

Abstract
Objective
Angiotensin converting enzyme 2 (ACE2) cleaves angiotensin II (AngII) to form angiotensin-(1-7) (Ang-(1-7)), which generally opposes effects of AngII. AngII infusion into hypercholesterolemic male mice induces formation of abdominal aortic aneurysms (AAAs). This study tests the hypothesis that deficiency of ACE2 promotes AngII-induced AAAs, while ACE2 activation suppresses aneurysm formation.
Approach and Results
ACE2 protein was detectable by immunostaining in mice and human AAAs. Whole body deficiency of ACE2 significantly increased aortic lumen diameters and external diameters of suprarenal aortas from AngII-infused mice. Conversely, ACE2 deficiency in bone marrow-derived cells had no effect on AngII-induced AAAs. In contrast to AngII-induced AAAs, ACE2 deficiency had no significant effect on external aortic diameters of elastase-induced AAAs. Since ACE2 deficiency promoted AAA formation in AngII-infused mice, we determined if ACE2 activation suppressed AAAs. ACE2 activation by administration of diminazine aceturate (DIZE, 30 mg/kg/day) to Ldlr−/− mice increased kidney ACE2 mRNA abundance and activity and elevated plasma Ang-(1-7) concentrations. Unexpectedly, administration of DIZE significantly reduced total sera cholesterol and VLDL-cholesterol concentrations. Notably, DIZE significantly decreased aortic lumen diameters and aortic external diameters of AngII-infused mice resulting in a marked reduction in AAA incidence (from 73 to 29%). None of these effects of DIZE were observed in the Ace2−/y mice.
Conclusions
These results demonstrate that ACE2 exerts a modulatory role in AngII-induced AAA formation, and that therapeutic stimulation of ACE2 could be a benefit to reduce AAA expansion and rupture in patients with an activated renin-angiotensin system.
Keywords: ACE2, AAAs, angiotensin II, hypercholesterolemia, angiotensin-(1-7)
Introduction
Abdominal aortic aneurysms (AAAs) affect more than 1 million people in the United States1, 2 There are no validated medical therapies that favorably impact aneurysm growth and rupture, making surgery the only available treatment option for AAAs. As AAA size increases, the risk of rupture increases, and ruptured aneurysms contribute to mortality rates of 60–80%.3–5 Therefore, there is a need for research to define mechanisms of AAA formation so that potential drug targets for this devastating disease can be identified.
Infusion of angiotensin II (AngII) into male mice is a commonly used animal model to gain insight into mechanisms of human AAAs.6, 7 Similar to the human disease, AAAs that develop in response to AngII exhibit progressive leukocyte accumulation, extracellular matrix degradation, luminal expansion and thrombus.8 Manipulation of the renin-angiotensin system (RAS) pharmacologically through use of angiotensin type 1 receptor (AT1R) antagonists,9 or genetically through AT1aR deficiency,10 substantially reduces AngII-induced AAA formation. Recent studies demonstrate that inhibition of the RAS also suppressed experimental aneurysms in an elastase-induced model.11 Currently, it is unclear whether manipulation of the RAS is an effective mode for AAA therapy in humans.12–16 However, several ongoing clinical trials (NCT01118520, NCT001904981) are either actively recruiting or in the process of evaluating efficacy of angiotensin converting enzyme inhibitors or angiotensin receptor antagonists on either the size or expansion rate of human AAAs. Moreover, a recent study demonstrated that long-term blockade of the RAS in hypertensive patients attenuated expansion of non-aneurysmal abdominal aorta, suggesting that RAS blockade given before advancement of aortic remodeling may slow the development of AAAs.17
Angiotensin converting enzyme-2 (ACE2) is a homolog of ACE that converts AngII to angiotensin-(1-7((Ang-(1-7)).18, 19 The ability of ACE2 to degrade a vasoconstrictor (AngII) and produce a vasodilator (Ang-(1-7)) provides rationale for this enzyme as a therapeutic target. Previous studies in our laboratory demonstrated that deficiency of ACE2 promoted hypercholesterolemia-induced atherosclerosis.20 Effects of ACE2 deficiency to promote atherosclerosis appear to result from elevations in AngII as well as from reductions in Ang-(1-7) concentrations, since co-infusion of Ang-(1-7) with AngII in Ldlr−/− mice reduced atherosclerosis.20 While these results suggest that manipulation of ACE2 influences atherosclerotic lesion formation, the role of ACE2 as a modulator of AAA formation and severity has not been defined.
Macrophages play an important role in the formation and progression of AngII-induced AAAs.10, 21 Previous studies demonstrated that bone marrow deficiency of ACE2 promoted diet-induced atherosclerosis, suggesting that leukocyte ACE2 suppresses atherosclerotic lesion formation.20 Several studies have shown that deletion of specific proteins in bone marrow-derived cells can enhance22–24 or attenuate10, 25, 26 the formation of AAAs. It is unclear if leukocyte ACE2 also modulates the susceptibility to AngII-induced AAAs.
Since ACE2 catabolizes AngII to Ang-(1-7), activators of ACE2 are potential therapeutics in treatment of AngII-induced diseases. Diminazene aceturate (DIZE) has been described as an activator of ACE227–29 that lowered blood pressure and endothelin-1-induced ischemic stroke when administered centrally to rats,30 and that attenuated pulmonary hypertension in rats.29 This compound has not been examined in AngII-induced AAAs. In this study, we first determined if ACE2 localizes to human and murine AAAs. Then, we determined effects of whole body ACE2 deficiency on AngII-induced AAAs in Ldlr−/− mice. As a potential therapeutic modality to suppress the RAS and thereby blunt AAA formation, we administered DIZE to ACE2 wild-type (Ace2+/y) or deficient mice (Ace2−/y) to determine if ACE2 activation reduced AngII-induced AAAs. We also examined effects of whole body ACE2 deficiency on elastase-induced AAAs in Ldlr−/− mice.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement (http://atvb.ahajournals.org).
Results
ACE2 Localized to Murine and Human AAAs
In serial sections of abdominal aortas from saline or AngII-infused Ldlr−/− mice, ACE2 immunostaining localized predominately to the intima and adventitia (Figure 1). In AAA regions exhibiting a break in medial elastin, ACE2 immunostaining was pronounced in adventitia. ACE2 immunostaining was performed on tissue sections from human abdominal aortas and in sections from patients with AAAs (Figure 2, Supplemental Figure I). ACE2 localized to the intima and media of both non-and aneurysmal abdominal aorta, and was also present in vaso vasorum (Supplemental Figure IA in the online-only Data Supplement). In human AAA tissue sections, ACE2 was present in intimal plaque (arrow, Figure 2) with abundant immunostaining in cells of inflammatory foci (dotted arrow, Figure 2, Supplemental Figure IB in the online-only Data Supplement). CD68 positive cells also stained positive for ACE2 in inflammatory foci of human AAAs (lower panels of Supplemental Figure I in the online-only Data Supplement).
