

Abstract
Background:
Progressive cerebellar ataxia inherited by autosomal dominant transmission is known as Spino Cerebellar Ataxia (SCA).
Aims and Objectives:
To look for various clinical profile and molecular genetics of patients with SCAs and their phenotype-genotype correlation of patients with SCAs.
Materials and Methods:
This was a cross-sectional study conducted at Bangur Institute of Neurosciences, Kolkata from June 2010 to April 2013. We selected patients from the neurogenetic clinic of our institute and performed genetic test for SCA 1, 2, 3, 6 and 12. The diagnosis was based on suggestive clinical features and positive genetic study, done by polymerase chain reaction.
Results:
83 patients were tested for trineucleotide repeats and turned out 45 positive for the mentioned SCAs. We found 13(28.9%) SCA-1, 18(40%) SCA-2, 7(15.6%) SCA-3, 6(13.3) SCA-6 and 1(2.2%) SCA-12 patients. Half of the remaining 38 patients had positive family history. The mean age of onset were 38.46 years in SCA-1, 29.55 years in SCA-2, 38.43 years in SCA-3, 47.33 years in SCA-6. Slow saccades were observed in 7(53.8%) SCA-1, 17(94.4%) SCA-2, 4(57.1%) SCA-3, 3(50%) SCA-6 patients. Hyporeflexia was noticed in 5(27.8%) SCA-2 patients. Pyramidal tract involvement was found in 8(61.5%) SCA-1, 4(22.2%) SCA-2, 4(57.1%) SCA-3 and 1(16.7%) SCA-6 patients.
Conclusion:
Our study showed SCA-2 is the most common variety of SCA and genotypic-phenotypic correlation was observed in SCA-1,2,6 and 12 patients.
Keywords: SCA, SCA genetics, spinocerebellar ataxia, spinocerebellar ataxia genetics
Introduction
Spinocerebellar ataxias (SCAs) are autosomal dominant progressive neurodegenerative disorders showing clinical and genetic heterogeneities.[1] SCAs usually manifest clinically in the third to fifth decade of life, although there is a wide variability in the age of onset. More than 36 different types of SCAs have been reported so far in the world and about half of them are caused by pathological expansion of the trinucleotide cytosine, adenine, guanine (CAG) repeat. The global prevalence of SCA is 0.3-2 per 100,000 population,[2] SCA3 being the most common variety, worldwide, accounting for 2050 per cent of all cases, though rare in India.[3] The prevalence varies significantly depending on race, place of birth and founder effect. One study conducted by Subramony et al.,[4] shows the prevalence to be 12 per 100,000 population, whereas a study in two villages comprising of ethnic Tamil community indicates that the prevalence is high, i.e. 7.2%.[5] Interest in the field of inherited ataxias in India has been kindled by the seminal work of Wadia et al., over a period of about three decades.[6,7] A number of other workers from India have reported the phenotype and genotype in various SCAs in their studies and showed that the clinical features and genotype varied in different geographical areas.[5,15] Variable phenotypic presentation and discovery of newer mutations in recent years need continued research on clinical and molecular aspects of SCAs, and the objective of the present study is to find out the clinical features and genetic pattern of different SCA patients seen in a tertiary care center in Eastern India.
Materials and Methods
This prospective study was carried out at Bangur Institute of Neurosciences, Kolkata, a tertiary referral center.
We selected patients from the general outpatient department and they were sent to the neurogenetic clinic of our hospital. Eightythree consecutive cases of suspected SCAs were primarily included for genetic study after history taking, analysis of familial pedigree and clinical examination. The clinical examination was carried out by senior neurologists and the findings were recorded in structured proforma of the neurogenetic clinic. Ethical clearances for the above study was obtained from the institutional ethical committee governed by Indian council of Medical Reasearch (ICMR) guidelines and written consent from the patients was obtained before the genetic study.
The inclusion criteria were cases with progressive degenerative cerebellar ataxia, familial or non-familial, who were negative for any known metabolic defect. In familial cases, one or more than one member had ataxia other than the proband. Non-familial or sporadic cases were individuals with features of primary degenerative cerebellar ataxia having no family history of similar illness. Sporadic cases could be the manifestation of new mutants or skipped generations in autosomal dominant inheritance pattern and possible unreliable family history about the previous generations.
