

Abstract
Over 70 different Charcot-Marie-Tooth disease (CMT)–associated genes have now been discovered and their number is growing. Conventional genetic testing for all CMT genes is cumbersome, expensive, and impractical in an individual patient. Next-generation sequencing (NGS) technology allows cost-effective sequencing of large scale DNA, even entire exome (coding sequences) or whole genome and thus, NGS platform can be employed to effectively target a large number or all CMT-related genes for accurate diagnosis. This overview discusses how NGS can be strategically used for genetic diagnosis in patients with CMT or unexplained neuropathy. A comment is made to combine simple clinical and electrophysiological algorithm to assign patients to major CMT subtypes and then employ NGS to screen for all known mutations in the subtype-specific CMT gene panel.
Keywords: CMT, exome, genetic testing, genome, next-generation sequencing
Introduction
Charcot-Marie-Tooth disease (CMT, also called hereditary sensory and motor neuropathy) is named after three physicians who first described the condition in 1886. CMT is one of the most common inherited neuromuscular diseases, with population prevalence of approximately 1 in 2500.[1] CMT is characterized clinically by distal weakness and atrophy and sensory loss that starts in feet and progresses slowly in a length-dependent manner. CMT is closely related to two other rare inherited neuropathies: The hereditary motor neuropathy (HMN), which only has motor involvement; and hereditary sensory and autonomic neuropathy (HSAN), which involves chiefly sensory nerves. These three disorders (CMT, HMN, and HSAN) form a continuum and one gene may cause more than one phenotype. Further, each of these CMT subtypes can be inherited in dominant and recessive manners involving autosomes or X-chromosome and each subtype can be caused by mutations in a number of different genes.[2,3,4,5]
With the advent of nerve electrophysiology and pathology in the twenteith century, it became possible to phenotype CMT into major subtypes—demyelinating and axonal forms, with a cutoff of motor conduction velocities (MCV) in forearm nerves at 38 m/s.[6] CMT cases with MCVs below 38 m/s are defined as demyelinating (CMT1) and those with MCVs above 38 m/s are defined as axonal subtypes (CMT2). A third group, intermediate CMT, also exists in which MCV lies between 25 m/s and 45 m/s. Several CMT genes, including GJB1, NEFL, and MPZ are associated with an MCV in this intermediate range.[5]
First breakthrough in the basis of CMT occurred in 1991 with the discovery that duplication in the short arm of chromosome 17, which contains the peripheral myelin protein 22 (PMP22) gene, is the most common cause of CMT and it was classified as CMT1A.[7] Over the ensuing 2 years, mutations in GJB1, PMP22, and MPZ were discovered. These four gene mutations collectively account for approximately one-third of all CMT cases and approximately two-third of CMT1 cases.[5,8] At the time of publication of the rough draft of human genome in 2001, a total pf 12 CMT genes were discovered.[5] In the past 5 years, the genetic landscape of CMT has dramatically changed. This rapid evolution in gene discovery is attributable to the development and incremental improvement and affordability of multiple-parallel or next-generation sequencing (NGS) technology, which enables sequencing of large stretches of DNA, including the entire genome or exome (entire protein-coding sequences) or parts thereof, in a matter of days.[9]
At the time of this writing, about 70 disease-causing genes have been described in CMT and related disorders[5] (see also: http://neuromuscular.wustl.edu/time/hmsn.html and www.molgen.ua.ac.be/CMTMutations/). This brief review discusses an emerging practical approach that combines clinical and electrophysiological features and NGS for rapid and efficient genetic diagnosis of CMT. This review does not cover in detail the use of NGS technology for whole genome sequencing (WGS) or whole exome sequencing (WES) which is currently being employed to discover novel CMT genes in families.[10,11]
Diagnosis of CMT
The diagnosis of inherited neuropathy may seem obvious in the context of a positive family history in a large pedigree or when multiple siblings are born to consanguineous parents. But in modern times, families are generally small and a clear family history of neuropathy may not be forthcoming.[12] In such cases, for example, in a single patient with neuropathy, a long and slowly progressive distal weakness and wasting, difficulty walking, and foot deformity are important indicators of a potential inherited neuropathy. Many CMT cases are described being poor in sports at school and may have required reconstructive foot surgery in childhood.