Figure 1.
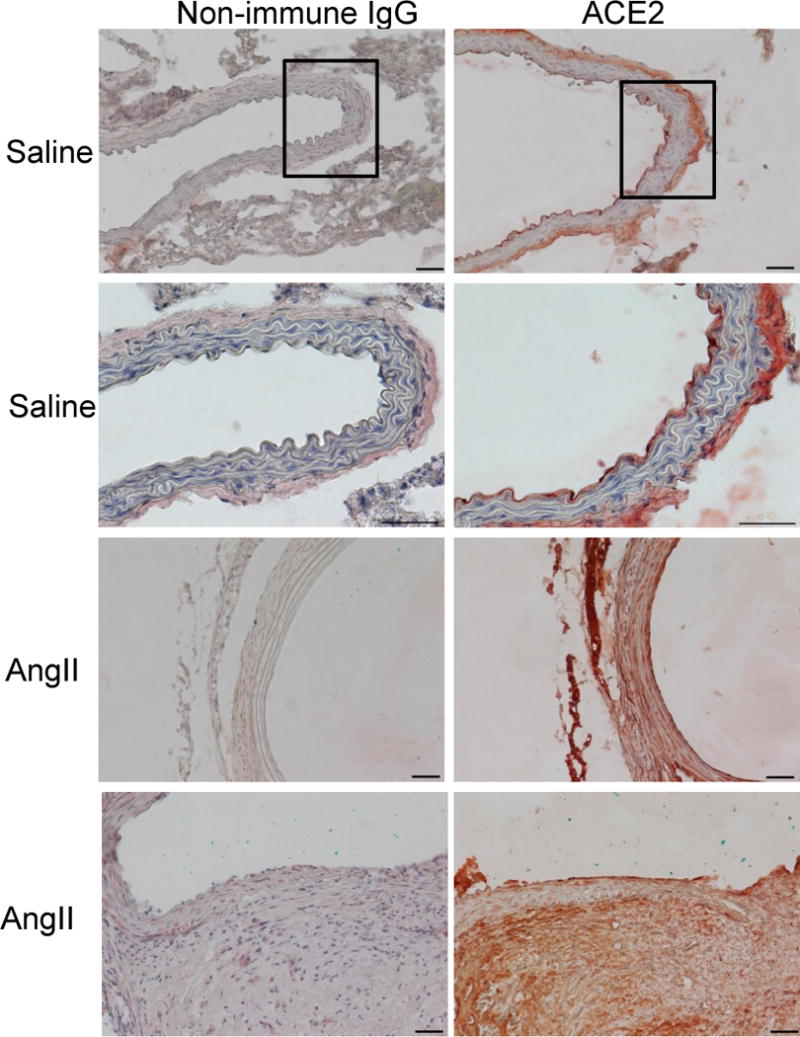
Localization of ACE2 to abdominal aortas of Ldlr−/− mice infused with either saline or AngII. Left, Non-immune IgG. Right, ACE2 immunostaining in abdominal aortic sections from saline (top four panels) versus AngII-infused mice (bottom four panels). ACE2 staining was present in all layers of the vascular wall in aortic sections from mice in each group. Black box represents area that magnified in bottom panels for saline-infused mice. Bottom panels for AngII-infused mice at medial break display ACE2 immunostaining. Scale bars represent 50 μm.
Figure 2.
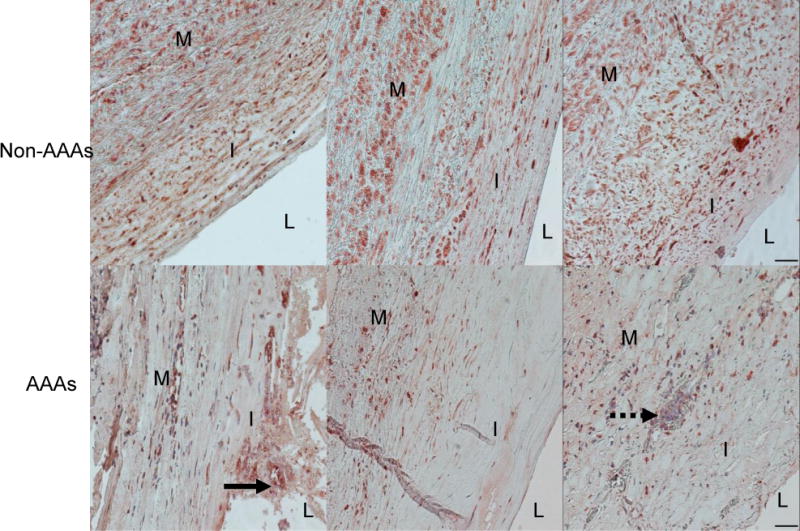
ACE2 localization to human abdominal aortas and AAAs. Top, ACE2 immunostaining of tissue sections from human abdominal aortas (N = 3 non-AAA aortas) where L = lumen, I = intima, and M = media. Bottom, ACE2 immunostaining of tissue sections from AAAs (N = 3). ACE2 staining was present in an atherosclerotic plaque (arrow) and in an inflammatory foci (dotted arrow) of AAA tissue sections. Scale bars represent 50 μm.
Whole Body ACE2 Deficiency Increased AngII-Induced AAAs
To determine the contribution of ACE2 to AngII-induced AAAs, male Ldlr−/− mice that were either Ace2+/y or Ace2−/y were infused with AngII. ACE2 deficiency had no significant effect on body weight, plasma renin concentrations, serum ACE activity, or sera lipid concentrations in AngII-infused mice (Table I in the online-only Data Supplement). However, plasma AngII concentrations were significantly increased in AngII-infused Ace2−/y compared to Ace2+/y mice (Table I in the online-only Data Supplement; P < 0.05). Systolic blood pressure was increased significantly by AngII infusions in both genotypes, with significantly increased pressures in Ace2−/y compared to Ace2+/y mice (Table I in the online-only Data Supplement). AngII infusion significantly increased suprarenal aortic lumen diameters at day 14 and 28 compared to baseline in both genotypes (Figure 3A; P < 0.05). In addition, lumen diameters were increased significantly in AngII-infused Ace2−/y compared to Ace2+/y mice on days 14 and 28 (P < 0.05). Similarly, external diameters of AAAs were significantly increased in AngII-infused Ace2−/y compared to Ace2+/y mice (Figure 3B; P < 0.05). An increase in AAA size is illustrated in representative aortas from Ace2−/y compared to Ace2+/y mice (Figure 3C); however, Gomori-trichrome staining was similar in AAA sections from either genotype (Supplemental Figure II in the online-only Data Supplement). We performed CD68 immunostaining in AAA sections from Ace2+/y and Ace2−/y mice (Supplemental Figure III in the online-only Data Supplement). At sites of medial degeneration, CD68 immunostaining was more pronounced in AAA sections from Ace2−/y compared to Ace2+/y mice.