Patients with metabolic, toxic, nutritional, infective, neoplastic, vascular and alcohol-related degeneration were excluded. Clinical assessment of eye movement in different gaze was done and slow saccades were recognized when eyes moved in a particular gaze taking more than one jump. Slit lamp examination was done is all patients for the presence of Kayser-Fleischer ring, andneuroimaging of the brain was performed in all patients. Routine blood biochemistry including blood glucose, lipid profile, thyroid profile and serum ceruloplasmin were performed in all cases. Serum lactate and serum vitamin E level were performed in selected cases to exclude progressive ataxia of known metabolic origin. Electrophysiological evaluation including nerve conduction study was done in all cases. Videography of patients was done for careful examination of the saccadic eye movements.
Molecular genetics study
The genetic study was aimed at detection of trinucleotiderepeat expansions in known SCA genes. DNA was isolated from patients’ blood sample by phenol: Chloroform method. Polymerase chain reaction was carried out using primers of SCA1, SCA 2, SCA 3, SCA6 and SCA12. Agarose gel (12.4%) electrophoresis was carried out followed by visualizations of bands using ethidium bromide. The number of CAG repeat was ascertained by comparing with a 100 bp DNA ladder.
Results and Discussion
We analyzed the genotype of 83 clinically suspected cases of SCAs for SCA1, SCA2, SCA3, SCA6 and SCA12. Forty-five cases were genetically positive for the above mentioned SCAs. Among genetically confirmed cases, we found 13 cases of SCA1, 18 cases of SCA2, 7 cases of SCA3, 6 cases of SCA6 and only one case of SCA12. Positive family history in different SCA patients is shown in Table 1. Demography and classification after genetic analysis of different SCA patients are shown in Table 2. Clinical and investigative parameters are shown in Table 3, while the relative frequency of different SCAs in India and abroad are shown in Table 4.
Table 1.
Family history

Table 2.
Demographic and classification after genotyping at SCA1, SCA2, SCA3, SCA6 and SCA12 loci
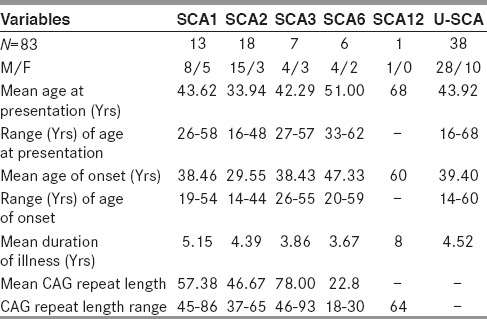
Table 3.
Clinical and investigative parameters of the study patients with SCA
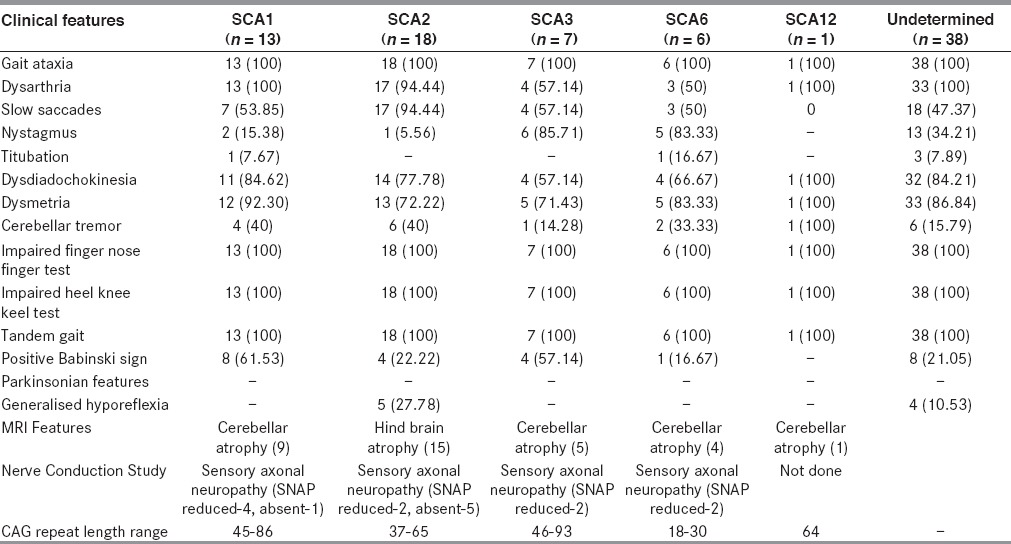
Table 4.
