

Abstract
In infants exposed to maternal stress in utero, phenotypic plasticity through epigenetic events may mechanistically explain increased risk of preterm birth (PTB), which confers increased risk for neurodevelopmental disorders, cardiovascular disease, and cancers in adulthood. We examined associations between prenatal maternal stress and PTB, evaluating the role of DNA methylation at imprint regulatory regions. We enrolled women from prenatal clinics in Durham, NC. Stress was measured in 537 women at 12 weeks of gestation using the Perceived Stress Scale. DNA methylation at differentially methylated regions (DMRs) associated with H19, IGF2, MEG3, MEST, SGCE/PEG10, PEG3, NNAT, and PLAGL1 was measured from peripheral and cord blood using bisulfite pyrosequencing in a sub-sample of 79 mother–infant pairs. We examined associations between PTB and stress and evaluated differences in DNA methylation at each DMR by stress. Maternal stress was not associated with PTB (OR = 0.98; 95% CI, 0.40–2.40; P = 0.96), after adjustment for maternal body mass index (BMI), income, and raised blood pressure. However, elevated stress was associated with higher infant DNA methylation at the MEST DMR (2.8% difference, P < 0.01) after adjusting for PTB. Maternal stress may be associated with epigenetic changes at MEST, a gene relevant to maternal care and obesity. Reduced prenatal stress may support the epigenomic profile of a healthy infant.
Keywords: epigenetics, intrauterine growth restriction (IUGR), perinatal, pregnancy, imprinting, perceived stress
Introduction
Infant mortality rates in the United States lag behind most other developed countries, especially within minority groups, and much of the mortality is attributed to high rates of preterm birth (PTB).1 Moreover, PTB has been associated with an increased risk of neurodevelopmental disorders, coronary artery disease, stroke, type 2 diabetes, and obesity in adulthood.2–5 Multiple factors may contribute to increasing risk of PTB, and epidemiological studies have shown that physiologic stressors are important risk factors of poor birth outcomes.6–9 This supports the early origins of disease hypothesis, which postulates that in utero exposures may affect adult health. More than 25 years ago, Barker and others hypothesized that poor conditions in utero lead to poor birth outcomes, which may increase susceptibility to cardiovascular disease in adulthood.2,10,11 In support of this hypothesis, victims of the 1944–1945 Dutch Hunger Winter and the 1959–1961 Chinese famine who were exposed to severe caloric restriction in utero had increased risk of poor birth outcomes, obesity, hypertension, dyslipidemia, and schizophrenia later in life.3,5
Epigenetic alterations because of adverse exposures in utero have been hypothesized to be linked with later risk of common chronic diseases.10,12 The underlying concept is based on the idea of phenotypic plasticity—the ability of cells to adapt in response to internal or external environmental cues.12,13 Imprinted genes are expressed from only a single parental allele, are important in regulating early growth and development including neural development, and rely on DNA methylation for their appropriate expression. As such, the epigenetic patterns regulating imprinted genes may function to adapt gene expression patterns in response to the perceived in utero environmental conditions.13,14 DNA methylation at imprinted loci is established differentially during gametogenesis in a sex-dependent and parent-of-origin-specific manner.13,14 The differentially methylated regions (DMRs) are thus entrenched prior to germ layer specification, and these methylation patterns are faithfully transmitted to daughter cells during somatic cell division and thereby perpetuated throughout life in all tissues.14 An example of this comes from the study of the Dutch famine survivors exposed early in gestation, who exhibited persistent lower levels of DNA methylation at the DMR regulating imprinted insulin-like growth factor II (IGF2) compared to unexposed same-sex siblings, six decades after the exposure.15 Lower methylation at this DMR has been associated with biallelic expression of this gene, leading to deregulated growth,16 elevated plasma IGF2 protein concentrations, and high birth weight,17 as well as increased risk of colon cancer.18
Animal studies have shown that stress during pregnancy can affect the epigenome and offspring physiology,19 and have found that high levels of maternal licking/grooming modified offspring stress responses by dampening hypothalamus–pituitary–adrenal (HPA) axis responses to stress through increased glucocorticoid receptor (GR) expression and negative feedback.20 These changes in the offspring occured as a result of epigenetic alterations, including DNA demethylation and increased histone acetylation.21
Our previous work showed that severe maternal depressed mood was associated with a three-fold increase in the risk of low birth weight and that DNA methylation levels at the offspring MEG3 DMR differed significantly by maternal depressed mood, measured by using the Centers for Epidemiologic Studies Depression Scale (CES-D) [22]. Moreover, maternal stress, especially in the first trimester, has previously been associated with an increased risk of PTB.6,7,22,23 However, the potential for an epigenetic response to be involved in mediating these effects has not been evaluated, and we hypothesized that epigenetic plasticity may mechanistically explain some of the association between prenatal stress and PTB. Herein, we evaluate the association between prenatal maternal stress, measured by using the Perceived Stress Scale (PSS10), and PTB and examine the role of DMR methylation at imprinted genes H19, IGF2, MEG3, MEST, SGCE/PEG10, PEG3, NNAT, and PLAGL1 in these associations.