Figure 3.
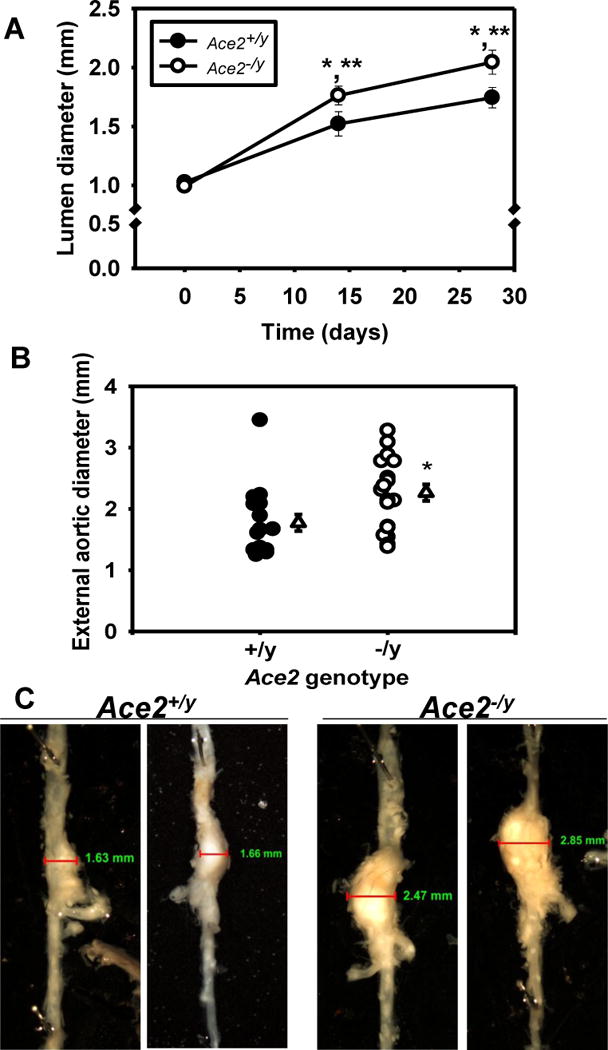
Whole body ACE2 deficiency promoted AngII-induced AAAs. A, Aortic lumen diameter was quantified by ultrasound in Ace2+/y or−/y Ldlr−/− mice on day 0, 14, and 27 of AngII-infusion (N = 17–19 mice/genotype). B, External diameters of suprarenal aortas. Circles represent individual mice of each genotype, while triangles represent mean ± SEM (N = 17–19 mice/genotype). C, Representative abdominal aortas from Ace2+/y and −/y Ldlr−/− mice. *, P < 0.05 compared to Day 0 ultrasound. **, P < 0.05 compared to Ace2+/y.
We also examined effects of ACE2 deficiency on dilations of the infrarenal aorta in response to intra-aortic elastase perfusion.31 The percent increase in aortic diameter, quantified 14 days after elastase perfusion, was not significantly different between Ace2+/y and Ace2−/y Ldlr−/− mice (Supplemental Figure IV in the online-only Data Supplement).
Leukocyte ACE2 Deficiency had no Effect on AngII-induced AAAs
Previous studies demonstrated that ACE2 deficiency in whole bone marrow cells augmented diet-induced atherosclerosis.20 Since macrophages are a prominent cell type in AngII-induced AAAs,8 we transplanted whole bone marrow cells from Ace2+/y and Ace2−/y mice into lethally irradiated Ldlr−/− recipients prior to infusing with AngII. Bone marrow from recipient mice exhibited donor mouse genotypes at study endpoint (Supplemental Figure V in the online-only Data Supplement). Deficiency of ACE2 in bone marrow-derived cells had no significant effect on body weight, systolic blood pressure, white blood cell count (WBC), plasma renin concentrations, or sera cholesterol concentrations in AngII-infused Ldlr−/− mice (data not shown on WBC and Table I in the online-only Data Supplement). In addition, deficiency of ACE2 in whole bone marrow cells had no effect on suprarenal aortic lumen diameters of AngII-infused Ldlr−/− mice (donor genotype: Ace2+/y, 1.31 ± 0.06; Ace2−/y, 1.32 ± 0.07 mm; P > 0.05). Similarly, external diameters of suprarenal aortas were not significantly different between donor genotypes (Supplemental Figure VI in the online-only Data Supplement; P > 0.05).
DIZE Reduced AngII-Induced AAAs in Ace2+/y, but not in Ace2−/y Mice
Since ACE2 deficiency promoted AngII-induced AAAs, this suggests that activation of ACE2 may reduce AAA formation and severity. We used DIZE as a previously described ACE2 activator27, 29, 30 to define if ACE2 activation blunts AAA formation and/or severity. Moreover, to address specificity of DIZE, we determined effects of the compound on AngII-induced AAAs in Ace2+/y and Ace2−/y Ldlr−/− mice. Pilot studies determined concentration-dependent effects of DIZE to activate ACE2 when infused subcutaneously by osmotic minipumps to Ldlr−/− mice (Supplemental Figure VII in the online-only Data Supplement). Following 7 days of infusion of 30 mg/kg/day of DIZE, plasma Ang-(1-7) concentrations, kidney ACE2 mRNA abundance and activity were increased significantly compared to vehicle controls (Supplemental Figure VII in the online-only Data Supplement; P < 0.05). Unfortunately, when DIZE was placed in 28 day osmotic minipumps, the stability of the compound decreased markedly after 7 days (data not shown). Therefore, we administered vehicle or DIZE with a drug stabilizer (anti-pyrine) by daily intramuscular injections at a dose of 30 mg/kg/day.
Administration of DIZE did not significantly influence systolic blood pressure, plasma renin or AngII concentrations or sera triglyceride concentrations in AngII-infused Ace2+/y or Ace2−/y mice (Table 1 in the online-only Data Supplement; P > 0.05). While DIZE administration had no significant effect on plasma Ang-(1-7) concentrations (data not shown), kidney ACE2 mRNA abundance and activity were increased significantly in Ace2+/y mice (Supplemental Figure VIII in the online-only Data Supplement). Body weight was decreased significantly by DIZE administration in Ace2+/y and Ace2−/y mice (VEH Ace2+/y 27 ± 1g; DIZE 30 mg/kg/day Ace2+/y, 23 ± 0.4g; DIZE Ace2−/y, 23 ± 1g ; P < 0.05 compared to VEH Ace2+/y). Unexpectedly, sera cholesterol concentrations were decreased significantly in Ace2+/y, but not in Ace2−/y mice administered DIZE compared to vehicle controls (Figure 4A; P < 0.05 compared to VEH Ace2+/y mice). The decrease in serum total cholesterol concentrations was attributable to decreased VLDL-cholesterol concentrations by DIZE administration in Ace2+/y, but not in Ace2−/y mice (Figure 4B,C; P < 0.05).