First step in diagnosis of suspected CMT and related disorders is the identification of the most likely mode of inheritance and phenotypic ascertainment of neuropathy to a major subtype (CMT1, CMT2, HMN, or HSAN). In a large family with multiple affected members, autosomal dominant or X-chromosome-linked inheritance may be apparent. In small family or apparently sporadic case, the mode of inheritance is difficult to determine. In most western countries, autosomal or de novo dominant inheritance is likely in single cases, but in other parts of world where consanguineous marriages are common (for example, middle east, European Gypsies, Pakistan, Indonesia), autosomal recessive inheritance is likely to be more common.
Determination of at least probable mode of inheritance remains crucial to efficiently use high throughput NGS protocols, such as WGS or WES, to identify a new disease gene, because “filtering” of large sequence data is dependent on the mode of inheritance. For example, WGS data in an individual can reveal nearly four million and WES data over 20,000 polymorphic sites.[5,9,10] Effective “filtering out” of innocuous polymorphic sites is needed to zero in to possible culprit gene.[9,10] The technical aspect of WGS and WES “filtering” is beyond the scope of this review.
Once the clinical diagnosis of potential inherited neuropathy has been made, the next step is to perform electrophysiology to differentiate CMT1 from CMT2. Electrophysiology is also useful to detect sensory involvement which may not be obvious on clinical assessment and thus to differentiate CMT2 from HMN. In recent years, neurophysiological features that were considered to be typical of acquired inflammatory neuropathy, chronic inflammatory demyelinating polyradiculoneuropathy, have been described in some rare forms of CMT, including CMT related to GJB1, SH3TC2, and FIG4 gene mutations.[5,13,14,15,16] These findings have led to consideration of inherited disorders in patients with a much broader spectrum of neurophysiological findings than would have previously been considered.
Third logical step after clinical and neurophysiological characterization is to utilize conventional Sanger sequencing to screen for relatively common candidate CMT genes that are known to cause a particular phenotype.[17] This step is to possibly rule in one of the relatively common mutations, before embarking on next NGS step in yet undiagnosed cases. It is now known from several large series that 60% of patients with CMT have CMT1 and 40% have an axonal neuropathy, of which approximately 50% have CMT2 and 25% each of distal HMN and HSAN.[5] In a patient with CMT1 and MCV below 38 m/s, therefore, a screening test for chromosome 17p duplication is recommended. Chromosome 17p duplication is found in two-third of patients with CMT1. In a male patient with CMT1 or intermediate range MCVs and in whom 17p duplication is absent, GJB1 and MPZ genes should be screened. GJB1 gene-related CMT is X-chromosome-linked and is present in 5-12% of CMT1 cases.[18] In CMT2 phenotype, conventional genetic testing for MFN2 gene should be performed; MFN2 gene mutation is present in up to 20% of CMT2 cases.[19]
Aside from testing for 17p duplication, PMP22, GJB1, MPZ, and MFN2 genes in select cases, the use of conventional Sanger sequencing for the remaining CMT genes is time consuming, cumbersome, expensive and unrewarding. This is particularly true for patients with CMT2 in which the other disease-causing genes are uncommon, that is, each one is individually rare. As mentioned before, as genetic testing has become more widely available, it is also becoming increasingly evident that a single gene on occasion can cause multiple phenotypes[12] and may be inherited in more than one modes of inheritance.[2,3,4,5] For example, mutations in HSPB1 can cause CMT2 and distal HMN,[20] and it can be inherited in both autosomal dominant and autosomal recessive manner.[5] A wider scale NGS can capture accurate genetic mutation in such confusing scenarios.
