 14:4575. [DOI] [PubMed] [Google Scholar]
- 99.Friedl P. and Gilmour D: Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 2009; 10:445. [DOI] [PubMed] [Google Scholar]
- 100.Hashmi S. and Marinkovich MP: Molecular organization of the basement membrane zone. Clin Dermatol 2011; 29:398. [DOI] [PubMed] [Google Scholar]
- 101.Clark RA, Ashcroft GS, Spencer MJ, Larjava H, and Ferguson MW: Re-epithelialization of normal human excisional wounds is associated with a switch from αvβ5 to αvβ6 integrins. Br J Dermatol 1996; 135:46. [PubMed] [Google Scholar]
- 102.Haapasalmi K, Zhang K, Tonnesen M, Olerud J, Sheppard D, Salo T, Kramer R, Clark RA, Uitto VJ, and Larjava H: Keratinocytes in human wounds express αvβ6 integrin. J Invest Dermatol 1996; 106:42. [DOI] [PubMed] [Google Scholar]
- 103.Pilcher BK, Dumin JA, Sudbeck BD, Krane SM, Welgus HG, and Parks WC: The activity of collagenase-1 is required for keratinocyte migration on a type I collagen matrix. J Cell Biol 1997; 137:1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Frank DE. and Carter WG: Laminin 5 deposition regulates keratinocyte polarization and persistent migration. J Cell Sci 2004; 117:1351. [DOI] [PubMed] [Google Scholar]
- 105.Sehgal BU, DeBiase PJ, Matzno S, Chew TL, Claiborne JN, Hopkinson SB, Russell A, Marinkovich MP, and Jones JC: Integrin β4 regulates migratory behavior of keratinocytes by determining laminin-332 organization. J Biol Chem 2006; 281:35487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Margadant C, Frijns E, Wilhelmsen K, and Sonnenberg A: Regulation of hemidesmosome disassembly by growth factor receptors. Curr Opin Cell Biol 2008; 20:589. [DOI] [PubMed] [Google Scholar]
- 107.Frijns E, Kuikman I, Litjens S, Raspe M, Jalink K, Ports M, Wilhelmsen K, and Sonnenberg A: Phosphorylation of threonine 1736 in the C-terminal tail of integrin β4 contributes to hemidesmosome disassembly. Mol Biol Cell 2012; 23:1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ngora H, Galli UM, Miyazaki K, and Zöller M: Membrane-bound and exosomal metastasis-associated C4.4A promotes migration by associating with the α6β4 integrin and MT1-MMP. Neoplasia 2012; 14:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kligys KR, Wu Y, Hopkinson SB, Kaur S, Platanias LC, and Jones JC: α6β4 integrin, a master regulator of expression of integrins in human keratinocytes. J Biol Chem 2012; 287:17975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Grose R, Hutter C, Bloch W, Thorey I, Watt FM, Fässler R, Brakebusch C, and Werner S: A crucial role of β1 integrins for keratinocyte migration in vitro and during cutaneous wound repair. Development 2002; 129:2303. [DOI] [PubMed] [Google Scholar]
- 111.Chen L, Hughes RA, Baines AJ, Conboy J, Mohandas N, and An X: Protein 4.1R regulates cell adhesion, spreading, migration and motility of mouse keratinocytes by modulating surface expression of β1 integrin. J Cell Sci 2011; 124:2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hodivala-Dilke KM, DiPersio CM, Kreidberg JA, and Hynes RO: Novel roles for α3β1 integrin as a regulator of cytoskeletal assembly and as a trans-dominant inhibitor of integrin receptor function in mouse keratinocytes. J Cell Biol 1998; 142:1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang Z, Morla AO, Vuori K, Bauer JS, Juliano RL, and Ruoslahti E: The αvβ1 integrin functions as a fibronectin receptor but does not support fibronectin matrix assembly and cell migration on fibronectin. J Cell Biol 1993; 122:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Patel GK, Wilson CH, Harding KG, Finlay AY, and Bowden PE: Numerous keratinocyte subtypes involved in wound re-epithelialization. J Invest Dermatol 2006; 126:497. [DOI] [PubMed] [Google Scholar]
- 115.Hosoya A, Lee JM, Cho SW, Kim JY, Shinozaki N, Shibahara T, Shimono M, and Jung HS: Morphological evidence of basal keratinocyte migration during the re-epithelialization process. Histochem Cell Biol 2008; 130:1165. [DOI] [PubMed] [Google Scholar]