Figure 4.
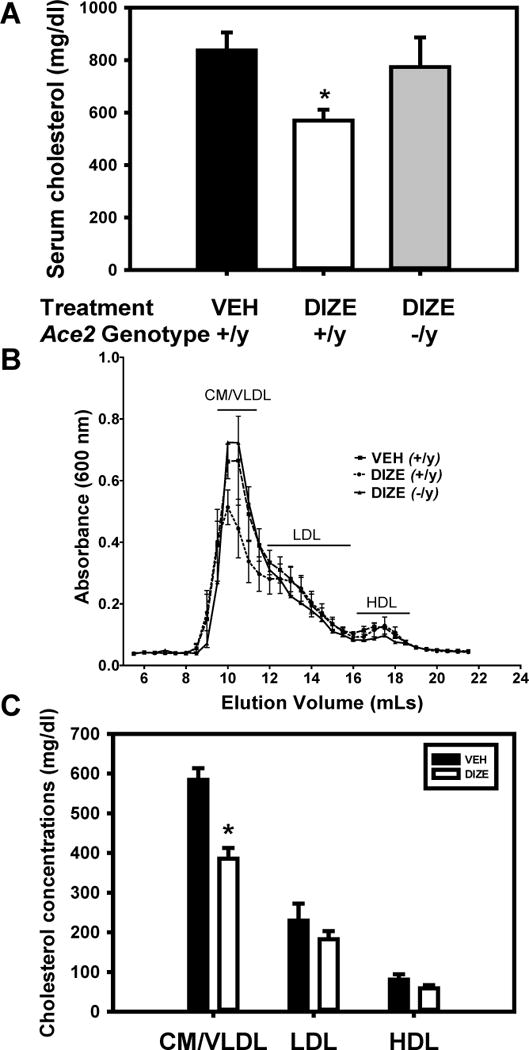
DIZE administration reduced VLDL-cholesterol concentrations in Ace2+/y Ldlr−/− male mice. A, Sera cholesterol concentrations of mice administered either vehicle (VEH, Ace2+/y) or ACE2 activator (DIZE, Ace2+/y or−/y). Data are mean ± SEM from N = 11–15 mice in each group. B, Size exclusion chromatographic resolution of serum lipoproteins from mice administered either VEH (Ace2+/y) or DIZE (Ace2+/y or −/y; N = 4 per group). C, Sera cholesterol concentrations in CM (chylomicrons)/VLDL, LDL, and HDL calculated from areas under the curve from B above. *, P < 0.05 compared to VEH.
DIZE administration decreased significantly lumen diameters (Figure 5A; P < 0.05) and external diameters of suprarenal aortas (Figure 5B; P < 0.05) and also decreased significantly AAA incidence (from 73 to 29%; Figure 5C; P < 0.05). In contrast, administration of DIZE to ACE2 deficient male mice infused with AngII had no effect on aortic lumen diameters (Ace2+/y, VEH: 1.77 ± 0.09, Ace2−/y, DIZE: 1.60 ± 0.11 mm; P > 0.05). Kaplan-Meier analysis revealed that DIZE administration to Ace2+/y mice prevented AAA ruptures in comparison to ruptures observed in vehicle Ace2+/y and DIZE Ace2−/y mice (Supplemental Figure IX in online-only Data Supplement; P < 0.05).
Figure 5.
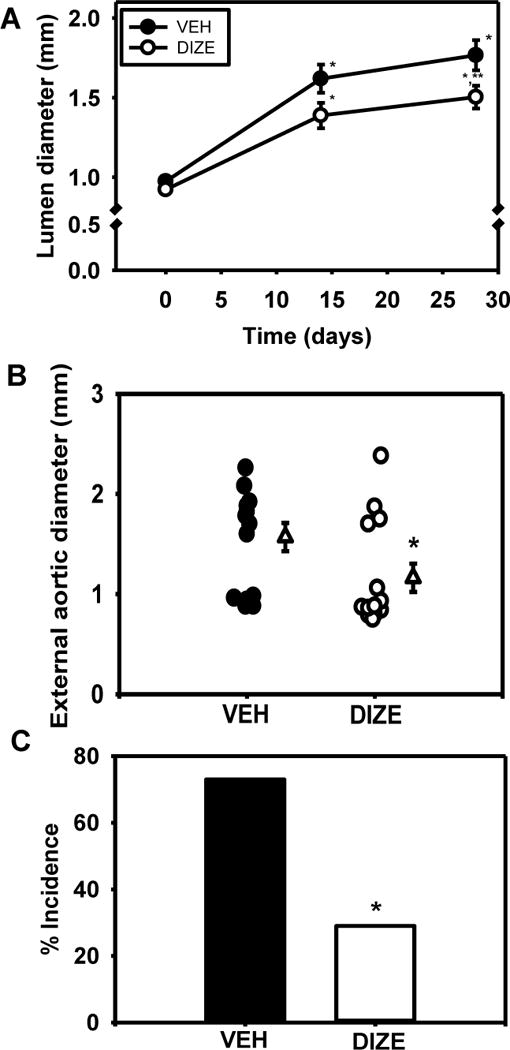
Activation of ACE2 by DIZE administration reduced AngII-induced AAAs. A, Aortic lumen diameter was quantified by ultrasound on day 0, 7, 14, and 28 of AngII infusion in mice administered vehicle (VEH) or diminazene aceturate (DIZE, 15 or 30 mg/kg/day). Data are mean ± SEM of N = 7–14 mice/group. *, P < 0.05 compared to Day 0. **, P < 0.05 compared to VEH. B, External diameters of suprarenal aortas from mice administered VEH or DIZE. Circles are individual mice while triangles represent mean ± SEM. C, AAA incidence in mice from each group. *, P<0.05 compared to VEH.