Methods
Study participants, data collection, and ethical approval
Participants were identified from the cohort of pregnant women recruited as part of the Newborn Epigenetics STudy (NEST), a prospective study of mother/infant dyads aimed at investigating the effects of in utero exposures on epigenetic profiles and phenotypes in children. The target population and methods for participant identification and enrollment are similar to those described in detail previously.24,25 Briefly, between 2009 and 2011, pregnant women were recruited during their first or second trimester visit to prenatal clinics serving Duke or Durham Regional Hospitals, the two obstetrics facilities serving the County of Durham, North Carolina. The gestational age at enrollment ranged from 4 to 32.5 weeks (mean and median of 12 and 11 weeks, respectively). The eligibility criteria included women of 18 years or older, having intention to use Duke Hospital or Durham Regional Hospital for delivery, and speaking English and/or Spanish. We excluded women who planned to give up custody of their children and women with HIV because of the limited research on the interaction of HIV with DNA methylation alterations in the offspring. This study was approved by the Duke University Institutional Review Board (IRB) and was conducted in accordance with the Declaration of Helsinki and guidelines established by the Federal Government for the protection of human subjects.
After providing written, informed consent, pregnant women either self-completed or had an interviewer-administered questionnaire at enrollment that solicited information on sociodemographic characteristics, maternal health, and lifestyle factors. Questionnaires were administered in both English and Spanish, and a native Spanish speaker provided forward and back translations, as is required by the Duke University IRB. The questionnaire included the Cohen Perceived Stress scale (PSS10).26 Women also self-reported age, race, ethnicity, level of education, income, parity, marital status, health status, cigarette smoking status, vitamin and mineral supplement use, height, weight at last menstrual period (LMP), alcohol use, psychotropic medication use, raised blood pressure during current pregnancy, and anemia during current pregnancy. Trained personnel abstracted data from medical records at delivery including gestational age at birth, infant sex, and birth weight. PTB was defined as gestational age <37 weeks, and low birth weight was defined as birth weight <2500 g.27
Between July 2009 and December 2011, 2548 women were approached and 1700 (66.6%) consented to participate. The 848 women who declined were similar to those who consented with respect to age (P = 0.70) but not race/ethnicity (P < 0.001), as Asian and Native American were more likely to decline participation, while other racial and ethnic groups were similar. Of the 1700 women, 396 were withdrawn because of miscarriage (n = 109), death of infant after birth (n = 4), illiteracy (n = 1), being under age 18 years (n = 1), moved from the region (n = 21), refusing further participation (n = 146), or giving birth at a hospital other than Duke or Durham Regional Hospital (n = 114). Thus, 1304 (76.7%) women remained enrolled in the study up to the time of analysis. These analyses are limited to the 537 women in whom stress and parturition data were available. These women were similar to those of the larger study with respect to percentages of PTB (P = 0.330) but were of younger maternal age (P = 0.0845) with lower numbers of Asian and Native American women than the entire cohort (P < 0.01).
Measurement of maternal stress during pregnancy
Stress was measured using the Cohen Perceived Stress Scale (PSS10), a well-validated scale with Cronbach’s alpha of 0.85.26,28 It has been found to correlate well with life event scales,28 depression scales,28 and predicted health outcomes (physical symptomatology and visits to a health care center), better than life event measures.28 Hedegaard et al found that the significant association between stressful life events and birth outcomes was dependent on the participant’s appraisal of that event and that there was no significant association between the stressful events alone on birth outcomes.29 We therefore chose PSS10 because it measures the perceived effect of stress as assessed by participants. PSS10 was scored according to established protocol.26,28 Questions 4 (in the last month, how often have you felt confident about your ability to handle your personal problems?), 5 (in the last month, how often have you felt that things were going your way?), 7 (in the last month, how often have you been able to control irritations in your life?), and 8 (in the last month, how often have you felt that you were on top of things?) were scored positively, while the rest were scored negatively. The sum of all 10 questions was calculated for the final PSS10 score. Stress was dichotomized at the 75th percentile, corresponding to a score of 19 to define high stress (top 25th percentile of PSS score), with the bottom 25th percentile of PSS score defining low stress. There is no recommended cut-point for high stress; researchers have stratified this continuous variable using study-specific thresholds.30
DNA methylation analysis
Methylation was evaluated in the context of stress for a subset of 79 mother–infant pairs where completed stress and methylation data were available (56% Black, 28% White, 13% Hispanic, and 3% Other; mean maternal age 27.71 years (SD = 5.87 years), 55% male infants, and 45% female infants). Infant cord blood specimens were collected at birth. Samples were collected in EDTA-containing vacutainer tubes and centrifuged using standard protocols to allow for collection of plasma and buffy coat, with buffy coat used for DNA extraction (Qiagen; Valencia, CA). Specimens were stored at −80°C until the time of analysis. DNA was extracted using Puregene reagents according to the manufacturer’s protocol (Qiagen), and quantity and quality were assessed using a Nanodrop 1000 Spectrophotometer (Thermo Scientific; Wilmington, DE). Maternal and infant genomic DNA (800 ng) were modified by treatment with sodium bisulfite using the EZ DNA Methylation kit (Zymo Research; Irvine, CA). Bisulfite treatment of denatured DNA converts all unmethylated cytosines to uracils, leaving methylated cytosines unchanged, allowing for quantitative measurement of cytosine methylation status. Pyrosequencing was performed using a PyroMark Q96 MD pyrosequencer (Qiagen). The bisulfite pyrosequencing assays were utilized to quantitatively measure the level of methylation at CpG sites contained within nine imprinted DMRs for both mothers and infants. DMRs analyzed were the paternally methylated IGF2 DMR, H19 DMR, MEG3-IG DMR (located intergenic to DLK1 and MEG3), and MEG3 DMR (promoter); and the maternally methylated PEG3 DMR, MEST DMR, SGCE/PEG10 DMR, NNAT DMR, and PLAGL1 DMR. Pyrosequencing assay design, genomic coordinates, assay conditions, and assay validation are described in detail elsewhere.14,31 Briefly, assays were designed to query established imprinted gene DMRs using the PyroMark Assay Design Software (Qiagen). PCR conditions were optimized to produce a single, robust amplification product. Defined mixtures of fully methylated and unmethylated control DNAs were used to show a linear increase in detection of methylation values as the level of input DNA methylation increased (Pearson r is >0.99 for all DMRs). Once optimal conditions were defined, each DMR was analyzed using the same amount of input DNA from each specimen (40 ng, assuming complete recovery following bisulfite modification of 800 ng DNA). Percentage of methylation for each CpG cytosine was determined using Pyro Q-CpG software (Qiagen). Pyrosequencing assays were performed in duplicate for all specimens whose values fell more than two standard deviations above or below the means in which case the average of the two runs was used. The values obtained represent the mean methylation for the CpG sites contained within the sequence being analyzed.