Relative frequencies of different SCAs in India and outside India
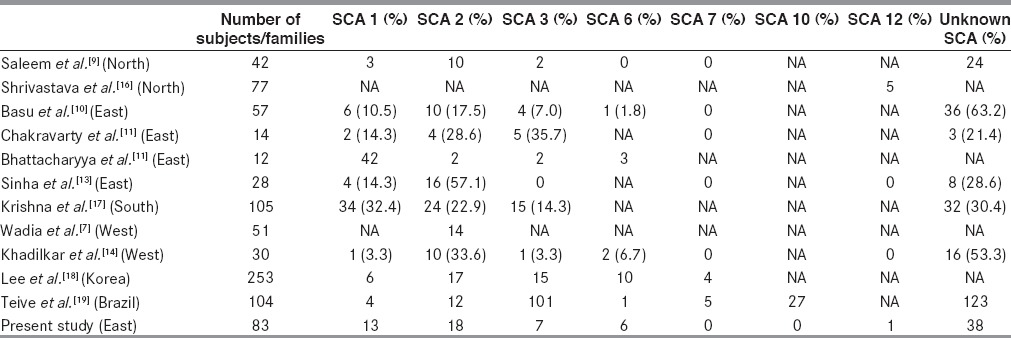
We describe the phenotypic features and genetic findings in patients of SCA and the estimated occurrence of ataxias in a tertiary care center in Eastern India. Total of 45 subjects out of the 83 investigated were positive for the mutation tested and SCA2, SCA1, SCA3, SCA6 and SCA12 were detected in descending order of frequency. SCA3 is the most common variety worldwide and it accounts for 2050 percent of all cases.[3] This is followed by SCA2, which accounts for 1020 percent of all cases and this is common in Europe, the USA and the UK.[20,21] SCA 1 is common in Italy, South Africa, Northern Japan and Russia.[20,22] In general, SCA1 and SCA2 are more frequent in populations of Caucasian ancestry, whereas SCA3 and Dentatorubropallidoluysian Atrophy are more common in population of Eastern ancestry.[23] In India, SCA2 is the most common form of ataxia in studies from northern,[9] eastern[10,13] and western[7,14] India. However, Chakravorty et al.,[11] and Bhattacharya et al.,[12] reported SCA3 to be the most common variety in ethnic Bengalee population. Rengaraj et al.,[5] Krishna et al.,[17] and Mittal et al.,[24] found very high prevalence of SCA-1 in south Indian population. Significant variations in the incidence of different types of SCA have been reported form countries like Japan,[25] China,[26] Finland,[27] Korea[18] and Brazil[19] where diverse ethnic communities exist.
India, another multiethnic country also shows variation in the occurrence of different types of SCA in different regions as mentioned earlier. Chakraborty et al.,[11] have reported 37 cases of SCA in 14 ethnic Bangalee families. They tested for SCA1, SCA2, and SCA3 only. Seventeen cases belonged to SCA3 genotype and six cases were SCA2, five cases were SCA1 and nine cases remained undetermined. Later on, Bhattacharyya et al.,[12] analyzed genotype of 35 individuals belonging to 18 families and they tested for SCA1, SCA 2, SCA3, SCA6, SCA12 and SCA17. They found 6 cases of SCA1, 8 cases of SCA2, 10 cases of SCA3 and 11 cases of undetermined variety.
In our study, we did not include family members of the patients and we found SCA2 in 18 cases, SCA1 in 13 cases, SCA3 in 7 cases, SCA6 in 6 cases and SCA12 in one case, whereas 38 cases were of undetermined variety. So, this present study shows that SCA 2 is the most common variety and SCA3 is of lower occurrence in the general population when ethnic factors are not taken into account. We have also shown significant occurrence of SCA6 (13.33%) patients in the study from Eastern India, which was not observed before. The apparent difference from the earlier study could be due to study on limited number of families (14 and 18 families) whereas this study included 83 probands. Clustering effect could also be present in the previous studies.