Discussion
We first demonstrated that ACE2 localized to murine AngII-induced AAAs and human AAAs. To determine if ACE2 modulates AAA formation, we examined the effects of ACE2 deficiency on formation and severity of AngII-induced AAAs. Whole body ACE2 deficiency promoted both formation and the severity of AngII-induced AAAs. In contrast, whole body ACE2 deficiency had no effect on elastase-induced AAAs. Leukocyte ACE2 appeared to play no role, as ACE2 deficiency in whole bone marrow cells had no effect on AngII-induced AAAs. Since ACE2 deficiency promoted AAA formation, we turned to therapeutic activation of ACE2 to suppress AAA formation and severity. Administration of the ACE2 activator, DIZE, reduced the size, severity and incidence of AngII-induced AAAs in wild type, but not in ACE2-deficient mice. These results suggest that ACE2 activation of ACE2 may serve as a novel therapeutic target in the treatment of AAAs in patients with an activated RAS.
An interesting feature of ACE2 is that it catabolizes AngII to form Ang-(1-7), two angiotensin peptides that counterbalance each other in vaso-contractile versus vaso-relaxation, respectively.32–40 This makes activation of ACE2 an attractive target to reduce the relative balance of AngII/Ang-(1-7) and thereby inhibit vascular disease.41 In this study, while whole body deficiency of ACE2 promoted AngII-induced AAAs, ACE2 activation inhibited AngII-induced AAAs. Beneficial effects of ACE2 manipulation against AAA formation could result from regulation of AngII concentrations (e.g., plasma concentrations increased in ACE2 deficient mice), or from regulation of Ang-(1-7) concentrations (e.g., plasma concentrations increased with DIZE administration). Plasma AngII concentrations were increased by ACE2 deficiency, but not altered in AngII-infused mice administered DIZE. However, both manipulations influenced AngII-induced AAAs, suggesting that plasma AngII concentrations are not the primary mechanism of action of ACE2. In contrast, whole body ACE2 deficiency had no effect on AAAs induced by intra-aortic elastase perfusion. Recent results demonstrate that blockade of AT1 receptors provides benefit against elastase-induced AAA formation.11 Moreover, whole body AT1a receptor deficiency totally abolished AngII-induced AAAs,10 demonstrating a critical role for AngII effects at AT1 receptors in both of these experimental AAA models. If ACE2 deficiency augmented AngII-induced AAAs by elevating AngII concentrations available to act at AT1 receptors, then we would have anticipated that ACE2 deficiency would have similar effects in both experimental AAA models (similar to AT1 receptor blockade). Given the diverging effects of ACE2 deficiency between the AngII- and elastase-induced AAAs, these results suggest that effects of ACE2 may be attributed to regulation of Ang-(1-7).
In contrast to previous findings demonstrating a role for leukocyte ACE2 in diet-induced atherosclerosis,20 results from this study demonstrate that leukocyte ACE2 deficiency had no effect on AngII-induced AAAs. However, AAAs from ACE2 deficient mice exhibited pronounced macrophage immunostaining at sites of medial degeneration. Since leukocyte ACE2 deficiency had no effect on AngII-induced AAAs, it is likely that ACE2 deficiency in other cell types (e.g., vascular wall cells) promoted macrophage recruitment to developing AAAs. It is clear that angiotensin type 1a receptors (AT1aR) are required for AngII-induced AAAs.10 Similar to the lack of effect of leukocyte ACE2 deficiency on AngII-induced AAAs, deficiency of AT1aR on leukocytes did not significantly influence AngII-induced AAAs in Ldlr−/− mice.10 In addition, recent studies demonstrated that neither endothelial nor smooth muscle cell AT1aR deficiency influenced AngII-induced AAAs in male Ldlr−/− mice.42 Thus, the primary cell type(s) responding to AngII to induce AAA formation have not been identified. Future studies should address cell type(s) expressing ACE2 that modulate local angiotensin peptide concentrations to promote AAA formation and severity.
Whole body deficiency of ACE2 in the present study resulted in an increased blood pressure response to AngII infusion. In mice, infusion of norepinephrine to hypercholesterolemic male mice at a rate that increased blood pressure to a similar extent as observed in AngII-infused mice did not result in AAA formation.43 Moreover, administration of the vasodilator hydralazine to AngII-infused mice had no significant effect on AAA formation. Given these findings, it is unlikely that blood pressure increases in ACE2 deficient mice infused with AngII contributed to increases in AAA severity.
Since there are no proven medical therapies that modify AAA growth and rupture, an important finding of the present study was the ability of ACE2 activation to reduce the formation and severity of AngII-induced AAAs. We used DIZE, a compound previously shown to activate ACE2 and lower blood pressure and endothelin-1-induced ischemic stroke when administered centrally to rats.30, 44 Recent studies demonstrated that administration of DIZE not only prevented the development of pulmonary hypertension in rats, but also arrests the progression of established pulmonary hypertension.29 However, recent studies indicate that DIZE may also exhibit ACE2-independent effects.45 We included the drug stabilizer, anti-pyrine to improve the stability of DIZE since the drug has a short half-life.46 Our results also demonstrate that DIZE increased ACE2 mRNA abundance and elevated plasma Ang-(1-7) concentrations. These findings are in agreement with results from studies using pulmonary hypertensive rats, where DIZE administration significantly increased the ACE2/ACE ratio.29 Importantly, administration of DIZE to ACE2 deficient mice had no effect on AngII-induced AAAs, supporting an ACE2-dependent mechanism of the compound. In addition to efficacy to decrease AAA formation and severity, DIZE administration resulted in other ACE2-dependent (sera cholesterol) as well as independent (body weight) effects, suggesting ancillary properties of the compound. These results support the need to include ACE2-deficient mice in studies examining novel ACE2 activators to define the specificity of drug action on the parameter of interest. In addition, these results support further studies examining efficacy of ACE2 activation to retard AAA progression.
It is noteworthy that DIZE administration reduced sera cholesterol concentrations, specifically VLDL-cholesterol, in wild type but not in ACE2-deficient mice. Mechanisms responsible for ACE2-mediated regulation of sera cholesterol concentrations are unclear. Recent studies identified novel effects of ACE2 to regulate dietary amino acid homeostasis in the gut, effects which were independent of ACE2-mediated regulation of the RAS.47 In addition to the gut, ACE2 is also expressed in liver48 where it has been suggested to modulate cirrhosis. It is conceivable that ACE2 exerts unidentified effects, potentially related to the gut and/or liver, to influence cholesterol absorption and/or metabolism. Effects of DIZE to reduce sera cholesterol concentrations may result from increased levels of systemic Ang-(1-7), as recent studies demonstrate that administration of an oral formulation of Ang-(1-7) to high fat-fed mice significantly reduced sera cholesterol concentrations.49 While beneficial in the treatment of atherosclerosis, reductions (32%) in sera cholesterol are unlikely to be the mediator of DIZE’s ability to decrease AAA formation in response to AngII, since sera cholesterol concentrations in mice administered DIZE (571 mg/dL) remained considerably higher than those typically present in C57BL/6 mice (< 80 mg/dL). Even at low levels of sera cholesterol concentration, male C57BL/6 mice continue to exhibit some susceptibility (20–30%) to AngII-induced AAAs.50, 51
In conclusion, results from these studies support a role for ACE2 as a modulator of the formation and severity of AngII-induced AAAs. Whole body, but not leukocyte deficiency of ACE2 promoted AngII-induced AAAs, suggesting that ACE2 in other cell types regulates AAA formation and severity. Finally, activation of ACE2 suppressed both the formation and severity of AngII-induced AAAs, identifying ACE2 as a potential novel target in the medical therapy of AAAs in patients with an activated RAS.