Statistical analysis
Pearson’s chi-squared tests were performed to compare the distribution of demographic and obstetric descriptors among women with high stress and no stress during pregnancy. Multivariable logistic regression models were fit to examine the relationship between maternal stress and the birth outcomes of PTB, adjusting for covariates. Initial models were fit using all variables considered clinically important, including maternal age, race/ethnicity, marital status, employment, parity, household income, education, maternal health status, maternal body mass index (BMI) at LMP, smoking, alcohol use, psychotropic medicine use, folic acid supplementation, infant sex, delivery mode, raised maternal blood pressure, maternal anemia, and trimester of enrollment. All non-binary variables were added to multivariable logistic regression models using indicator variables. A backward stepwise approach (exclusion P > 0.10 and inclusion P < 0.09) was used to refine the model, and log-likelihood tests were used to create the final parsimonious model, which was adjusted for BMI, income, and raised blood pressure.
Among the 79 mother–infant pairs where maternal and infant methylation data at the nine DMRs and stress data were available, we also examined the extent to which DMR methylation in infants was associated with increased stress. These mother–infant pairs were similar to the larger sample of 591 women with respect to distribution of high (top 25th percentile PSS) and low stresses (P = 0.87) and mean stress score (P = 0.29) during pregnancy.
We first assessed each infant DMR for normality using the Kolmogorov–Smirnov test. We found that with the exception of PLAGL1 (P < 0.01), PEG3 (P < 0.01), MEG3-IG (P < 0.01), and SGCE/PEG10 (P = 0.03), all other infant DMRs were normally distributed (P > 0.05). Confirmatory factor analysis of individual infant CpGs revealed that Cronbach’s alphas for all DMRs were >0.83 suggesting that mean methylation levels for each DMR could be used in models.
T-tests were used to compare infant DNA methylation differences at DMRs by high and low stresses. Wilcoxon-rank sum tests were used for DMRs that were not normally distributed. In addition, methylation values for the DMRs that were significantly associated with stress were added individually to the overall multivariate models of stress and PTB described above. DMRs that resulted in attenuation of the odds ratio (ORs) between stress and PTB by >10% and significantly changed the overall model before and after inclusion (log-likelihood P-value <0.05) were considered possible effect modifiers of the association between stress and PTB. We repeated these analyses in DMRs where significant differences were noted, restricted by sex to explore whether these associations varied by sex. All analyses were conducted using STATA 12.0 (StataCorp College Station, TX), and a P-value <0.05 was set as the threshold for statistical significance.
Results
Maternal demographics
Of the 537 women, 122 had high stress (Table 1). Scores on the Perceived Stress Scale (PSS10) were normally distributed (P = 0.35) and ranged from 0 to 40 with a median of 15 and a mean of 14.8 (SD = 6.81). Women with high stress were more likely to be Black (P = 0.05). Women with high stress were also more likely to be from low-income households (P = 0.01) and were likely to smoke during pregnancy (P < 0.01). Participants were comparable with respect to maternal age (P = 0.11), parity (P = 0.14), raised blood pressure (P = 0.15), maternal BMI at LMP (P = 0.58), folic acid use (P = 0.20), infant sex (P = 0.66), and delivery mode (P = 0.51).
Table 1.