Next-Generation Sequencing in CMT
NGS is a high-throughput DNA sequencing technology that employs parallel sequencing of small DNA segments from whole genome, whole exome or from gene-enriched genomic regions. The basic principle of NGS is the simultaneous amplification and sequencing of small sections of DNA (amplicons) that are subsequently realigned to construct larger DNA sequences.[9]
Up until recently, WGS and WES have been employed in CMT families only as a research tool to identify new disease causing genes. NGS has obvious advantage over older genetic linkage and Sanger sequencing in that it can be scaled up to screen whole genome or exome efficiently and quickly, and it requires less informative family than is required in conventional linkage analysis. However, one of the drawbacks with WES and WGS has been the insufficient depth and coverage in some cases, although this situation is expected to change in near future. Read-depth refers to the number of target DNA sequence (in genome, exome or individual genes) that has been amplified and sequenced. That is, the number of measured nucleotide at a given location is the basis of determining the genotype in NGS. Thus, higher the read-depth (> 10 sequencing reads) more the sequence becomes error-free. Read-depth and coverage can be an issue for certain sites in genome-wide NGS. For example, the X-chromosome and many GC-rich and nucleotide repeat regions are poorly covered in WGS.[5,9] But targeting and sequencing only the previously defined select CMT-related genes obviates this hurdle, thus providing the solution to the issue of low read-depth and coverage.
The reasons why NGS technology is particular suitable for CMT diagnostics include: (i) CMT is a relatively common neuropathy, (ii) several dozen genes cause CMT phenotype, and (iii) Both dominant and recessive alleles on autosomal and X-chromosome are known to cause CMT. Rapidly evolving NGS technology and more importantly, rapidly falling cost of NGS protocols is making it an affordable and practical way for genetic testing; that is, for testing of a larger set or all genes that are reported to cause CMT.[5,11] Even better, it can be accomplished by gene screening in customized CMT subtype-specific panels in which only a selective list of CMT genes are screened.
CMT Subtype-Specific NGS Panels
CMT testing panels apply NGS to regions of the genome that contain coding sequences and flanking regions of CMT genes. For a major CMT subtype, for example CMT2, appropriate “NGS panel” can be designed with good read-depth and coverage for effective use in genetic diagnosis [Figure 1].
Figure 1.
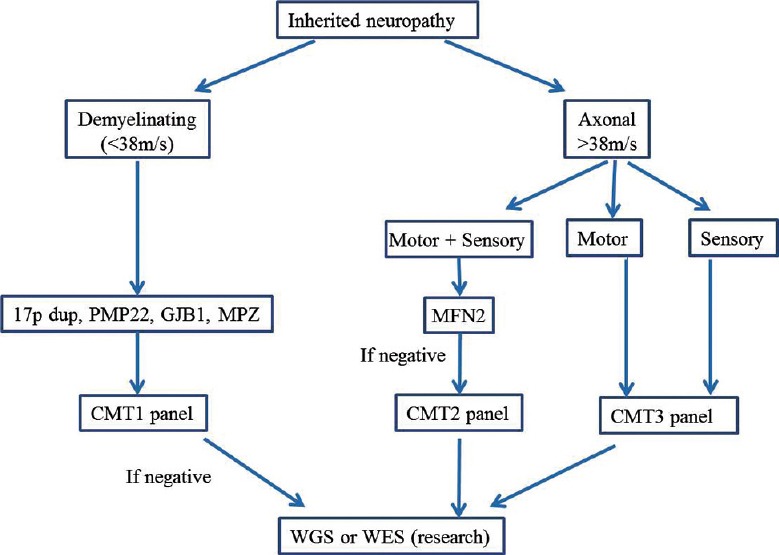
An algorithm for genetic testing of CMT using clinical and electrophysiological features and disease subtype-specific NGS panels. CMT, Charcot-Marie-Tooth disease; HMN, hereditary motor neuropathy; HSAN, hereditary sensory and autonomic neuropathy
One may surmise that an easy way is just to develop one “panel” that encompasses the largest number or all currently known 70 or so CMT genes, covering all disease phenotypes. This may look attractive to clinicians who are not specialists in neuropathic disorders. This also has advantage that all patients with CMT and related disorders can be screened using the same “panel”. However, there are major disadvantages of this method and these include high cost and difficulty in result interpretation. For example, when upward of 65 genes are targeted in an individual patient, many polymorphic nucleotide changes of dubious significance will be encountered. The challenge, then, is to work out which mutation is likely to be relevant. No reliable functional tests currently exist to prove the pathogenic nature of a polymorphism in patients with neuropathy.