Supplementary Material
Significance.
Abdominal aortic aneurysms (AAAs) and ruptures are associated with significant mortality. Currently, there are no drug therapies that are effective to slow the progression of AAAs. Infusion of angiotensin II (AngII) induces AAA formation in experimental mice. Angiotensin converting enzyme-2 (ACE2) catabolizes AngII to form its functional antagonist, angiotensin-(1-7). Our results demonstrate that ACE2 localizes to murine and human AAAs. To determine the functional role of ACE2 in AAA formation and severity, we examined effects of ACE2 deficiency versus ACE2 activation on AngII-induced AAAs. Deficiency of ACE2 augmented AAA formation and severity, while increasing ACE2 activity therapeutically suppressed AAA formation and severity. However, ACE2 deficiency had no effect on elastase-induced AAAs. These results suggest that ACE2 activators may serve as a novel therapeutic modality to reduce the formation and severity of AAAs in patients with an activated renin-angiotensin system.
Acknowledgments
We acknowledge Victoria English for quantification of plasma renin concentrations. We acknowledge human aorta samples (source of tissue, National Heart, Lung, and Blood Institute K08 HL84004 (JAC) and P50 HL083762 (RWT)). We acknowledge Dr. William O’Connor (University of Kentucky, Lexington, KY) for help with analysis of human aorta samples.
Funding Sources
Funding for this project was through NIH R01 HL107326 (LAC), NIH R01 HL62846 (AD), Alaska Kidney Foundation Research Grant (SBG), through cores supported by NIH 8 P20 GM103527 (LAC), and a Ruth L. Kirschstein National Research Service Award (F32 HL095281, SET).
Nonstandard Abbreviations and Acronyms
- AAA
Abdominal aortic aneurysm
- AT1aR
Angiotensin receptor 1a
- AngII
Angiotensin II
- ACE2
Angiotensin-converting enzyme 2
- Ang-(1-7)
angiotensin-(1-7)
- CD68
Macrosialin
- CM
Chylomicron
- DIZE
Diminazene aceturate
- LDLR
Low-density lipoprotein receptor
- RAS
Renin-angiotensin system
- VLDL
Very low-density lipoprotein
Footnotes
Disclosures
None
References
- 1.Baxter BT, Terrin MC, Dalman RL. Medical management of small abdominal aortic aneurysms. Circulation. 2008;117:1883–1889. doi: 10.1161/CIRCULATIONAHA.107.735274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freestone T, Turner RJ, Coady A, Higman DJ, Greenhalgh RM, Powell JT. Inflammation and matrix metalloproteinases in the enlarging abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 1995;15:1145–1151. doi: 10.1161/01.atv.15.8.1145. [DOI] [PubMed] [Google Scholar]
- 3.Acosta S, Ogren M, Bergqvist D, Lindblad B, Dencker M, Zdanowski Z. The hardman index in patients operated on for ruptured abdominal aortic aneurysm: A systematic review. J Vasc Surg. 2006;44:949–954. doi: 10.1016/j.jvs.2006.07.041. [DOI] [PubMed] [Google Scholar]
- 4.Dobrin PB, Baker WH, Gley WC. Elastolytic and collagenolytic studies of arteries. Implications for the mechanical properties of aneurysms. Arch Surg. 1984;119:405–409. doi: 10.1001/archsurg.1984.01390160041009. [DOI] [PubMed] [Google Scholar]
- 5.Powell JT, Brown LC. The natural history of abdominal aortic aneurysms and their risk of rupture. Adv Surg. 2001;35:173–185. [PubMed] [Google Scholar]
- 6.Daugherty A, Manning MW, Cassis LA. Angiotensin ii promotes atherosclerotic lesions and aneurysms in apolipoprotein e-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daugherty A, Rateri DL, Lu H, Inagami T, Cassis LA. Hypercholesterolemia stimulates angiotensin peptide synthesis and contributes to atherosclerosis through the at1a receptor. Circulation. 2004;110:3849–3857. doi: 10.1161/01.CIR.0000150540.54220.C4. [DOI] [PubMed] [Google Scholar]
- 8.Saraff K, Babamusta F, Cassis LA, Daugherty A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin ii-infused, apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:1621–1626. doi: 10.1161/01.ATV.0000085631.76095.64. [DOI] [PubMed] [Google Scholar]
- 9.Daugherty A, Manning MW, Cassis LA. Antagonism of at2 receptors augments angiotensin ii-induced abdominal aortic aneurysms and atherosclerosis. Br J Pharmacol. 2001;134:865–870. doi: 10.1038/sj.bjp.0704331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cassis LA, Rateri DL, Lu H, Daugherty A. Bone marrow transplantation reveals that recipient at1a receptors are required to initiate angiotensin ii-induced atherosclerosis and aneurysms. Arterioscler Thromb Vasc Biol. 2007;27:380–386. doi: 10.1161/01.ATV.0000254680.71485.92. [DOI] [PubMed] [Google Scholar]
- 11.Iida Y, Xu B, Schultz GM, Chow V, White JJ, Sulaimon S, Hezi-Yamit A, Peterson SR, Dalman RL. Efficacy and mechanism of angiotensin ii receptor blocker treatment in experimental abdominal aortic aneurysms. PLoS One. 2012;7:e49642. doi: 10.1371/journal.pone.0049642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hackam DG, Thiruchelvam D, Redelmeier DA. Angiotensin-converting enzyme inhibitors and aortic rupture: A population-based case-control study. Lancet. 2006;368:659–665. doi: 10.1016/S0140-6736(06)69250-7. [DOI] [PubMed] [Google Scholar]
- 13.Railton CJ, Wolpin J, Lam-McCulloch J, Belo SE. Renin-angiotensin blockade is associated with increased mortality after vascular surgery. Can J Anaesth. 2010;57:736–744. doi: 10.1007/s12630-010-9330-4. [DOI] [PubMed] [Google Scholar]
- 14.Samson R. Can pharmacologic agents slow abdominal aortic aneurysm growth? Semin Vasc Surg. 2012;25:25–28. doi: 10.1053/j.semvascsurg.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Sweeting MJ, Thompson SG, Brown LC, Greenhalgh RM, Powell JT. Use of angiotensin converting enzyme inhibitors is associated with increased growth rate of abdominal aortic aneurysms. J Vasc Surg. 2010;52:1–4. doi: 10.1016/j.jvs.2010.02.264. [DOI] [PubMed] [Google Scholar]