- 116.Morasso MI. and Tomic-Canic M: Epidermal stem cells: the cradle of epidermal determination, differentiation and wound healing. Biol Cell 2005; 97:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bata-Csorgo Z, Cooper KD, Ting KM, Voorhees JJ, and Hammerberg C: Fibronectin and α5 integrin regulate keratinocyte cell cycling. A mechanism for increased fibronectin potentiation of T cell lymphokine-driven keratinocyte hyperproliferation in psoriasis. J Clin Invest 1998; 101:1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Eslami A, Gallant-Behm CL, Hart DA, Wiebe C, Honardoust D, Gardner H, Häkkinen L, and Larjava HS: Expression of integrin αvβ6 and TGF-β in scarless vs scar-forming wound healing. J Histochem Cytochem 2009; 57:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Curnis F, Gasparri AM, Longhi R, Colombo B, D'Alessio S, Pastorino F, Ponzoni M, and Corti A: Chromogranin A binds to αvβ6-integrin and promotes wound healing in mice. Cell Mol Life Sci 2012; 69:2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chanson M, Derouette JP, Roth I, Foglia B, Scerri I, Dudez T, and Kwak BR: Gap junctional communication in tissue inflammation and repair. Biochim Biophys Acta 2005; 1711:197. [DOI] [PubMed] [Google Scholar]
- 121.Wong VW, Akaishi S, Longaker MT, and Gurtner GC: Pushing back: wound mechanotransduction in repair and regeneration. J Invest Dermatol 2011; 131:2186. [DOI] [PubMed] [Google Scholar]
- 122.Lokmic Z, Musyoka J, Hewitson TD, and Darby IA: Hypoxia and hypoxia signaling in tissue repair and fibrosis. Int Rev Cell Mol Biol 2012; 296:139. [DOI] [PubMed] [Google Scholar]
- 123.Xueyong L, Shaozong C, Wangzhou L, Yuejun L, Xiaoxing L, Jing L, Yanli W, and Jinqing L: Differentiation of the pericyte in wound healing: The precursor, the process, and the role of the vascular endothelial cell. Wound Repair Regen 2008; 16:346. [DOI] [PubMed] [Google Scholar]
- 124.Ghersi G, Dong H, Goldstein LA, Yeh Y, Häkkinen L, Larjava HS, and Chen WT: Regulation of fibroblast migration on collagenous matrix by a cell surface peptidase complex. J Biol Chem 2002; 277:29231. [DOI] [PubMed] [Google Scholar]
- 125.Hinz B: Formation and function of the myofibroblast during tissue repair. J Invest Dermatol 2007; 127:526. [DOI] [PubMed] [Google Scholar]
- 126.Kato A, Okamoto O, Ishikawa K, Sumiyoshi H, Matsuo N, Yoshioka H, Nomizu M, Shimada T, and Fujiwara S: Dermatopontin interacts with fibronectin, promotes fibronectin fibril formation, and enhances cell adhesion. J Biol Chem 2011; 286:14861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brooks R, Williamson R, and Bass M: Syndecan-4 independently regulates multiple small GTPases to promote fibroblast migration during wound healing. Small GTPases 2012; 3:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Häkkinen L, Heino J, Koivisto L, and Larjava H: Altered interaction of human granulation-tissue fibroblasts with fibronectin is regulated by α5β1 integrin. Biochim Biophys Acta 1994; 1224:33. [DOI] [PubMed] [Google Scholar]
- 129.Liu S, Xu SW, Blumbach K, Eastwood M, Denton CP, Eckes B, Krieg T, Abraham DJ, and Leask A: Expression of integrin β1 by fibroblasts is required for tissue repair in vivo. J Cell Sci 2010; 123:3674. [DOI] [PubMed] [Google Scholar]
- 130.Lygoe KA, Wall I, Stephens P, and Lewis MP: Role of vitronectin and fibronectin receptors in oral mucosal and dermal myofibroblast differentiation. Biol Cell 2007; 99:601. [DOI] [PubMed] [Google Scholar]
- 131.Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, and Thomas PE: Myofibroblast differentiation by transforming growth factor-β1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem 2003; 278:12384. [DOI] [PubMed] [Google Scholar]
- 132.Vi L, de Lasa C, DiGuglielmo GM, and Dagnino L: Integrin-linked kinase is required for TGF-β1 induction of dermal myofibroblast differentiation. J Invest Dermatol 2011; 131:586. [DOI] [PubMed] [Google Scholar]