To overcome these drawbacks, a more logical approach is to initially break down the CMT phenotypic spectrum into small number of major subtypes and design smaller NGS panels that cover individual major subtypes. As mentioned before, a wealth of experience currently exists in clinical and electrophysiological phenotyping of patients with CMT, and not to use this information in genetic testing seems wasteful. An approach based on three broad CMT NGS panels is shown in Figure 1.
The CMT phenotype-specific panels are the most closely aligned to the traditional method of genetic testing for a single gene, and this is obviously a preferred choice for multi-gene testing as well. There are several reasons why CMT subtype-specific panels are more useful. First, restricted gene panel is cheaper than the complete CMT gene panel. Second, the number of dubious or irrelevant variants that are likely to be discovered in single smaller panel is much reduced. These two related issues are not trivial as the time and financial cost of confirming segregation of new mutation site in family members is considerable. Finally, as new gene is discovered it will be added to the phenotype-specific panel and the upgrading of relatively smaller NGS panel will be less cumbersome.
The labor-intensity and cost of an NGS panel will depend on the number of genes included in the panel and panel's self-life, that is, the time before the number of newly discovered genes will reduce the panel's clinical utility. Since the production of the panel accounts for the bulk of expenditure for commercial laboratories, the need to continually update CMT panels to include new genes incurs additional cost. Despite this shortcoming, disease subtype-specific NGS panels seem to be the best method for simultaneous screening of CMT-related genes and, until the read-depth, coverage and cost of WES and WGS improve, NGS panels are likely to remain a comprehensive tool for genetic testing in patients with CMT.
Conclusion
Although there is no curative treatment of CMT, the implications of accurate genetic testing cannot be overemphasized. The precise diagnosis allows better counseling regarding the disease prognosis and cogent advice for preventive antenatal or pre-implantation genetics. Genetic test also helps assign correct diagnosis is some cases that were previously thought to have other forms of neuropathy. As the cost of NGS is falling, customized CMT diagnostic panels may become the standard of care in a foreseeable future. Revolutionary technology as it is, NGS platform is set to become a routine diagnostic tool in earlier stage evaluation of patients with CMT and related neuropathies.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
References
- 1.Reilly MM, Murphy SM, Laurá M. Charcot-Marie-Tooth disease. J Peripher Nerv Syst. 2011;16:1–14. doi: 10.1111/j.1529-8027.2011.00324.x. [DOI] [PubMed] [Google Scholar]
- 2.Vallat JM, Mathis S, Funalot B. The various Charcot-Marie-Tooth diseases. Curr Opin Neurol. 2013;26:473–80. doi: 10.1097/WCO.0b013e328364c04b. [DOI] [PubMed] [Google Scholar]
- 3.Azzedine H, Senderek J, Rivolta C, Chrast R. Molecular genetics of Charcot-Marie-Tooth disease: From genes to genomes. Mol Syndromol. 2012;3:204–14. doi: 10.1159/000343487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Züchner S, Vance JM. Mechanisms of disease: A molecular genetic update on hereditary axonal neuropathies. Nat Clin Pract Neurol. 2006;2:45–53. doi: 10.1038/ncpneuro0071. [DOI] [PubMed] [Google Scholar]
- 5.Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol. 2013;9:562–71. doi: 10.1038/nrneurol.2013.179. [DOI] [PubMed] [Google Scholar]