Maternal demographics by stress in the NEST cohort.
| VARIABLE | LOW STRESS (n = 415) | HIGH STRESS (TOP 25th PERCENTILE) (n = 122) | P-VALUE* |
|---|---|---|---|
| Maternal age (years) | |||
| 18 to <20 | 9 (2%) | 1 (1%) | 0.11 |
| 20 to 29 | 221 (53%) | 78 (65%) | |
| 30 to 39 | 167 (40%) | 38 (32%) | |
| 40+ | 18 (5%) | 2 (2%) | |
| Mean age (SD) | 29 (5.86) | 27.5 (5.45) | |
| Maternal Race/Ethnicity* | |||
| White | 127 (31%) | 25 (21%) | 0.051 |
| Black | 194 (47%) | 65 (56%) | |
| Hispanic | 81 (19%) | 24 (20%) | |
| Other | 13 (3%) | 3 (3%) | |
| Parity | |||
| Nulliparous | 141 (35%) | 41 (36%) | 0.14 |
| Multiparous | 264 (65%) | 74 (64%) | |
| Household income* | |||
| <$10,000 | 69 (20%) | 32 (33%) | 0.014 |
| $10,000–24,999 | 88 (25%) | 29 (30%) | |
| $25,000–$49,999 | 58 (17%) | 16 (17%) | |
| $50,000–$100,000 | 83 (24%) | 15 (15%) | |
| >$100,000 | 48 (14%) | 5 (5%) | |
| Raised blood pressure | |||
| Yes | 26 (6%) | 8 (7%) | 0.15 |
| No | 389 (94%) | 105 (93%) | |
| Maternal BMI at LMP | |||
| <18.5 | 16 (5%) | 3 (3%) | 0.58 |
| 18.5–<25 | 132 (38%) | 37 (39%) | |
| 25–<30 | 89 (25%) | 25 (26%) | |
| 30–<35 | 64 (18%) | 14 (15%) | |
| 35–<40 | 27 (8%) | 7 (7%) | |
| 40+ | 21 (6%) | 9 (10%) | |
| Mean (SD) | 29 (7.46) | 30 (7.96) | |
| Maternal smoking* | |||
| Smoking prior to pregnancy | 37 (9%) | 11 (9%) | <0.01 |
| Smoking during pregnancy | 70 (17%) | 30 (26%) | |
| No smoking | 304 (74%) | 76 (65%) | |
| Folic acid supplementation | |||
| Yes | 297 (72%) | 75 (64%) | 0.20 |
| No | 115 (28%) | 43 (36%) | |
| Infant sex | |||
| Male | 197 (54%) | 58 (59%) | 0.66 |
| Female | 171 (46%) | 41 (41%) | |
| Delivery mode | |||
| Vaginal | 251 (68%) | 69 (69%) | 0.51 |
| C-section | 117 (32%) | 31 (31%) | |
Notes:
Chi-squared P-value. Numbers do not necessarily match due to missing values. This table describes the demographic characteristics of the mother–infant dyads included in our study grouped by the exposures of prenatal high versus low stress. Chi-squared tests were used to assess any differences in the distribution of these demographic variables between the two groups.
Associations between stress and birth outcomes
For infants of women with high stress, birth weight ranged from 1425 to 4370 g (mean 3294 g) and gestational age ranged from 24 to 41 weeks with a mean of 39 weeks. For infants of women without high stress, birth weight ranged from 999 to 5025 g (mean 3260 g) and gestational age ranged from 29 to 42 weeks (mean 39 weeks). Although stress was moderately correlated with PTB (correlation coefficient 0.24, P < 0.001), it was not associated with risk of PTB (OR = 1.10; 95% CI, 0.57–2.15; P = 0.96) (Table 2) compared to women with low stress, after adjustment for maternal BMI, income, and raised blood pressure.
Table 2.
Associations between stress and PTB in NEST.
| TERM BIRTH (≥37 WEEKS GESTATION) | PTB (<37 WEEKS GESTATION) | OR | ADJUSTED OR* | |
|---|---|---|---|---|
| High stress | 109 (89%) | 13 (11%) | 1.10 (0.57–2.15), p = 0.77 | 0.98 (0.40–2.40), p = 0.96 |
| Low stress | 352 (90%) | 38 (10%) | 1 | 1 |
Notes:
Model adjusted for maternal BMI, income, and raised blood pressure. This table describes the logistic regression models of stress with PTB. No associations were found between maternal stress during pregnancy and PTB, even after adjustment for maternal BMI, income, and raised blood pressure.
DNA methylation at imprint regulatory regions
High maternal stress was associated with a 2.8% increase in infant DNA methylation at the MEST DMR as compared to infants of women with low stress (44.2% (n = 19) vs. 41.4% (n = 60); P < 0.01) (Fig. 1). This difference was maintained in female infants (n = 35) born to women with high stress as compared to those born to women with low stress (45.9% (n = 6) vs. 40.9% (n = 29); P < 0.01), but there was no significant difference in the 43 male infants analyzed (43.7% (n = 12) vs. 41.8% (n = 31); P = 0.09) (Fig. 2). However, the cross-product term for the relationship between stress and DNA methylation at MEST by infant sex was not statistically significant (interaction term P-value = 0.12). To minimize the possibility that our findings were wholly influenced by extreme values, we repeated the analyses with four outliers excluded (27.2, 32.4, 34.3, and 34.3%), and the pattern of these differences was unchanged with the exception of a detectable significant difference in methylation at the MEST DMR in the males born to stressed vs. non-stressed women (42.4 vs. 44.6%, P = 0.01). All of these methylation differences persisted even after adjustment for PTB. However, adding the MEST DMR into final models of maternal stress and PTB with the covariates maternal BMI, income, and raised blood pressure suggested that DNA methylation levels at MEST did not significantly contribute to models of stress (log-likelihood P = 0.40). The Pearson correlation coefficient between stress score as a continuous variable and infant DNA methylation at MEST was 0.20, P = 0.08. There were no differences in infant DNA methylation at any of the other eight DMRs examined by maternal stress (Table 3).