- 16.Thompson A, Cooper JA, Fabricius M, Humphries SE, Ashton HA, Hafez H. An analysis of drug modulation of abdominal aortic aneurysm growth through 25 years of surveillance. J Vasc Surg. 2010;52:55–61 e52. doi: 10.1016/j.jvs.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 17.Silverberg D, Younis A, Savion N, Harari G, Yakubovitch D, Sheick Yousif B, Halak M, Grossman E, Schneiderman J. Long-term renin-angiotensin blocking therapy in hypertensive patients with normal aorta may attenuate the formation of abdominal aortic aneurysms. Journal of the American Society of Hypertension: JASH. 2014;8:571–577. doi: 10.1016/j.jash.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 18.Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ace2) converts angiotensin i to angiotensin 1–9. Circ Res. 2000;87:E1–9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 19.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 20.Thatcher SE, Zhang X, Howatt DA, Lu H, Gurley SB, Daugherty A, Cassis LA. Angiotensin-converting enzyme 2 deficiency in whole body or bone marrow-derived cells increases atherosclerosis in low-density lipoprotein receptor−/− mice. Arterioscler Thromb Vasc Biol. 2011;31:758–765. doi: 10.1161/ATVBAHA.110.221614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rateri DL, Howatt DA, Moorleghen JJ, Charnigo R, Cassis LA, Daugherty A. Prolonged infusion of angiotensin ii in apoe(−/−) mice promotes macrophage recruitment with continued expansion of abdominal aortic aneurysm. Am J Pathol. 2011;179:1542–1548. doi: 10.1016/j.ajpath.2011.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaneko H, Anzai T, Horiuchi K, Kohno T, Nagai T, Anzai A, Takahashi T, Sasaki A, Shimoda M, Maekawa Y, Shimizu H, Yoshikawa T, Okada Y, Yozu R, Fukuda K. Tumor necrosis factor-alpha converting enzyme is a key mediator of abdominal aortic aneurysm development. Atherosclerosis. 2011;218:470–478. doi: 10.1016/j.atherosclerosis.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 23.Tang EH, Shvartz E, Shimizu K, Rocha VZ, Zheng C, Fukuda D, Shi GP, Sukhova G, Libby P. Deletion of ep4 on bone marrow-derived cells enhances inflammation and angiotensin ii-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2011;31:261–269. doi: 10.1161/ATVBAHA.110.216580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tazume H, Miyata K, Tian Z, Endo M, Horiguchi H, Takahashi O, Horio E, Tsukano H, Kadomatsu T, Nakashima Y, Kunitomo R, Kaneko Y, Moriyama S, Sakaguchi H, Okamoto K, Hara M, Yoshinaga T, Yoshimura K, Aoki H, Araki K, Hao H, Kawasuji M, Oike Y. Macrophage-derived angiopoietin-like protein 2 accelerates development of abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2012;32:1400–1409. doi: 10.1161/ATVBAHA.112.247866. [DOI] [PubMed] [Google Scholar]
- 25.Findeisen HM, Gizard F, Zhao Y, Cohn D, Heywood EB, Jones KL, Lovett DH, Howatt DA, Daugherty A, Bruemmer D. Telomerase deficiency in bone marrow-derived cells attenuates angiotensin ii-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2011;31:253–260. doi: 10.1161/ATVBAHA.110.218545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owens AP, 3rd, Rateri DL, Howatt DA, Moore KJ, Tobias PS, Curtiss LK, Lu H, Cassis LA, Daugherty A. Myd88 deficiency attenuates angiotensin ii-induced abdominal aortic aneurysm formation independent of signaling through toll-like receptors 2 and 4. Arterioscler Thromb Vasc Biol. 2011;31:2813–2819. doi: 10.1161/ATVBAHA.111.238642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foureaux G, Nogueira JC, Nogueira BS, Fulgencio GO, Menezes GB, Fernandes SO, Cardoso VN, Fernandes RS, Oliveira GP, Franca JR, Faraco AA, Raizada MK, Ferreira AJ. Antiglaucomatous effects of the activation of intrinsic angiotensin-converting enzyme 2. Invest Ophthalmol Vis Sci. 2013 doi: 10.1167/iovs.12-11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kulemina LV, Ostrov DA. Prediction of off-target effects on angiotensin-converting enzyme 2. J Biomol Screen. 2011;16:878–885. doi: 10.1177/1087057111413919. [DOI] [PubMed] [Google Scholar]
- 29.Shenoy V, Gjymishka A, Jarajapu YP, Qi Y, Afzal A, Rigatto K, Ferreira AJ, Fraga-Silva RA, Kearns P, Douglas JY, Agarwal D, Mubarak KK, Bradford C, Kennedy WR, Jun JY, Rathinasabapathy A, Bruce E, Gupta D, Cardounel AJ, Mocco J, Patel JM, Francis J, Grant MB, Katovich MJ, Raizada MK. Diminazene attenuates pulmonary hypertension and improves angiogenic progenitor cell functions in experimental models. Am J Respir Crit Care Med. 2013;187:648–657. doi: 10.1164/rccm.201205-0880OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mecca AP, Regenhardt RW, O’Connor TE, Joseph JP, Raizada MK, Katovich MJ, Sumners C. Cerebroprotection by angiotensin-(1-7) in endothelin-1-induced ischaemic stroke. Exp Physiol. 2011;96:1084–1096. doi: 10.1113/expphysiol.2011.058578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin J, Arif B, Garcia-Fernandez F, Ennis TL, Davis EC, Thompson RW, Curci JA. Novel mechanism of aortic aneurysm development in mice associated with smoking and leukocytes. Arterioscler Thromb Vasc Biol. 2012;32:2901–2909. doi: 10.1161/ATVBAHA.112.300208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark MA, Diz DI, Tallant EA. angiotensin-(1-7) downregulates the angiotensin ii type 1 receptor in vascular smooth muscle cells. Hypertension. 2001;37:1141–1146. doi: 10.1161/01.hyp.37.4.1141. [DOI] [PubMed] [Google Scholar]
- 33.Ferrario CM, Martell N, Yunis C, Flack JM, Chappell MC, Brosnihan KB, Dean RH, Fernandez A, Novikov SV, Pinillas C, Luque M. Characterization of angiotensin-(1-7) in the urine of normal and essential hypertensive subjects. Am J Hypertens. 1998;11:137–146. doi: 10.1016/s0895-7061(97)00400-7. [DOI] [PubMed] [Google Scholar]