- 6.Ouvrier R. What can we learn from the history of Charcot-Marie-Tooth disease? Dev Med Child Neurol. 2010;52:405–6. doi: 10.1111/j.1469-8749.2010.03675.x. [DOI] [PubMed] [Google Scholar]
- 7.Raeymaekers P, Timmerman V, Nelis E, De Jonghe P, Hoogendijk JE, Baas F, et al. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). The HMSN Collaborative Research Group. Neuromuscul Disord. 1991;1:93–7. doi: 10.1016/0960-8966(91)90055-w. [DOI] [PubMed] [Google Scholar]
- 8.Murphy SM, Laura M, Fawcett K, Pandraud A, Liu YT, Davidson GL, et al. Charcot-Marie-Tooth disease: Frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry. 2012;83:706–10. doi: 10.1136/jnnp-2012-302451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rehm HL. Disease-targeted sequencing: A cornerstone in the clinic. Nat Rev Genet. 2013;14:295–300. doi: 10.1038/nrg3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tucci A, Liu YT, Preza E, Pitceathly RD, Chalasani A, Plagnol V, et al. Novel C12orf65 mutations in patients with axonal neuropathy and optic atrophy. J Neurol Neurosurg Psychiatry. 2014;85:486–92. doi: 10.1136/jnnp-2013-306387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montenegro G, Powell E, Huang J, Speziani F, Edwards YJ, Beecham G, et al. Exome sequencing allows for rapid gene identification in a Charcot-Marie-Tooth family. Ann Neurol. 2011;69:464–70. doi: 10.1002/ana.22235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Østern R, Fagerheim T, Hjellnes H, Nygård B, Mellgren SI, Nilssen Ø. Segregation analysis in families with Charcot-Marie-Tooth disease allows reclassification of putative disease causing mutations. BMC Med Genet. 2014;15:12. doi: 10.1186/1471-2350-15-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cottenie E, Menezes MP, Rossor AM, Morrow JM, Yousry TA, Dick DJ, et al. Rapidly progressive asymmetrical weakness in Charcot-Marie-Tooth disease type 4J resembles chronic inflammatory demyelinating polyneuropathy. Neuromuscul Disord. 2013;23:399–403. doi: 10.1016/j.nmd.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 14.Houlden H, Laura M, Ginsberg L, Jungbluth H, Robb SA, Blake J, et al. The phenotype of Charcot-Marie-Tooth disease type 4C due to SH3TC2 mutations and possible predisposition to an inflammatory neuropathy. Neuromuscul Disord. 2009;19:264–9. doi: 10.1016/j.nmd.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 15.Michell AW, Laura M, Blake J, Lunn MP, Cox A, Gibbons VS, et al. GJB1 gene mutations in suspected inflammatory demyelinating neuropathies not responding to treatment. J Neurol Neurosurg Psychiatry. 2009;80:699–700. doi: 10.1136/jnnp.2008.150557. [DOI] [PubMed] [Google Scholar]
- 16.Verma A. Neuropathic scapuloperoneal syndrome (Davidenkow's syndrome) with chromosome 17p11.2 deletion. Muscle Nerve. 2005;32:668–71. doi: 10.1002/mus.20402. [DOI] [PubMed] [Google Scholar]
- 17.Tousignant R, Trepanier A, Shy ME, Siskind CE. Genetic testing practices for Charcot-Marie-Tooth type 1A disease. Muscle Nerve. 2014;49:478–82. doi: 10.1002/mus.23991. [DOI] [PubMed] [Google Scholar]
- 18.Keckarevic-Markovic M, Milic-Rasic V, Mladenovic J, Dackovic J, Kecmanovic M, Keckarevic D, et al. Mutational analysis of GJB1, MPZ, PMP22, EGR2, and LITAF/SIMPLE in Serbian Charcot-Marie-Tooth patients. J Peripher Nerv Syst. 2009;14:125–36. doi: 10.1111/j.1529-8027.2009.00222.x. [DOI] [PubMed] [Google Scholar]
- 19.Cartoni R, Martinou JC. Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp Neurol. 2009;218:268–73. doi: 10.1016/j.expneurol.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Houlden H, Laura M, Wavrant-De Vrièze F, Blake J, Wood N, Reilly MM. Mutations in the HSP27 (HSPB1) gene cause dominant, recessive, and sporadic distal HMN/CMT type 2. Neurology. 2008;71:1660–8. doi: 10.1212/01.wnl.0000319696.14225.67. [DOI] [PubMed] [Google Scholar]