Figure 1.
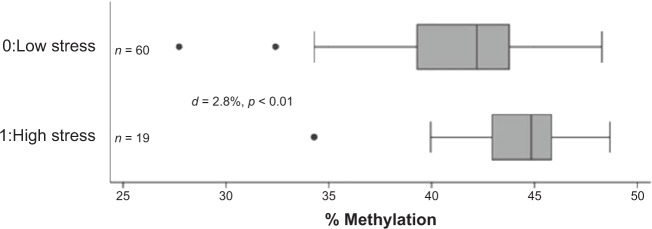
Infant DNA methylation at MEST by stress. Median and interquartile ranges of infant DNA methylation levels at MEST by high and low stress. Overall, high stress was associated with increased infant methylation (mean 44.2 vs. 41.4%, P < 0.01) using t-test.
Abbreviation: d, difference in percentage of methylation levels.
Figure 2.
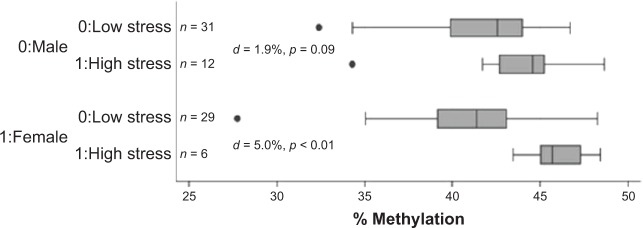
Infant DNA methylation at MEST by stress and infant gender. Median and interquartile ranges of infant DNA methylation levels at MEST by high and low stress. When analyses were stratified by infant gender, high stress was associated with increased infant methylation in female (45.9 vs. 40.9%, P < 0.01) but not in male infants (43.7 vs. 41.8%, P = 0.09).
Abbreviation: d, difference in percentage of methylation levels.
Table 3.
Infant DNA methylation by high vs. low stress.
| DMR | HIGH STRESS | LOW STRESS |
t-TEST P-VALUE |
||
|---|---|---|---|---|---|
| n | MEAN (SD) | n | MEAN (SD) | ||
| IGF2 | 18 | 50.2 (4.9) | 59 | 49.2 (3.9) | 0.39 |
| H19 | 16 | 46.5 (2.6) | 60 | 45.3 (3.8) | 0.25 |
| MEG3-IG | 15 | 49.6 (2.5) | 54 | 48.4 (3.7) | 0.24 |
| MEG3 | 16 | 72.2 (6.0) | 52 | 71.5 (5.1) | 0.67 |
| MEST | 19 | 44.2 (3.3) | 60 | 41.4 (3.6) | 0.0028 |
| PEG3 | 17 | 35.6 (1.7) | 58 | 35.5 (4.9) | 0.94 |
| NNAT | 17 | 53.1 (6.1) | 56 | 54.9 (5.6) | 0.25 |
| PLAGL1 | 20 | 55.6 (4.9) | 69 | 55.4 (4.5) | 0.85 |
| SGCE/PEG10 | 18 | 45.1 (10.8) | 62 | 41.4 (6.8) | 0.08 |
| Bonferroni correction = 0.05/9 = 0.006 | |||||
Notes: This table shows the differences in infant mean methylation levels at the nine DMRs by stress, as assessed by t-tests. DNA methylation levels are higher at the DMR regulating MEST in infants exposed to stress as compared to those not exposed to stress in pregnancy, even after Bonferroni correction.
Discussion
We examined the association between maternal stress during pregnancy and PTB to determine whether certain imprint regulatory regions played a role in this association. We found maternal stress during pregnancy was not a risk predictor for PTB, after adjustment for maternal BMI, household income, and raised blood pressure. However, high levels of maternal stress during pregnancy were associated with increased infant methylation at the MEST DMR. No evidence of a direct mediating effect of methylation at MEST was found, as addition of the MEST DMR variable into crude or unadjusted models of stress and PTB did not show significant contributions.
Prior studies in human cohorts revealed an association between perceived stress, distress, and stressful life events in pregnancy and increased risk of PTB, irrespective of race.9,22,23,32 Stress-related changes in endocrine and immune function may play a role in how stress alters the course of pregnancy.23 Psychosocial stressors may be associated with higher circulating levels of inflammatory markers, potentially increasing the risk of poor birth outcomes.23 Additionally, stress may be an important risk factor for poor birth outcomes in minority populations.33 Geronimus34 found that Black women on average and those with low incomes in particular had higher risk of LBW and very low birth weight (VLBW) infants. Although some studies have shown that stress and distressful life events predict LBW,7,35 our study did not confirm these findings, as any associations were attenuated after adjustment for PTB in models. This may be a result of the overlap between PTB and LBW in many infants, as well as a limited sample size.