- 133.He Y, Esser P, Schacht V, Bruckner-Tuderman L, and Has C: Role of kindlin-2 in fibroblast functions: implications for wound healing. J Invest Dermatol 2011; 131:245. [DOI] [PubMed] [Google Scholar]
- 134.Blumbach K, Zweers MC, Brunner G, Peters AS, Schmitz M, Schulz JN, Schild A, Denton CP, Sakai T, Fässler R, Krieg T, and Eckes B: Defective granulation tissue formation in mice with specific ablation of integrin-linked kinase in fibroblasts—role of TGFβ1 levels and RhoA activity. J Cell Sci 2010; 123:3872. [DOI] [PubMed] [Google Scholar]
- 135.Carracedo S, Lu N, Popova SN, Jonsson R, Eckes B, and Gullberg D: The fibroblast integrin α11β1 is induced in a mechanosensitive manner involving activin A and regulates myofibroblast differentiation. J Biol Chem 2010; 285:10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Talior-Volodarsky I, Connelly KA, Arora PD, Gullberg D, and McCulloch CA: α11 integrin stimulates myofibroblast differentiation in diabetic cardiomyopathy. Cardiovasc Res 2012; 96:265. [DOI] [PubMed] [Google Scholar]
- 137.Lygoe KA, Norman JT, Marshall JF, and Lewis MP: αv integrins play an important role in myofibroblast differentiation. Wound Repair Regen 2004; 12:461. [DOI] [PubMed] [Google Scholar]
- 138.Asano Y, Ihn H, Yamane K, Jinnin M, and Tamaki K: Increased expression of integrin αvβ5 induces the myofibroblastic differentiation of dermal fibroblasts. Am J Pathol 2006; 168:499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang L, Pedroja BS, Meyers EE, Garcia AL, Twining SS, and Bernstein AM: Degradation of internalized αvβ5 integrin is controlled by uPAR bound uPA: effect on β1 integrin activity and α-SMA stress fiber assembly. PLoS One 2012; 7:e33915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jones C. and Ehrlich HP: Fibroblast expression of α-smooth muscle actin, α2β1 integrin and αvβ3 integrin: influence of surface rigidity. Exp Mol Pathol 2011; 91:394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sarrazy V, Billet F, Micallef L, Coulomb B, and Desmoulière A: Mechanisms of pathological scarring: role of myofibroblasts and current developments. Wound Repair Regen 2011; 19Suppl 1:s10. [DOI] [PubMed] [Google Scholar]
- 142.Jun JI. and Lau LF: Cellular senescence controls fibrosis in wound healing. Aging (Albany, NY) 2010; 2:627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mak K, Manji A, Gallant-Behm C, Wiebe C, Hart DA, Larjava H, and Häkkinen L: Scarless healing of oral mucosa is characterized by faster resolution of inflammation and control of myofibroblast action compared to skin wounds in the red Duroc pig model. J Dermatol Sci 2009; 6:168. [DOI] [PubMed] [Google Scholar]
- 144.van der Flier A, Badu-Nkansah K, Whittaker CA, Crowley D, Bronson RT, Lacy-Hulbert A, and Hynes RO: Endothelial α5 and αv integrins cooperate in remodeling of the vasculature during development. Development 2010; 137:2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Robinson SD. and Hodivala-Dilke KM: The role of β3-integrins in tumor angiogenesis: context is everything. Curr Opin Cell Biol 2011; 23:630. [DOI] [PubMed] [Google Scholar]
- 146.Widgerow AD: Chronic wounds—is cellular ‘reception’ at fault? Examining integrins and intracellular signalling. Int Wound J 2013; 10:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Häkkinen L, Koivisto L, Gardner H, Saarialho-Kere U, Carroll JM, Lakso M, Rauvala H, Laato M, Heino J, and Larjava H: Increased expression of β6-integrin in skin leads to spontaneous development of chronic wounds. Am J Pathol 2004; 164:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Morgan MR, Thomas GJ, Russell A, Hart IR, and Marshall JF: The integrin cytoplasmic-tail motif EKQKVDLSTDC is sufficient to promote tumor cell invasion mediated by matrix metalloproteinase (MMP)-2 or MMP-9. J Biol Chem 2004; 279:26533. [DOI] [PubMed] [Google Scholar]
- 149.Minhas U, Martin TA, Ruge F, Harding KG, and Jiang WG: Pattern of expression of CCN family members Cyr61, CTGF and NOV in human acute and chronic wounds. Exp Ther Med 2011; 2:641. [DOI] [PMC free article] [PubMed] [Google Scholar]