We observed clinical heterogeneity among different genotypes of SCA, which was also found in previous studies. All but one patient with SCA1 genotype presented with gait ataxia and dysarthria initially. One patient of SCA1 had tremulousness of upper limbs as the initial presentation. Slow saccadic eye movement was noted in 7 (53.85%) of 13 SCA1 patients. Among the less frequently present signs were tremor in limbs (38.46%) and pyramidal signs (23.08%)
All except one in SCA2 (94.44%) category showed typical cerebellar signs and slow saccadic eye movements. In nine out of 18 cases, we found obligatory blink with head thrust while testing for saccadic eye movement. Though slow saccades are characteristic of SCA2, they were also observed in other varieties as described by Wadia et al.[7] We found slow saccade in 4 patients (57%) of SCA3, three patients (50%) of SCA6 and in the single SCA-12 patient. Hyporeflexia caused by peripheral neuropathy is an important phenotypic clue for diagnosis of SCA2.[5,6,9,11] Four (22.22%) of our patients of SCA 2 and none from other SCA types showed clinical and or electrophysiological features suggestive of peripheral neuropathy and distal amyotrophy. Bhattacharyya et al.,[12] also found peripheral neuropathy in half of their SCA2 patients. None of our patients had bulbar features including lingual atrophy and perioral fasciculation, which are common in SCA 3 patients. We also performed Mini Mental Status examination in all SCA patients but cognitive decline was not noted in any of our patients. Bhattacharyya et al., found cognitive abnormality in none, but in one SCA1 patient. Thus, our findings corroborate with that of Bhattacharyya et al.[12] On the contrary Chakravarty et al.,[11] found significant cognitive decline in SCA3 and SCA2 patients in their study. Though we have noted slow saccadic movement in 53.85 % of SCA1, 94.44% of SCA2 and 57.14% of SCA3, gross ophthalmoplegia was observed in none of the SCA patients as was suggested by Chakraborty et al., in their study. SCA6 is highly frequent in Japan (11%)[23] and Taiwan (10.8%),[28] whereas this is relatively less common in Caucasians (5%).[23] Interestingly, no case of SCA 6 has been reported from mainland China before Jiang et al.,[29] reported SCA6 in four families. Basu et al.,[10] reported one family with SCA6 from India. We have found 6 (13.33 %) patients of SCA6 in this present study. SCA12 was initially reported in an American family of German descent but most of the other families have been reported from Indian subcontinent. Srivastava et al.,[16] and Fujigasaki et al.,[30] reported cases of SCA12 from India, whereas we found only one case of SCA12 in our study.
Conclusion
In our study, SCA2 outnumbered other varieties, when only ethnic Bengalees were not considered in the study. To the best of our knowledge, the largest number of patients from Eastern India was recruited in this study till date. There was significant overlap of clinical signs among different SCA types. No clinical sign could be found exclusive for any particular type of SCA. We also observed significant difference in phenotypical expression among individuals with same number of CAG repeats.
Limitations
The limitations in our study were the lack of primers for the detection of other varieties of SCAs. That is the reason we could not find the genetic locus in 38 out of 83 cases studied in our series, which thus remain as “unidentified”. Additionally, sequencing or capillary electrophoresis based sizing analysis could not be carried out for lack of facilities in our center for identical reasons. We could not carry out graphic recordings of slow eye movements and present them in our paper.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Zuhlke C, Dalski A, Hellenbroich Y, Bubel S, Schwinger E, Bürk K. Spinocerebellar ataxia type 1 (SCA1): Phenotype-genotype correlation studies in intermediate alleles. Eur J Hum Genet. 2002;10:204–9. doi: 10.1038/sj.ejhg.5200788. [DOI] [PubMed] [Google Scholar]