The developmental origins, of disease hypothesis suggests that early exposures, such as stress, may affect birth and long-term health outcomes through epigenetic changes and consequent phenotypic plasticity.2,10 In support of this hypothesis, Mulligan et al found that maternal prenatal stress was significantly correlated with infant birth weight and methylation at the promoter of the non-imprinted GRNR3C1, a region associated with growth.36 We found that high maternal stress was associated with an increase in infant DNA methylation at the MEST DMR as compared to infants of women with low stress. MEST is a maternally methylated imprinted gene on chromosome 7 that has been implicated in Silver–Russell syndrome (SRS), a syndrome of growth retardation.37 Mest deficiency in mice is associated with abnormal maternal behavior, including deficiencies in rearing of pups as well as growth retardation.38 The biological function of the MEST protein however remains unclear; it may be involved in the metabolism pathways that affect growth and maintenance of mesodermal cells via its hydrolase activity.37–39 Recent evidence from studies in mice suggests that Mest is up-regulated in response to a high fat diet regimen, and that this increase foretells expansion of adipose tissue.40 In the offspring exposed to stress, increased methylation may alter adipocyte expansion.41,42 Additionally, in infants of women with insulin- and non-insulin-dependent gestational diabetes mellitus, decreased levels of methylation (4–7 percentage points) at MEST were found in cord blood and placental tissue samples as compared to control infants of mothers without diabetes.43 Decreased levels of methylation at MEST were also found in adults with morbid obesity as compared to those of normal-weight controls.43 Maternal stress may impact methylation at the MEST DMR and consequently affect growth and development of the infant. Our study did not find a direct mediating effect, and the role of methylation in this association may be complex and may involve higher order regulatory networks.44
Study strengths include the usage of a combination of validated scales and self-reported responses to characterize our exposure. Multiple comparisons were made, Bonferroni corrections were used, and significant differences at MEST in infants exposed to high stress as compared to those who were not exposed to stress were still maintained. We found sex-specific DNA methylation differences at MEST for infants born to women with high and low stress, which is consistent with other studies.24,45–47
Our study is limited by a modest sample size, which while adequate for psychosocial measures may have been insufficient for evaluating DNA methylation. Furthermore, sample size was limited to stratify analyses by race. Although we only measured stress at one point during gestation, prior research indicates that stress during months 5 and 6 of gestation is associated with the greatest risk of PTB,6 and our measurement of stress during the first trimester may be an underestimate of exposure. In addition, we examined nine imprinted regulatory regions implicated as important in growth and development; this does not encompass the entirety of the imprintome.48 Finally, cord blood may not be optimal for epigenetic studies, although our group has shown that methylation patterns are relatively consistent across tissue types and within cord blood fractions.14 Other studies assessing methylation levels are needed to corroborate our work.
Conclusions
We found methylation alterations at the DMRs regulating MEST in infants were associated with increased prenatal stress. Future studies, including genome-wide analysis, are needed to replicate these findings and to further elucidate these complex regulatory networks of imprinted genes and how they interact to affect health outcomes.
Acknowledgments
We are especially grateful to the women and families involved in the NEST, and acknowledge the expert contributions of study coordinator Stacy Murray, research nurse Tammy Bishop, and laboratory technicians Carole Grenier, Erin Erginer, Cara Davis, and Allison Barratt.
Glossary
Abbreviations
- DMR
differentially methylated region
- PSS10
Perceived Stress Scale
- PTB
preterm birth
- BMI
body mass index
- LMP
last menstrual period
- NEST
Newborn Epigenetics STudy
- IQR
interquartile range
- C-section
cesarean section
Footnotes
Author Contributions
Conceived and designed the experiments: SKM, CH. Analyzed the data: YL, ESI, ACV. Wrote the first draft of the manuscript: YL, AMT, SKM. Contributed to the writing of the manuscript: SEBN, BFF, APM, JS, RLJ. Agree with manuscript results and conclusions: ACV, SEBN, YL, AMT, BFF, CH, APM, ZH, JS, FO, JK, RLJ, ESI, SKM. Jointly developed the structure and arguments for the paper: SKM, CH. Made critical revisions and approved final version: ACV, SKM. All authors reviewed and approved of the final manuscript.
ACADEMIC EDITOR: Christian Bronner, Editor in Chief
FUNDING: This work was funded through the US National Institutes of Health (NIH) (agreements R01ES016772 and P01ES022831) and by the USEPA (RD-83543701). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the USEPA. Further, the NIH and the USEPA do not endorse the purchase of any commercial products or services mentioned in the publication.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
This paper was subject to independent, expert peer review by a minimum of two blind peer reviewers. All editorial decisions were made by the independent academic editor. All authors have provided signed confirmation of their compliance with ethical and legal obligations including (but not limited to) use of any copyrighted material, compliance with ICMJE authorship and competing interests disclosure guidelines and, where applicable, compliance with legal and ethical guidelines on human and animal research participants.