- 34.Grobe JL, Mecca AP, Lingis M, Shenoy V, Bolton TA, Machado JM, Speth RC, Raizada MK, Katovich MJ. Prevention of angiotensin ii-induced cardiac remodeling by angiotensin-(1-7) Am J Physiol Heart Circ Physiol. 2007;292:H736–742. doi: 10.1152/ajpheart.00937.2006. [DOI] [PubMed] [Google Scholar]
- 35.Hayashi N, Yamamoto K, Ohishi M, Tatara Y, Takeya Y, Shiota A, Oguro R, Iwamoto Y, Takeda M, Rakugi H. The counterregulating role of ace2 and ace2-mediated angiotensin 1-7 signaling against angiotensin ii stimulation in vascular cells. Hypertens Res. 2010;33:1182–1185. doi: 10.1038/hr.2010.147. [DOI] [PubMed] [Google Scholar]
- 36.Iyer SN, Averill DB, Chappell MC, Yamada K, Allred AJ, Ferrario CM. Contribution of angiotensin-(1-7) to blood pressure regulation in salt-depleted hypertensive rats. Hypertension. 2000;36:417–422. doi: 10.1161/01.hyp.36.3.417. [DOI] [PubMed] [Google Scholar]
- 37.Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, Heringer-Walther S, Pinheiro SV, Lopes MT, Bader M, Mendes EP, Lemos VS, Campagnole-Santos MJ, Schultheiss HP, Speth R, Walther T. angiotensin-(1-7) is an endogenous ligand for the g protein-coupled receptor mas. Proc Natl Acad Sci U S A. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Diez-Freire C, Dooies A, Jun JY, Sriramula S, Mariappan N, Pourang D, Venugopal CS, Francis J, Reudelhuber T, Santos RA, Patel JM, Raizada MK, Katovich MJ. The angiotensin-converting enzyme 2/angiogenesis-(1-7)/mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2010;182:1065–1072. doi: 10.1164/rccm.200912-1840OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tesanovic S, Vinh A, Gaspari TA, Casley D, Widdop RE. Vasoprotective and atheroprotective effects of angiotensin (1-7) in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol. 2010;30:1606–1613. doi: 10.1161/ATVBAHA.110.204453. [DOI] [PubMed] [Google Scholar]
- 40.Sampaio WO, Henrique de Castro C, Santos RA, Schiffrin EL, Touyz RM. angiotensin-(1-7) counterregulates angiotensin ii signaling in human endothelial cells. Hypertension. 2007;50:1093–1098. doi: 10.1161/HYPERTENSIONAHA.106.084848. [DOI] [PubMed] [Google Scholar]
- 41.Ferreira AJ, Bader M, Santos RA. Therapeutic targeting of the angiotensin-converting enzyme 2/angiotensin-(1-7)/mas cascade in the renin-angiotensin system: A patent review. Expert Opin Ther Pat. 2012;22:567–574. doi: 10.1517/13543776.2012.682572. [DOI] [PubMed] [Google Scholar]
- 42.Rateri DL, Moorleghen JJ, Knight V, Balakrishnan A, Howatt DA, Cassis LA, Daugherty A. Depletion of endothelial or smooth muscle cell-specific angiotensin ii type 1a receptors does not influence aortic aneurysms or atherosclerosis in ldl receptor deficient mice. PLoS One. 2012;7:e51483. doi: 10.1371/journal.pone.0051483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cassis LA, Gupte M, Thayer S, Zhang X, Charnigo R, Howatt DA, Rateri DL, Daugherty A. Ang ii infusion promotes abdominal aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. Am J Physiol Heart Circ Physiol. 2009;296:H1660–1665. doi: 10.1152/ajpheart.00028.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hernandez Prada JA, Ferreira AJ, Katovich MJ, Shenoy V, Qi Y, Santos RA, Castellano RK, Lampkins AJ, Gubala V, Ostrov DA, Raizada MK. Structure-based identification of small-molecule angiotensin-converting enzyme 2 activators as novel antihypertensive agents. Hypertension. 2008;51:1312–1317. doi: 10.1161/HYPERTENSIONAHA.107.108944. [DOI] [PubMed] [Google Scholar]
- 45.Haber PK, Ye M, Wysocki J, Maier C, Haque SK, Batlle D. Angiotensin-converting enzyme 2-independent action of presumed angiotensin-converting enzyme 2 activators: Studies in vivo, ex vivo, and in vitro. Hypertension. 2014;63:774–782. doi: 10.1161/HYPERTENSIONAHA.113.02856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peregrine AS, Mamman M. Pharmacology of diminazene: A review. Acta tropica. 1993;54:185–203. doi: 10.1016/0001-706x(93)90092-p. [DOI] [PubMed] [Google Scholar]
- 47.Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, Sigl V, Hanada T, Hanada R, Lipinski S, Wild B, Camargo SM, Singer D, Richter A, Kuba K, Fukamizu A, Schreiber S, Clevers H, Verrey F, Rosenstiel P, Penninger JM. Ace2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature. 2012;487:477–481. doi: 10.1038/nature11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paizis G, Tikellis C, Cooper ME, Schembri JM, Lew RA, Smith AI, Shaw T, Warner FJ, Zuilli A, Burrell LM, Angus PW. Chronic liver injury in rats and humans upregulates the novel enzyme angiotensin converting enzyme 2. Gut. 2005;54:1790–1796. doi: 10.1136/gut.2004.062398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feltenberger JD, Andrade JM, Paraiso A, Barros LO, Filho AB, Sinisterra RD, Sousa FB, Guimaraes AL, de Paula AM, Campagnole-Santos MJ, Qureshi M, Dos Santos RA, Santos SH. Oral formulation of angiotensin-(1-7) improves lipid metabolism and prevents high-fat diet-induced hepatic steatosis and inflammation in mice. Hypertension. 2013 doi: 10.1161/HYPERTENSIONAHA.111.00919. [DOI] [PubMed] [Google Scholar]
- 50.Police SB, Thatcher SE, Charnigo R, Daugherty A, Cassis LA. Obesity promotes inflammation in periaortic adipose tissue and angiotensin ii-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2009;29:1458–1464. doi: 10.1161/ATVBAHA.109.192658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Ait-Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S, Huang J, Offenstadt G, Combadiere C, Renia L, Johnson JL, Tharaux PL, Tedgui A, Mallat Z. Tgf-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin ii-infused mice. J Clin Invest. 2010;120:422–432. doi: 10.1172/JCI38136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.