- 2.van de Warrenburg BP, Sinke RJ, Verschuuren-Bemelmans CC, Scheffer H, Brunt ER, Ippel PF, et al. Spinocerebellar ataxias in the Netherlands: Prevalence and age at onset variance analysis. Neurology. 2002;58:702–8. doi: 10.1212/wnl.58.5.702. [DOI] [PubMed] [Google Scholar]
- 3.Mittal U, Srivastava AK, Jain S, Jain S, Mukerji M. Founder haplotype for Machado-Joseph disease in the Indian population: Novel insights from history and polymorphism studies. Arch Neurol. 2005;62:637–40. doi: 10.1001/archneur.62.4.637. [DOI] [PubMed] [Google Scholar]
- 4.Subramony SH. Disorders of the cerebellum including the degenerative ataxias in Neurology in clinical practice. In: Bradley WG, Daroff RB, Fenichel GM, Jankovic J, editors. 4th ed. Philadelphia: Butterworth Heinemann; 2004. pp. 2169–84. [Google Scholar]
- 5.Rengaraj R, Dhanaraj M, Arulmozhi T, Chattopadhyay B, Battacharyya NP. High prevalence of spinocerebellar ataxia type 1 in an ethnic Tamil community in India. Neurol India. 2005;53:308–10. doi: 10.4103/0028-3886.16929. [DOI] [PubMed] [Google Scholar]
- 6.Wadia NH, Swami RK. A new form of heredo-familial spinocerebellar degeneration with slow eye movements (nine families) Brain. 1971;94:359–74. doi: 10.1093/brain/94.2.359. [DOI] [PubMed] [Google Scholar]
- 7.Wadia N, Pang J, Desai J, Mankodi A, Desai M, Chamberlain S. A clinicogenetic analysis of six Indian spinocerebellar ataxia (SCA2) pedigress. The significance of slow saccades in diagnosis. Brain. 1998;121:2341–55. doi: 10.1093/brain/121.12.2341. [DOI] [PubMed] [Google Scholar]
- 8.Wadia NH, Chamberlain S, Desai J, Mankodi AK, Desai M, Twells R. J Neurol Sci. First International Symposium on Inherited Ataxias, Montreal Canada; 1997. Hereditary Cerebellar ataxia with slow saccades- (SCA2) more prevalent in India; pp. 150–96. [Google Scholar]
- 9.Saleem Q, Choudhry S, Mukerji M, Bashyam L, Padma MV, Chakravarthy A, et al. Molecular analysis of autosomal dominant hereditary ataxias in the Indian population: High frequency of SCA2 and evidence for a common founder mutation. Hum Genet. 2000;106:179–87. doi: 10.1007/s004390051026. [DOI] [PubMed] [Google Scholar]
- 10.Basu P, Chattopadhyay B, Gangopadhaya PK, Mukherjee SC, Sinha KK, Das SK, et al. Analysis of CAG repeats in SCA1, SCA2, SCA3, SCA6, SCA7 and DRPLA loci in spinocerebellar ataxia patients and distribution of CAG repeats at the SCA1, SCA2 and SCA6 loci in nine ethnic populations of eastern India. Hum Genet. 2000;106:597–604. doi: 10.1007/s004390000320. [DOI] [PubMed] [Google Scholar]
- 11.Chakravarty A, Mukherjee SC. Autosomal dominant cerebellar ataxias in ethnic Bengalees in West Bengal – an Eastern Indian state. Acta Neurol Scand. 2002;105:202–8. doi: 10.1034/j.1600-0404.2002.1o054.x. [DOI] [PubMed] [Google Scholar]
- 12.Bhattacharyya KB, Hire B, Misra A, Bose P, Basu S, Seshadri M. Clinical features and molecular genetics of adult onset dominant cerebellar ataxias in ethnic Bengalees of India Basal Ganglia (2012) Contents lists available at SciVerse ScienceDirect Basal Ganglia journal homepage. 2012;2:109–13. Available from: http://www.elsevier.com/locate/baga . [Google Scholar]
- 13.Sinha KK, Worth PF, Jha DK, Sinha S, Stinton VJ, Davis MB, et al. Autosomal dominant cerebellar ataxia: SCA2 is the most frequent mutation in eastern India. J Neurol Neurosurg Psychiatry. 2004;75:448–52. doi: 10.1136/jnnp.2002.004895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khadilkar SV, Dabi R, Dhonde P, Nadkarni N, Kulkarni S, Sarnath D. Trinucleotide repeat spinocerebellar ataxias: Experience of a tertiary care centre in Western India with review of Indian literature. Neurology Asia. 2012;17:213–7. [Google Scholar]
- 15.Wadia RS, Amin RB, Divate UP, Divate PG, Sainani GS, Sardesai HV. Autosomal dominant spinocerebellar ataxia with slow eye movements-a common hereditary ataxia in Western India. J Assoc Physicians India. 1976;24:367–71. [PubMed] [Google Scholar]
- 16.Srivastava AK, Choudhry S, Gopinath MS, Roy S, Tripathi M, Brahmachari SK, et al. Molecular and clinical correlation in five Indian families with spinocerebellar ataxia 12. Ann Neurol. 2001;50:796–800. doi: 10.1002/ana.10048. [DOI] [PubMed] [Google Scholar]