REFERENCES
- 1.Mathews TJ, MacDorman MF. Infant mortality statistics from the 2007 period linked birth/infant death data set. Natl Vital Stat Rep. 2011;59:1–31. [PubMed] [Google Scholar]
- 2.Barker DJ. The origins of the developmental origins theory. J Intern Med. 2007;261:412–417. doi: 10.1111/j.1365-2796.2007.01809.x. [DOI] [PubMed] [Google Scholar]
- 3.Lumey LH, Stein AD, Kahn HS, Romijn JA. Lipid profiles in middle-aged men and women after famine exposure during gestation: the Dutch Hunger Winter Families Study. Am J Clin Nutr. 2009;89:1737–1743. doi: 10.3945/ajcn.2008.27038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ravelli GP, Stein ZA, Susser MW. Obesity in young men after famine exposure in utero and early infancy. N Engl J Med. 1976;295:349–353. doi: 10.1056/NEJM197608122950701. [DOI] [PubMed] [Google Scholar]
- 5.Huang C, Li Z, Wang M, Martorell R. Early life exposure to the 1959–1961 Chinese famine has long-term health consequences. J Nutr. 2010;140:1874–1878. doi: 10.3945/jn.110.121293. [DOI] [PubMed] [Google Scholar]
- 6.Class QA, Lichtenstein P, Langstrom N, D’Onofrio BM. Timing of prenatal maternal exposure to severe life events and adverse pregnancy outcomes: a population study of 2.6 million pregnancies. Psychosom Med. 2011;73:234–241. doi: 10.1097/PSY.0b013e31820a62ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Copper RL, Goldenberg RL, Das A, et al. The preterm prediction study: maternal stress is associated with spontaneous preterm birth at less than thirty-five weeks’ gestation. National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network. Am J Obstet Gynecol. 1996;175:1286–1292. doi: 10.1016/s0002-9378(96)70042-x. [DOI] [PubMed] [Google Scholar]
- 8.Tegethoff M, Greene N, Olsen J, Meyer AH, Meinlschmidt G. Maternal psychosocial adversity during pregnancy is associated with length of gestation and offspring size at birth: evidence from a population-based cohort study. Psychosom Med. 2010;72:419–426. doi: 10.1097/PSY.0b013e3181d2f0b0. [DOI] [PubMed] [Google Scholar]
- 9.Lobel M, Cannella DL, Graham JE, DeVincent C, Schneider J, Meyer BA. Pregnancy-specific stress, prenatal health behaviors, and birth outcomes. Health Psychol. 2008;27:604–615. doi: 10.1037/a0013242. [DOI] [PubMed] [Google Scholar]
- 10.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wadhwa PD, Buss C, Entringer S, Swanson JM. Developmental origins of health and disease: brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod Med. 2009;27:358–368. doi: 10.1055/s-0029-1237424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr. 2007;27:363–388. doi: 10.1146/annurev.nutr.27.061406.093705. [DOI] [PubMed] [Google Scholar]
- 13.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy SK, Huang Z, Hoyo C. Differentially methylated regions of imprinted genes in prenatal, perinatal and postnatal human tissues. PLoS One. 2012;7:e40924. doi: 10.1371/journal.pone.0040924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith FM, Garfield AS, Ward A. Regulation of growth and metabolism by imprinted genes. Cytogenet Genome Res. 2006;113:279–291. doi: 10.1159/000090843. [DOI] [PubMed] [Google Scholar]
- 17.Hoyo C, Fortner K, Murtha AP, et al. Association of cord blood methylation fractions at imprinted insulin-like growth factor 2 (IGF2), plasma IGF2, and birth weight. Cancer Causes Control. 2012;23:635–645. doi: 10.1007/s10552-012-9932-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui H, Cruz-Correa M, Giardiello FM, et al. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. 2003;299:1753–1755. doi: 10.1126/science.1080902. [DOI] [PubMed] [Google Scholar]
- 19.Weaver IC, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 20.Weaver IC, D’Alessio AC, Brown SE, et al. The transcription factor nerve growth factor-inducible protein a mediates epigenetic programming: altering epigenetic marks by immediate-early genes. J Neurosci. 2007;27:1756–1768. doi: 10.1523/JNEUROSCI.4164-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weaver IC, Meaney MJ, Szyf M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci U S A. 2006;103:3480–3485. doi: 10.1073/pnas.0507526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hedegaard M, Henriksen TB, Sabroe S, Secher NJ. The relationship between psychological distress during pregnancy and birth weight for gestational age. Acta Obstet Gynecol Scand. 1996;75:32–39. doi: 10.3109/00016349609033280. [DOI] [PubMed] [Google Scholar]
- 23.Coussons-Read ME, Lobel M, Carey JC, et al. The occurrence of preterm delivery is linked to pregnancy-specific distress and elevated inflammatory markers across gestation. Brain Behav Immun. 2012;26:650–659. doi: 10.1016/j.bbi.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Murphy SK, Murtha AP, et al. Depression in pregnancy, infant birth weight and DNA methylation of imprint regulatory elements. Epigenetics. 2012;7:735–746. doi: 10.4161/epi.20734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoyo C, Murtha AP, Schildkraut JM, et al. Folic acid supplementation before and during pregnancy in the Newborn Epigenetics STudy (NEST) BMC Public Health. 2011;11:46. doi: 10.1186/1471-2458-11-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen S, Kamarck T, Mermelstein R. A global measure of perceived stress. J Health Soc Behav. 1983;24:385–396. [PubMed] [Google Scholar]
- 27.Kramer MS. The epidemiology of adverse pregnancy outcomes: an overview. J Nutr. 2003;133:1592S–1596S. doi: 10.1093/jn/133.5.1592S. [DOI] [PubMed] [Google Scholar]
- 28.Cohen S. Perceived stress in a probability sample of the United States. In: Spacapan S, Oskamp S, editors. The Social Psychology of Health. Thousand Oaks, CA, USA: Sage Publications, Inc; 1988. pp. 31–67. [Google Scholar]
- 29.Hedegaard M, Henriksen TB, Secher NJ, Hatch MC, Sabroe S. Do stressful life events affect duration of gestation and risk of preterm delivery? Epidemiology. 1996;7:339–345. doi: 10.1097/00001648-199607000-00001. [DOI] [PubMed] [Google Scholar]
- 30.Krabbendam L, Smits L, de Bie R, Bastiaanssen J, Stelma F, van Os J. The impact of maternal stress on pregnancy outcome in a well-educated Caucasian population. Paediatr Perinat Epidemiol. 2005;19:421–425. doi: 10.1111/j.1365-3016.2005.00679.x. [DOI] [PubMed] [Google Scholar]
- 31.Nye MD, Hoyo C, Huang Z, et al. Associations between methylation of paternally expressed gene 3 (PEG3), cervical intraepithelial neoplasia and invasive cervical cancer. PLoS One. 2013;8:e56325. doi: 10.1371/journal.pone.0056325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dunkel Schetter C. Psychological science on pregnancy: stress processes, biopsychosocial models, and emerging research issues. Annu Rev Psychol. 2011;62:531–558. doi: 10.1146/annurev.psych.031809.130727. [DOI] [PubMed] [Google Scholar]
- 33.Rosenthal L, Lobel M. Explaining racial disparities in adverse birth outcomes: unique sources of stress for Black American women. Soc Sci Med. 2011;72:977–983. doi: 10.1016/j.socscimed.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 34.Geronimus AT. Black/white differences in the relationship of maternal age to birthweight: a population-based test of the weathering hypothesis. Soc Sci Med. 1996;42:589–597. doi: 10.1016/0277-9536(95)00159-x. [DOI] [PubMed] [Google Scholar]
- 35.Sable MR, Wilkinson DS. Impact of perceived stress, major life events and pregnancy attitudes on low birth weight. Fam Plann Perspect. 2000;32:288–294. [PubMed] [Google Scholar]
- 36.Mulligan CJ, D’Errico NC, Stees J, Hughes DA. Methylation changes at NR3C1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics. 2012;7:853–857. doi: 10.4161/epi.21180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kobayashi S, Kohda T, Miyoshi N, et al. Human PEG1/MEST, an imprinted gene on chromosome 7. Hum Mol Genet. 1997;6:781–786. doi: 10.1093/hmg/6.5.781. [DOI] [PubMed] [Google Scholar]
- 38.Lefebvre L, Viville S, Barton SC, Ishino F, Keverne EB, Surani MA. Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat Genet. 1998;20:163–169. doi: 10.1038/2464. [DOI] [PubMed] [Google Scholar]
- 39.Ineson J, Stayner C, Hazlett J, et al. Somatic reactivation of expression of the silent maternal Mest allele and acquisition of normal reproductive behaviour in a colony of Peg1/mest mutant mice. J Reprod Dev. 2012;58(4):490–500. doi: 10.1262/jrd.11-115a. [DOI] [PubMed] [Google Scholar]
- 40.Voigt A, Agnew K, van Schothorst EM, Keijer J, Klaus S. Short-term, high fat feeding-induced changes in white adipose tissue gene expression are highly predictive for long-term changes. Mol Nutr Food Res. 2013;57(8):1423–1434. doi: 10.1002/mnfr.201200671. [DOI] [PubMed] [Google Scholar]
- 41.Kadota Y, Yanagawa M, Nakaya T, Kawakami T, Sato M, Suzuki S. Gene expression of mesoderm-specific transcript is upregulated as preadipocytes differentiate to adipocytes in vitro. J Physiol Sci. 2012;62:403–411. doi: 10.1007/s12576-012-0217-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi M, Kamei Y, Ezaki O. Mest/Peg1 imprinted gene enlarges adipocytes and is a marker of adipocyte size. Am J Physiol Endocrinol Metab. 2005;288:E117–E124. doi: 10.1152/ajpendo.00244.2004. [DOI] [PubMed] [Google Scholar]
- 43.El Hajj N, Pliushch G, Schneider E, et al. Metabolic programming of MEST DNA methylation by intrauterine exposure to gestational diabetes mellitus. Diabetes. 2013;62:1320–1328. doi: 10.2337/db12-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Varrault A, Gueydan C, Delalbre A, et al. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev Cell. 2006;11:711–722. doi: 10.1016/j.devcel.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 45.Tobi EW, Lumey LH, Talens RP, et al. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18:4046–4053. doi: 10.1093/hmg/ddp353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y, Hoyo C, Murphy S, et al. DNA methylation at imprint regulatory regions in preterm birth and infection. Am J Obstet Gynecol. 2013;208(5):395.e1–395.e7. doi: 10.1016/j.ajog.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murphy SK, Adigun A, Huang Z, et al. Gender-specific methylation differences in relation to prenatal exposure to cigarette smoke. Gene. 2012;494:36–43. doi: 10.1016/j.gene.2011.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skaar DA, Li Y, Bernal AJ, Hoyo C, Murphy SK, Jirtle RL. The human imprintome: regulatory mechanisms, methods of ascertainment, and roles in disease susceptibility. ILAR J. 2012;53:337–354. doi: 10.1093/ilar.53.3-4.341. [DOI] [PMC free article] [PubMed] [Google Scholar]