- 17.Krishna N, Mohan S, Yashavantha BS, Rammurthy A, Kiran Kumar HB, Mittal U, et al. SCA 1, SCA 2 and SCA 3/MJD mutations in ataxia syndromes in southern India. Indian J Med Res. 2007;126:465–70. [PubMed] [Google Scholar]
- 18.Lee WY, Jin DK, Oh MR, Lee JE, Song SM, Lee EA, et al. Frequency analysis and clinical characterization of spinocerebellar ataxia types 1, 2, 3, 6 and 7 in Korean Patients. Arch Neurol. 2003;60:858–63. doi: 10.1001/archneur.60.6.858. [DOI] [PubMed] [Google Scholar]
- 19.Teive HA, Munhoz RP, Arruda WO, Lopes-Cendes I, Raskin S, Werneck LC, et al. Spinocerebellar ataxias: Genotype-phenotype correlations in 104 Brazilian families. Clinics (Sao Paulo) 2012;67:443–9. doi: 10.6061/clinics/2012(05)07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kraft S, Furtado S, Ranawaya R, Parboosingh J, Bleoo S, McElligott K, et al. Adult onset spinocerebellar ataxia in a Canadian movement disorders clinic. Can J Neurol Sci. 2005;32:450–8. doi: 10.1017/s0317167100004431. [DOI] [PubMed] [Google Scholar]
- 21.Leggo J, Dalton A, Morrison PJ, Dodge A, Connarty M, Kotze MJ, et al. Analysis of spinocerebellar ataxia types 1, 2, 3, and 6, dentatorubral-pallidoluysian atrophy, and Friedreich's ataxia genes in spinocerebellar ataxia patients in the UK. J Med Genet. 1997;34:982–5. doi: 10.1136/jmg.34.12.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bryer A, Krause A, Bill P, Davids V, Bryant D, Butler J, et al. The hereditary adult-onset ataxias in South Africa. J Neurol Sci. 2003;216:47–54. doi: 10.1016/s0022-510x(03)00209-0. [DOI] [PubMed] [Google Scholar]
- 23.Takano H, Cancel G, Ikeuchi T, Lorenzetti D, Mawad R, Stevanin G, et al. Close associations between prevalences of dominantly inherited spinocerebellar ataxias with CAG-repeat expansions and frequencies of large normal CAG alleles in Japanese and Caucasian populations. Am J Hum Genet. 1998;63:1060–6. doi: 10.1086/302067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mittal U, Sharma S, Chopra R, Dheeraj K, Pal PK, Srivastava AK, et al. Insights into the mutational history and prevalence of SCA1 in the Indian population through anchored polymorphisms. Hum Genet. 2005;118:107–14. doi: 10.1007/s00439-005-0018-8. [DOI] [PubMed] [Google Scholar]
- 25.Matsuyama Z, Izumi Y, Kameyama M, Kawakami H, Nakamura S. The effect of CAT trinucleotide Interruptions on the age at onset of spinocerebellar ataxia type 1 (SCA1) J Med Genet. 1999;36:546–8. [PMC free article] [PubMed] [Google Scholar]
- 26.Tang B, Liu C, Shen L, Dai H, Pan Q, Jing L, et al. Frequency of SCA1, SCA2, SCA3/ MJD, SCA6, SCA7, and DRPLA CAG trinucleotide repeat expansion in patients with hereditary spinocerebellar ataxia from Chinese kindreds. Arch Neurol. 2000;57:540–4. doi: 10.1001/archneur.57.4.540. [DOI] [PubMed] [Google Scholar]
- 27.Juvonen V, Hietala M, Kairisto V, Savontaus ML. The occurrence of dominant spinocerebellar ataxias among 251 Finnish ataxia patients and the role of predisposing large normal alleles in a genetically isolated population. Acta Neurol Scand. 2005;111:154–62. doi: 10.1111/j.1600-0404.2005.00349.x. [DOI] [PubMed] [Google Scholar]
- 28.Soong BW, Lu YC, Choo KB, Lee HY. Frequency analysis of autosomal dominant cerebellar ataxias in Taiwanese patients and clinical and molecular characterization of spinocerebellar ataxia type 6. Arch Neurol. 2001;58:1105–9. doi: 10.1001/archneur.58.7.1105. [DOI] [PubMed] [Google Scholar]
- 29.Jiang H, Tang B, Xia K, Zhou Y, Xu B, Zhao G, et al. Spinocerebellar ataxia type 6 in Mainland China: Molecular and clinical features in four families. J Neuroi Sci. 2005;236:25–9. doi: 10.1016/j.jns.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 30.Fujigasaki H, Verma IC, Camuzat A, Margolis RL, Zander C, Lebre AS, et al. SCA12 is a rare locus for autosomal dominant cerebellar ataxia: A study of an Indian family. Ann Neurol. 2001;49:117–21. [PubMed] [Google Scholar]